
Abstract
Advances in medicine have led to more patients being at risk of fungal infections. Diagnostic tools are limited, but early antifungal treatment is crucial to improve outcome. Hence, not a few patients receive empirical antifungals with the disadvantage of increasing the burden of antifungal drug resistance. From a clinical point of view, it is of interest to understand how commonly resistance occurs, how easy it is induced through therapy, and how often it results in clinical treatment failure. The answer differs and depends on the clinical setting, the type of fungal disease, the class of antifungal agent, and treatment duration. This review provides a comprehensive overview on cross-resistance (CR) and multidrug resistance (MR) occurring in Candida species. Known amino acid substitutions are listed which lead to CR (resistance against ≥two azoles or echinocandins), pan-azole resistance (against all systemically applied azoles), pan-echinocandin resistance (against all echinocandins), or MR (polyene-azole resistance, 5-fluorouracil-azole resistance, and azole-echinocandin resistance). Data are supplemented with treatment results from animal studies and experiences from various case reports. An appraisal will be made based on the current frequency of CR and MR reported in the literature, and subsequently, the impact of CR and MR on patient management will be discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungi have become major human pathogens, and the isolation of Candida species less susceptible to current therapies and the recovery of increasingly resistant isolates are growing problems [1•]. Several factors contribute to this epidemiological situation with advances made in medical care resulting in sicker patients being susceptible to fungi [2]. Candida albicans still remains most important, but other non-C. albicans species may result from selective pressures associated with the increased administration of antifungal agents [3••]. Antifungal drug resistance is characterized as microbiological or clinical. Microbiological resistance displays the non-susceptibility of a fungal pathogen to an antifungal agent determined by in vitro susceptibility testing when compared with isolates of the same species. Primary, or intrinsic, resistance refers to an organism’s natural susceptibility to an antimicrobial and reflects to be a predictable trait. This innate level of susceptibility is thought to be a drug-organism characteristic and independent of drug exposure such as given for Candida krusei and fluconazole [4, 5]. Secondary or acquired resistance is much less predictable and potentially more problematic. Under the exposure of antifungal agents, a fungal population initially susceptible may begin to express resistance. It is likely that resistance occurs as the result of several processes, including the emergence of a resistant variant from a common genotype [6], the selection of resistant strains from a mixed population [7], and reinfection with a new resistant strain [8]. The antifungal susceptibility patterns and frequencies of various Candida species isolated vary considerably among institutions and even among units in the same institution. In southern countries such as Italy, Spain, and South America, Candida parapsilosis [9] ranks second, while in northern countries, Candida glabrata takes this position [10]. Clinical resistance refers to infection persistence despite treatment with adequate therapy. Although microbiological resistance can contribute to the development of clinical resistance, other factors may also be involved, such as impaired immune function, underlying disease, reduced drug bioavailability, biofilm formation, and increased drug metabolism [11]. Hence, microbiological resistance is one of the factors underlying clinical resistance but not the most important one. The controlled studies of clinical importance of cross-resistance (CR) and multidrug resistance (MR) are lacking, but irrespective of the pathogen, the issue of CR and MR is most likely to be considered in seriously compromised individuals with invasive fungal infection and extensive exposure to antifungal drugs. This review will focus on clinical relevant ascomycete yeasts and their tendencies to develop antifungal resistance against commonly used compounds for systemic therapy. The term Candida is used in this review in its “applied clinical sense” (were all ascomycete yeasts are pooled in an artificial genus called Candida). The authors are aware that the genus Candida (based on morphological features only) was split up in several taxonomical valid genera which are distantly related [12]. Giving some examples, Candida lusitaniae was renamed to Clavispora lusitaniae and C. krusei to Issatchenkia orientalis, respectively. Further information on up-to-date nomenclature of yeast can be found in a recent publication by Schmalreck et al. [3••]. In the current review, we will mainly focus on MR and CR of the two major species C. albicans and C. glabrata. It is important for clinicians to be aware of trends and mechanisms responsible for the expression of resistance to incorporate this knowledge into up-to-date patient management.
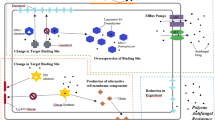
Figure 1 gives an overview on antimycotics, their sites of action, and mechanism of resistance.

Primary targets and resistance mechanisms of major systemic antifungal drugs
Microbial Resistance—a Clinical Issue?
From a clinical point of view, it is of interest to give answers on how commonly resistance occurs, how easy it is induced through therapy, and how often it results in the clinical failure of treatment? The answer differs within the clinical setting [9, 13, 14], type of fungal disease [15, 16], class of antifungal agents [17], and treatment duration [18, 19]. Clearly, we know that exposure to azoles is a significant risk factor for resistance development and that azole treatment leads to the selection of less susceptible species such as C. glabrata and C. krusei; in the past, the latter species predominated superficial infections [20] whereas in these days shift to blood stream infections [10, 21••]. In addition, appearance or disappearance of azole resistance depends on the rate of fungal growth, the number of mutations, or phenotypic changes necessary for resistance [6]. Fluconazole resistance remains uncommon in C. albicans (<5 %) but is more prevalent in C. parapsilosis (4–10 %) and Candida tropicalis (4–9 %) [22]. C. glabrata is a haploid species of Candida that has emerged as the second most common Candida organism associated with fungemia [23]. A likely contributing factor to the rapid growth (2 % in the 1970s to 20 % now) is the robust ability of C. glabrata to acquire tolerance to commonly deployed antifungal agents. Breakthrough fungal infections in bone marrow transplant patients receiving fluconazole prophylaxis [24] were attributed to C. glabrata displaying CR to fluconazole, voriconazole, itraconazole, and posaconazole [8]. Not only does C. glabrata relatively easily converts to an azole-resistant pathogen, but also becomes simultaneously MR.
Despite more than 30 years of clinical use, minimal resistance has developed to amphotericin B, and the drug continues to be important in the treatment of a variety of fungal pathogens. This may be due to its inherently fungicidal effect, limiting the selection of mutants. However, some Candida species including C. lusitaniae, C. glabrata, and Candida guilliermondii are capable of expressing resistance to amphotericin B [1•].
Echinocandins have established themselves as valuable agents for the treatment of candidiasis, and data show resistance to occur primary and secondary to mutation of the FKS1 gene [25]. One survey showed the frequency of C. parapsilosis, a species known for its reduced susceptibility to the echinocandins, to be increased after treatment with caspofungin (13 to 31 %) [13]. Clinical studies display Candida species less susceptible or resistant to caspofungin being more prevalent following treatment (30 days) with the drug (P < 0.001) in the ICU setting [14] and in patients suffering from hematological malignancies [13]; 7 days of exposure to echinocandin is sufficient to induce FKS mutations in C. glabrata [26], whereby the nature and/or the number of FKS mutations in C. glabrata and C. albicans influences in vivo resistance [27••]. FKS mutations were found in 7.9 % of 313 C. glabrata isolates from blood samples, and up to 80 % of patients infected with strains with both FKS mutations and high minimum inhibitory concentrations (MICs) for caspofungin experienced clinical failure or recurrent infection [28].
Epidemiological Cut-off Values and Clinical Breakpoints for Candida Species
The Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) have developed standard susceptibility testing methods for Candida species based on broth microdilution [29–31]. Epidemiological cut-off values (ECOFF) and clinical breakpoints (CBPs) have been developed for MIC interpretation. ECOFF is defined as the upper limit of the wild-type population, thereof discriminates wild-type from resistant strains and is useful to monitor MIC trends. CLSI and EUCAST introduced species-specific clinical breakpoints for azoles and echinocandins, and the classification covers strains being susceptible, susceptible dose dependent, or resistant [32]. However, in vivo and in vitro outcome still is not perfect [33], which in turn renders CBP setting into to a permanent changing process taking into account latest news on MICs and clinical outcome data to optimize patient management. So far, the last correction of CLSI CBPs [5, 4] caused an increase of micafungin-resistant C. glabrata isolates from 0.8 to 7.6 % and of voriconazole-resistant isolates from 6.1 to 18.4 % [34]. Overall, these changes resulted in 5.7 % instead of 2.1 % of all isolates being resistant [34]. Whether the use of revised CBPs may improve the clinical predictive value of in vitro susceptibility tests needs to be validated in more detail.
Cross-Resistance and Multidrug Resistance in Candida Species
CR is defined as resistance that occurs for two or more antifungal substances of one similar chemical class with a similar mode of action, e.g., resistance against ≥two azoles or echinocandins [6]. CR might develop in organisms which have been exposed to the same or similar substance. MR is defined as resistance against structurally unrelated antifungal agents with different cellular targets. MR may emerge by the long-term exposure of structurally unrelated antifungals (e.g., simultaneous azole and echinocandin resistance) or by the interaction of two structurally unrelated agents with linked cellular mechanism (e.g., simultaneous azole and amphotericin B resistance) [6]. CR and MR both are specified on in vitro phenotypes and may be associated with in vivo outcome or therapeutic failures. The terms pan-azole and pan-echinocandin cover resistance against all systemically applied azoles and echinocandins [27••]. Unfortunately, the clinical impact of all various drug-bug profiles is not known due to limited data available.
Cross-Resistance Among Azoles and Echinocandins
CR against the various systemically applied triazole agents such as fluconazole, itraconazole, posaconazole, and voriconazole is well known for Candida species [35] and has been described for anidulafungin, caspofungin, and micafungin [36]. CR was found to be associated with various molecular mechanisms, and most frequently, triazole resistance is associated with point mutations (single nucleotide polymorphisms (SNPs)) in the ERG11 gene, while echinocandin resistance is frequently connected with SNPs in the FKS1p gene [37–39]. Whether SNPs cause silent or missense amino acid mutations is essential for resistance characteristics, as silent mutations to not lead to a change in the amino acid (aa) substitution, while missense mutations do. Also, the differentiation between haploid and diploid yeasts is key for the interpretation of SNPs; as for diploid organisms such as C. albicans, mutations are either heterozygous (affecting only one of both alleles) or homozygous (affecting both alleles). Haploid Candida species (e.g., C. glabrata) mutate more frequently than diploid Candida species (e.g., C. albicans). In practice, a diploid strain carrying a heterozygous mutation still has the capacity to produce the wild-type (WT) protein, while strains with homozygous mutations exclusively produce the mutated protein [27••].
Fluconazole is frequently used for the treatment of invasive candidiasis and candidemia and for prophylaxis in non-neutropenic patients [18]. An intrinsic resistance against fluconazole, e.g., for C. krusei, does not necessarily result in CR against other triazoles, as the majority of this species are susceptible to voriconazole. In contrast, acquired resistance is to a greater extent associated with CR. Pfaller et al. [10] reports of approximately 9.5 % of C. glabrata causing blood stream infections being resistant against fluconazole and voriconazole. Such findings are proved by numerous cases of breakthrough infections and therapeutic failures under azoles [40–44], and a switch to echinocandins was found to be successful in several cases [41, 42]. Resistance against azoles and echinocandins is rare and so far only observed in chronically infected patients receiving antifungal long-term treatment [27••]. A switch to amphotericin B might be less successful as azole-resistant isolates may carry simultaneously resistance against amphotericin B (see chapter azole-amphotericin B MR) [45–48].
For C. albicans, various combinations of CR exist, and among them, ketoconazole/fluconazole, itraconazole/miconazole, fluconazole/clotrimazole/itraconazole, and itraconazole/ketoconazole are most important [25]. Pan-azole resistance is associated with CDR1 and CDR2 overexpression and SNPs in the ERG11 gene encoding for the 14α-sterol demethylase [49, 50]. Other mechanisms are ERG11 overexpression, upregulation of multidrug efflux transporters (including ATP-binding cassette and major facilitator superfamily (MFS) transporters), and a bypass of the ergosterol pathway via accompanying mutations in the Δ5,6-desaturase gene (erg3). Prasad and Singh [51] reviewed the role of lipids involved in cross talks between different cellular circuits that influence the acquisition of multidrug resistance in Candida species. They summarized how the lipid composition of the cell membrane impacts on the localization and function of multidrug transporter proteins (CDR1). Notable is the fact that not all multidrug resistance transporters are affected by cell membrane lipid imbalance; MDR1 remains correctly localized and shows no functional lost. New regulatory circuitries potential impacting the development of multidrug resistance are identified by Dhamgaye et al. [52] using gene profiling and RNA-Seq data.
Only limited information is known for C. tropicalis, but it is speculated that the molecular mechanisms of azole CR are highly similar to those described for C. albicans. Forastiero et al. [46] demonstrated azole CR being related to coding mutations in the ERG11p with or without alternations in the ergosterol biosynthesis pathway. In a case report, Couzigou et al. [37] describes a pan-azole-resistant isolate of Candida kefyr which was found to carry two coding mutations in the ERG11. An overview of ERG11 point mutations that cause pan-azole resistance in Candida isolates is given in Table 1. Pan-azole resistance is not rare to see accompanied by amphotericin B resistance (see chapter azole-amphotericin B multi-resistances).
Breakthrough fungal infections during echinocandin treatment are mainly caused by C. albicans, C glabrata, and C. parapsilosis [19, 21••, 53–57]. An overview of echinocandin resistance and potential treatment strategies was recently published by Beyda et al. [20]. Herein, the authors provide a comprehensive overview on species involved, underlying molecular resistance mechanisms being present and patient’s related outcome [20]. Point mutations in the FKS1p (encoded by the genes FKS1, FKS2, and FKS3) are mainly responsible for echinocandin resistance. Together with the regulatory protein RHO1p, FKS1p (catalytic subunit) forms the 1,3-β-d-glucan synthase [58]. In C. albicans, C. tropicalis, and C. krusei, echinocandin resistance is associated with mutations in the two FKS1 hot spot (hs) regions, hs 1 stretching from aa 641 to 649 and hs 2 stretching from aa 1345 to 1365 [59]. While for C. glabrata in addition to FKS1, also FKS2 hs 1 (aa 659–667) and hs 2 (1374–1381) are involved [59, 54, 60]. One of the most commonly found FKS1p mutation that leads to pan-echinocandin resistance is S645P; this mutation was reported from C. albicans [16] and C. kefyr [61]. In addition, substitutions of F641 [62, 57] or a loss of aa F641 (F641Δ) [61] are among the top mutations in the FKS1p in C. albicans, C. glabrata, and C. kefyr. Heat shock protein 90 upregulation was associated with enhanced echinocandin resistance, especially when accompanied with FKS1 mutation [20]. An overview on currently known FKS1 and FKS2 aa substitutions and their impact on echinocandin resistance is given in Table 2.
Multidrug Resistance Against Azoles and Echinocandins
The most commonly found MR is fluconazole resistance occurring simultaneously with echinocandin resistance. Pfaller et al. [10] found that about 11 % of all fluconazole-resistant C. glabrata isolates were also resistant against echinocandins, all of these carried FKS1 (S629P, R631G, D632Y, or D648E) or FKS2 (F659V, F659Y, S663P, or S663F) mutations. Bizerra et al. [54] reported an isolate being pan-echinocandin, fluconazole, and voriconazole resistant after approximately 3 weeks of exposure to fluconazole and micafungin. Lackner et al. [27••] detected a pan-azole and pan-echinocandin resistant C. albicans from a patient suffering from CMC and receiving azole and echinocandin therapy for longer than 1 year. C. glabrata collected from patients receiving multiple antifungal treatment regimens display major therapeutic challenges [15, 28, 63, 64]. To the best of our knowledge so far, no Candida isolates were described that exhibit both amphotericin B and echinocandin resistance.
Multiresistance Against Azoles and Amphotericin B
In contrast to azoles, amphotericin B targets membrane-bound ergosterol; its high affinity to ergosterol, but low affinity to ergosterol’s precursors (such as lanosterol, fecosterol, lichesterol, and episterol) favors the replacement of ergosterol in the fungal cell membrane by its precursors and thus led to the development of polyene resistance [26]. Major resistance mechanisms of amphotericin B are quantitative and qualitative changes of the ergosterol cell membrane composition; enzyme activity of ERG2, ERG3, and ERG5, or mutations in these ERG2, ERG3, and ERG5, respectively, regulate the ergosterol content [65]. The potential to develop amphotericin B resistance depends on the species but is higher for C. glabrata and C. parapsilosis [26]. In contrast to amphotericin B, azoles inhibit a key enzyme in the biosynthetic pathway of ergosterol, namely lanosterol 14-α demethylase. This enzyme belongs to the P-450 cytochromes, and its catalytic site is the primary target of azoles. The inhibition of this enzyme results in the accumulation of ergosterol precursors in the plasma membrane with the subsequent hampering of the integrity and cellular processes. This azole and amphotericin B action results in reduced cell membrane ergosterol, which in turn explains at least partially MR of Candida to structurally unrelated substances. Other MR mechanisms found to be the upregulation of stress response and transporter and efflux pumps.
The first clinical pan-azole- and polyene-resistant C. albicans with mutations in ERG5 and ERG11 was reported in 2010 by Martel et al. [65]. The isolate showed an aa substitution in ERG11p gene at position A114S and a sequence repetition of 10 nucleotides in ERG5p gene. A nucleotide repetition in ERG5p led to nullified C22 desaturase; as a consequence, fungal cell membrane contained no ergosterol but >80 % of total sterol fraction consisted of ergosta-5,7-dienol [65]. In 2012, a clinical C. glabrata was identified with a missense mutation in ERG11p which leads to CR against fluconazole and voriconazole; in addition, a shift in the sterol composition favored accompanying amphotericin B resistance [47]. C. albicans and C. tropicalis clinical isolates with resistance against amphotericin B and azoles were discovered during a screening study by Eddouzi et al. [66]. The underlying resistance mechanism of C. albicans was explained by the overexpression of a multidrug efflux pump of the major facilitator superfamily Mdr1. C. tropicalis lacked ergosterol in its cell membrane, instead 14α-methyl-fecosterol was accumulated which indicates the functional perturbation of at least two main ergosterol biosynthesis proteins (ERG11 and ERG3).
Multi-Resistance Against Azoles and 5-Fluorouracil
Gabriel et al. [67] showed that the simultaneous application of fluorinated nucleotides (e.g., 5-fluorouracil) at subinhibitory doses and fluconazole triggers resistance against fluconazole in vitro. The authors speculated that intracellular fluorinated nucleotides may play a role in azole resistance by either preventing azoles to target the lanosterol 14-alpha-demethylase, or by preventing azoles to bind to the lanosterol 14-alpha-demethylase catalytic site, or by acting as molecular switch for triggering the efflux transport. These data remain to be verified in greater detail to finally state the molecular mechanism behind. Moreover, these findings need to be validated in murine studies.
Conclusion
Reports of antifungal drug resistance are emerging and a matter of serious concern (see Fig. 2). Positive is the fact that antifungal treatment is still successful, as 80 % of C. albicans infections in ICUs are cleared with echinocandins [9]. However, CR as well as MR may be associated with worse clinical outcome, breakthrough fungal infections [68], multiple changes of treatment regimens and increased health care costs. Clinical improvement failed in patients infected with fluconazole and voriconazole-resistant Candida isolates when compared to susceptible strains [8]; similar findings are valid for resistance to echinocandins among various Candida species [69–71]. Most worrying is the emergence of acquired resistance of C. glabrata against the azoles and echinocandins; the limited number of antifungals renders these phenotypes to an emerging pathogen.

Common problems associated with antifungal drug resistant pathogens
Limited data are available on the economic impact of resistant Candida infections. However, it has been calculated that fungal infections add a total of US$8 billion to annual health care costs [72]. Resistant infections are thought to substantially increase these expenditures because of reinforced patients’ management consisting of a prolonged therapy, change of drug regimen applied, or rather using a combination, intense diagnostic procedures such as biopsies, as well as isolation procedures. Strategies for preventing the emergence and spread of antifungal drug resistance include the implementation of Antimicrobial Stewardship Programs covering (i) local fungal epidemiology and antifungal resistance rates, (ii) establishing therapeutic guidelines, (iii) implementation of treatment strategies for empirical and preemptive therapy including PK/PD data, (iv) catheter management, and (v) selection of adequate diagnostic assays.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Arendrup MC. Update on antifungal resistance in Aspergillus and Candida. Clin Microbiol Infect. 2014;20 Suppl 6:42–8. doi:10.1111/1469-0691.12513. This article is of special interest as it provides a comprehensive overview on modified clinical breakpoints for Aspergillus and Candida.
Glockner A. Treatment and prophylaxis of invasive candidiasis with anidulafungin, caspofungin and micafungin: review of the literature. Eur J Med Res. 2011;16(4):167–79.
Schmalreck AF, Lackner M, Becker K, et al. Phylogenetic relationships matter: antifungal susceptibility among clinically relevant yeasts. Antimicrob Agents Chemother. 2014;58(3):1575–85. This article summarizes all currently accepted changes in nomenclature among clinically relevant ascomycetous yeasts. Moreover, a correlation between the evolutionary relatedness of yeast and their antifungal susceptibility profiles.
Pfaller MA, Diekema DJ, Andes D, et al. Clinical breakpoints for the echinocandins and Candida revisited: integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat. 2011;14(3):164–76.
Pfaller MA, Andes D, Arendrup MC, et al. Clinical breakpoints for voriconazole and Candida spp. revisited: review of microbiologic, molecular, pharmacodynamic, and clinical data as they pertain to the development of species-specific interpretive criteria. Diagn Microbiol Infect Dis. 2011;70(3):330–43.
White TC, Marr KA, Bowden RA. Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin Microbiol Rev. 1998;11:382–402.
Arendrup MC, Garcia-Effron G, Buzina W, et al. Breakthrough Aspergillus fumigatus and Candida albicans double infection during caspofungin treatment: laboratory characteristics and implication for susceptibility testing. Antimicrob Agents Chemother. 2009;53(3):1185–93.
Pfaller MA. Antifungal drug resistance: mechanisms, epidemiology, and consequences for treatment. Am J Med. 2012;125:S3–13.
Maubon D, Garnaud C, Calandra T, et al. Resistance of Candida spp. to antifungal drugs in the ICU: where are we now? Intensiv Care Med. 2014;40(9):1241–55.
Pfaller MA, Castanheira M, Lockhart SR, et al. Frequency of decreased susceptibility and resistance to echinocandins among fluconazole-resistant bloodstream isolates of Candida glabrata. J Clin Microbiol. 2012;50(4):1199–203.
Rex JH, Pfaller MA, Galgiani JN, et al. Development of interpretive breakpoints for antifungal susceptibility testing: conceptual framework and analysis of in vitro-in vivo correlation data for fluconazole, itraconazole, and Candida infections. Subcommittee on Antifungal Susceptibility Testing of the National Committee for Clinical Laboratory Standards. Clin Infect Dis. 1997;24(2):235–47.
de Hoog GS et al. Name changes in medically important fungi and their implication on clinical practice. J Clin Microbiol. 2014; accepted for publication.
Lortholary O, Desnos-Ollivier M, Sitbon K, et al. Recent exposure to caspofungin or fluconazole influences the epidemiology of candidemia: a prospective multicenter study involving 2,441 patients. Antimicrob Agents Chemother. 2011;55:532–8.
Blanchard E, Lortholary O, Boukris-Sitbon K, et al. Prior caspofungin exposure in patients with hematological malignancies is a risk factor for subsequent fungemia due to decreased susceptibility in Candida spp.: a case-control study in Paris, France. Antimicrob Agents Chemother. 2011;55:5358–61.
Pfeiffer CD, Garcia-Effron G, Zaas AK, et al. Breakthrough invasive candidiasis in patients on micafungin. J Clin Microbiol. 2010;48(7):2373–80.
Slater JL, Howard SJ, Sharp A, et al. Disseminated Candidiasis caused by Candida albicans with amino acid substitutions in FKS1 at position Ser645 cannot be successfully treated with micafungin. Antimicrob Agents Chemother. 2011;55(7):3075–83.
Spreghini E, Orlando F, Sanguinetti M, et al. Comparative effects of micafungin, caspofungin, and anidulafungin against a difficult-to-treat fungal opportunistic pathogen, Candida glabrata. Antimicrob Agents Chemother. 2012;56(3):1215–22.
Tripathi N, Watt K, Benjamin Jr DK. Treatment and prophylaxis of invasive candidiasis. Semin Perinatol. 2012;36(6):416–23.
Ruggero MA, Topal JE. Development of echinocandin-resistant Candida albicans candidemia following brief prophylactic exposure to micafungin therapy. Transpl Infect Dis. 2014;16(3):469–72.
Beyda ND, Lewis RE, Garey KW. Echinocandin resistance in Candida species: mechanisms of reduced susceptibility and therapeutic approaches. Ann Pharmacother. 2012;46(7–8):1086–96.
Fekkar A, Dannaoui E, Meyer I, et al. Emergence of echinocandin-resistant Candida spp. in a hospital setting: a consequence of 10 years of increasing use of antifungal therapy? Eur J Clin Microbiol Infect Dis. 2014;33(9):1489–96. The article provides real-life clinical experience on fungal prophylaxis and its association with the development of antifungal resistances among Candida species.
Cavling Arendrup M, Cuenca-Estrella M, Lass-Florl C, Hope WW. European Committee on Antimicrobial Susceptibility Testing—Subcommittee on Antifungal Susceptibility T. EUCAST technical note on Candida and micafungin, anidulafungin and fluconazole. Mycoses. 2014;57(6):377–9.
Bizerra FC, Jimenez-Ortigosa C, Souza ACR, et al. Breakthrough candidemia due to multidrug resistant C. glabrata during prophylaxis with low dose of micafungin. Antimicrob Agents Chemother. 2014;58:2438–40.
Alexander BD, Schell WA, Miller JL, et al. Candida glabrata fungemia in transplant patients receiving voriconazole after fluconazole. Transplantation. 2005;80:868–71.
White TC, Holleman S, Dy F, et al. Resistance mechanisms in clinical isolates of Candida albicans. Antimicrob Agents Chemother. 2002;46(6):1704–13.
Sheikh N, Jahagirdar V, Kothadia S, Nagoba B. Antifungal drug resistance in Candida species. Eur J Gen Med. 2013;10(4):254–8.
Lackner M, Tscherner M, Schaller M, et al. Positions and numbers of FKS mutations in Candida albicans selectively influence in vitro and in vivo susceptibilities to echinocandin treatment. Antimicrob Agents Chemother. 2014;58(7):3626–35. This article shows the difference between heterozygote and homozygote mutations and their impact on in vitro and in vivo resistance. Moreover, it demonstrates that resistance is acquired during long-term therapy in chronically infected patients.
Pfaller MA, Messer SA, Moet GJ, et al. Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in Intensive Care Unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008–2009). Int J Antimicrob Agents. 2011;38(1):65–9.
Pfaller MA, Diekema DJ. Progress in antifungal susceptibility testing of Candida spp. by use of Clinical and Laboratory Standards Institute broth microdilution methods, 2010 to 2012. J Clin Microbiol. 2012;50(9):2846–56.
Subcommittee on Antifungal Susceptibility Testing (AFST) of the ESCMID European Committee for Antimicrobial Susceptibility Testing (EUCAST). EUCAST definitive document EDef 7.1: method for the determination of broth dilution MICs of antifungal agents for fermentative yeasts. Clin Microbiol Infect. 2008;14(4):398–405.
Clinical and Laboratory Standards Institute. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard. 3rd ed. CLSI document M27-A3. Wayne: Clinical and Laboratory Standards Institute; 2008
van Hal SJ, Chen SC, Sorrell TC, et al. Support for the EUCAST and revised CLSI fluconazole clinical breakpoints by Sensititre® YeastOne® for Candida albicans: a prospective observational cohort study. J Antimicrob Chemother. 2014;69(8):2210–4.
Eschenauer GA, Nguyen MH, Shoham S, et al. Real-world experience with echinocandin MICs against Candida species in a multicenter study of hospitals that routinely perform susceptibility testing of bloodstream isolates. Antimicrob Agents Chemother. 2014;58(4):1897–906.
Fothergill AW, Sutton DA, McCarthy DI, Wiederhold NP. Impact of new antifungal breakpoints on antifungal resistance in Candida species. J Clin Microbiol. 2014;52(3):994–7.
Chen TC, Chen YH, Chen YC, Lu PL. Fluconazole exposure rather than clonal spreading is correlated with the emergence of Candida glabrata with cross-resistance to triazole antifungal agents. Kaohsiung J Med Sci. 2012;28(6):306–15.
Pham CD, Iqbal N, Bolden CB, et al. Role of FKS Mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob Agents Chemother. 2014;58(8):4690–6.
Couzigou C, Gabriel F, Biteau N, et al. Two missense mutations, E123Q and K151E, identified in the ERG11 allele of an azole-resistant isolate of Candida kefyr recovered from a stem cell transplant patient for acute myeloid leukemia. Med Mycol Case Rep. 2014;5:12–5.
Ricardo E, Miranda IM, Faria-Ramos I, et al. In vivo and in vitro acquisition of resistance to voriconazole by Candida krusei. Antimicrob Agents Chemother. 2014;58(8):4604–11.
Strzelczyk JK, Slemp-Migiel A, Rother M, et al. Nucleotide substitutions in the Candida albicans ERG11 gene of azole-susceptible and azole-resistant clinical isolates. Acta Biochim Pol. 2013;60(4):547–52.
Chong Y, Shimoda S, Yakushiji H, et al. Fatal candidemia caused by azole-resistant Candida tropicalis in patients with hematological malignancies. J Infect Chemother. 2012;18(5):741–6.
Fanci R. Breakthrough Candida dubliniensis fungemia in an acute myeloid leukemia patient during voriconazole therapy successfully treated with caspofungin. J Chemother. 2009;21(1):105–7.
Krcmery V, Demitrovicova A, Kisac P. Breakthrough fungemia due to Candida glabrata during posaconazole prophylaxis in hematology patients treated with anidulafungin—report of 5 cases. J Chemother. 2011;23(5):310–1.
Myoken Y, Kyo T, Sugata T, et al. Breakthrough fungemia caused by fluconazole-resistant Candida albicans with decreased susceptibility to voriconazole in patients with hematologic malignancies. Haematologica. 2006;91(2):287–8.
Trifilio S, Singhal S, Williams S, et al. Breakthrough fungal infections after allogeneic hematopoietic stem cell transplantation in patients on prophylactic voriconazole. Bone Marrow Transplant. 2007;40(5):451–6.
Linares CE, Giacomelli SR, Altenhofen D, et al. Fluconazole and amphotericin-B resistance are associated with increased catalase and superoxide dismutase activity in Candida albicans and Candida dubliniensis. Rev Soc Bras Med Trop. 2013;46(6):752–8.
Forastiero A, Mesa-Arango AC, Alastruey-Izquierdo A, et al. Candida tropicalis antifungal cross-resistance is related to different azole target (ERG11p) modifications. Antimicrob Agents Chemother. 2013;57(10):4769–81.
Hull CM, Parker JE, Bader O, et al. Facultative sterol uptake in an ergosterol-deficient clinical isolate of Candida glabrata harboring a missense mutation in ERG11 and exhibiting cross-resistance to azoles and amphotericin B. Antimicrob Agents Chemother. 2012;56(8):4223–32.
Sanglard D, Ischer F, Monod M, Bille J. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob Agents Chemother. 1996;40(10):2300–5.
Dogra S, Krishnamurthy S, Gupta V, et al. Asymmetric distribution of phosphatidylethanolamine in C. albicans: possible mediation by CDR1, a multidrug transporter belonging to ATP binding cassette (ABC) superfamily. Yeast. 1999;15(2):111–21.
Maesaki S, Marichal P, Vanden Bossche H, et al. Rhodamine 6G efflux for the detection of CDR1-overexpressing azole-resistant Candida albicans strains. J Antimicrob Chemother. 1999;44(1):27–31.
Prasad R, Singh A. Lipids of Candida albicans and their role in multidrug resistance. Curr Genet. 2013;59(4):243–50.
Dhamgaye S, Bernard M, Lelandais G, et al. RNA sequencing revealed novel actors of the acquisition of drug resistance in Candida albicans. BMC Genomics. 2012;13:396.
Wilke M. Treatment and prophylaxis of invasive candidiasis with anidulafungin, caspofungin and micafungin and its impact on use and costs: review of the literature. Eur J Med Res. 2011;16(4):180–6.
Bizerra FC, Jimenez-Ortigosa C, Souza AC, et al. Breakthrough candidemia due to multidrug-resistant Candida glabrata during prophylaxis with a low dose of micafungin. Antimicrob Agents Chemother. 2014;58(4):2438–40.
Chan TS, Gill H, Hwang YY, et al. Breakthrough invasive fungal diseases during echinocandin treatment in high-risk hospitalized hematologic patients. Ann Hematol. 2014;93(3):493–8.
Chrenkova V, Hubacek P, Sedlacek P, et al. Post-mortem analysis of Candida albicans breakthrough infection during echinocandin treatment in haematopoietic stem cell transplant recipient. Epidemiol Mikrobiol Imunol. 2014;63(2):121–4.
Fekkar A, Meyer I, Brossas JY, et al. Rapid emergence of echinocandin resistance during Candida kefyr fungemia treatment with caspofungin. Antimicrob Agents Chemother. 2013;57(5):2380–2.
Garcia-Effron G, Lee S, Park S, et al. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-D-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob Agents Chemother. 2009;53(9):3690–9.
Park S, Kelly R, Kahn JN, et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother. 2005;49(8):3264–73.
Katiyar SK, Alastruey-Izquierdo A, Healey KR, et al. FKS1 and FKS2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob Agents Chemother. 2012;56(12):6304–9.
Staab JF, Neofytos D, Rhee P, et al. Target enzyme mutations confer differential echinocandin susceptibilities in Candida kefyr. Antimicrob Agents Chemother. 2014;58(9):5421–7.
Garcia-Effron G, Katiyar SK, Park S, Edlind TD, Perlin DS. A naturally occurring proline-to-alanine amino acid change in FKS1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob Agents Chemother. 2008;52(7):2305–12.
Chapeland-Leclerc F, Hennequin C, Papon N, et al. Acquisition of flucytosine, azole, and caspofungin resistance in Candida glabrata bloodstream isolates serially obtained from a hematopoietic stem cell transplant recipient. Antimicrob Agents Chemother. 2010;54(3):1360–2.
Sun HY, Singh N. Characterisation of breakthrough invasive mycoses in echinocandin recipients: an evidence-based review. Int J Antimicrob Agents. 2010;35(3):211–8.
Martel CM, Parker JE, Bader O, et al. A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14alpha-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob Agents Chemother. 2010;54(9):3578–83.
Eddouzi J, Parker JE, Vale-Silva LA, et al. Molecular mechanisms of drug resistance in clinical Candida species isolated from Tunisian hospitals. Antimicrob Agents Chemother. 2013;57(7):3182–93.
Gabriel F, Sabra A, El-Kirat-Chatel S, et al. Deletion of the uracil permease gene confers cross-resistance to 5-fluorouracil and azoles in Candida lusitaniae and highlights antagonistic interaction between fluorinated nucleotides and fluconazole. Antimicrob Agents Chemother. 2014;58(8):4476–85.
Pfeiffer CD, Garcia-Effron G, Zaas AK, et al. Breakthrough invasive candidiasis in patients on micafungin. J Clin Microbiol. 2010;48:2373–80.
Hakki M, Staab JF, Marr KA. Emergence of a Candida krusei isolate with reduced susceptibility to caspofungin during therapy. Antimicrob Agents Chemother. 2006;50:2522–4.
Krogh-Madsen M, Arendrup MC, Heslet L, Knudsen JD. Amphotericin B and caspofungin resistance in Candida glabrata isolates recovered from a critically ill patient. Clin Infect Dis. 2006;42:938–44.
Cleary JD, Garcia-Effron G, Chapman SW, Perlin DS. Reduced Candida glabrata susceptibility secondary to an FKS1 mutation developed during candidemia treatment. Antimicrob Agents Chemother. 2008;52:2263–5.
Cleveland AA, Farley MM, Harrison LH, et al. Changes in incidence and antifungal drug resistance in candidemia: results from population-based laboratory surveillance in Atlanta and Baltimore, 2008–2011. Clin Infect Dis. 2012;55:1352–61.
Cernicka J, Subik J. Resistance mechanisms in fluconazole-resistant Candida albicans isolates from vaginal candidiasis. Int J Antimicrob Agents. 2006;27(5):403–8.
Chau AS, Mendrick CA, Sabatelli FJ, et al. Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azoles. Antimicrob Agents Chemother. 2004;48(6):2124–31.
Wang H, Kong F, Sorrell TC, et al. Rapid detection of ERG11 gene mutations in clinical Candida albicans isolates with reduced susceptibility to fluconazole by rolling circle amplification and DNA sequencing. BMC Microbiol. 2009;14(9):167.
Feng LJ, Wan Z, Wang XH, et al. Relationship between antifungal resistance of fluconazole resistant Candida albicans and mutations in ERG11 gene. Chin Med J (Engl). 2010;123(5):544–8.
Favre B, Didmon M, Ryder NS. Multiple amino acid substitutions in lanosterol 14alpha-demethylase contribute to azole resistance in Candida albicans. Microbiology. 1999;145(10):2715–25.
Löffler J, Kelly SL, Hebart H, et al. Molecular analysis of CYP51 from fluconazole-resistant Candida albicans strains. FEMS Microbiol Lett. 1997;151(2):263–8.
Xiang MJ, Liu JY, Ni PH, et al. ERG11 mutations associated with azole resistance in clinical isolates of Candida albicans. FEMS Yeast Res. 2013;13(4):386–93.
Oliveira Carvalho V, Okay TS, Melhem MS, et al. The new mutation L321F in Candida albicans ERG11 gene may be associated with fluconazole resistance. Rev Iberoam Micol. 2013;30(3):209–12.
Morio F, Loge C, Besse B, et al. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn Microbiol Infect Dis. 2010;66(4):373–84.
Marichal P, Koymans L, Willemsens S, et al. Contribution of mutations in the cytochrome P450 14alpha-demethylase (ERG11p, CYP51p) to azole resistance in Candida albicans. Microbiology. 1999;145(Pt10):2701–13.
Manastir L, Ergon MC, Yücesoy M. Investigation of mutations in ERG11 gene of fluconazole resistant Candida albicans isolates from Turkish hospitals. Mycoses. 2011;54(2):99–104.
Goldman GH, Da Silva Ferreira ME, dos Reis Marques E, et al. Evaluation of fluconazole resistance mechanisms in Candida albicans clinical isolates from HIV-infected patients in Brazil. Diagn Microbiol Infect Dis. 2004;50(1):25–32.
Sanglard D, Ischer F, Koymans L, Bille J. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob Agents Chemother. 1998;42(2):241–53.
Ying Y, Zhao Y, Hu X, et al. In vitro fluconazole susceptibility of 1,903 clinical isolates of Candida albicans and the identification of ERG11 mutations. Microb Drug Resist. 2013;19(4):266–73.
Arendrup MC, Cuenca-Estrella M, Lass-Flörl C, Hope WW. Breakpoints for antifungal agents: an update from EUCAST focussing on echinocandins against Candida spp. and triazoles against Aspergillus spp. Drug Resist Updat. 2013;16(6):81–95.
Garcia-Effron G, Chua DJ, Tomada JR, et al. Novel FKS mutations associated with echinocandin resistance in Candida species. Antimicrob Agents Chemother. 2010;54(5):2225–7.
Jensen RH, Johansen HK, Arendrup MC. Stepwise development of a homozygous S80P substitution in FKS1p, conferring echinocandin resistance in Candida tropicalis. Antimicrob Agents Chemother. 2013;57(1):614–7.
Pfaller MA, Diekema DJ, Jones RN, Castanheira M. Use of anidulafungin as a surrogate marker to predict susceptibility and resistance to caspofungin among 4,290 clinical isolates of Candida using CLSI methods and interpretive criteria. J Clin Microbiol. 2014;52(9):3223–9.
Dannaoui E, Desnos-Ollivier M, Garcia-Hermoso D, et al. Candida spp. with acquired echinocandin resistance, France, 2004–2010. Emerg Infect Dis. 2012;18(1):86–90.
Desnos-Ollivier M, Bretagne S, Raoux D, et al. Mutations in the FKS1 gene in Candida albicans, C. tropicalis, and C. krusei correlate with elevated caspofungin MICs uncovered in AM3 medium using the method of the European Committee on Antibiotic Susceptibility Testing. Antimicrob Agents Chemother. 2008;52(9):3092–8.
Zimbeck AJ, Iqbal N, Ahlquist AM, et al. FKS mutations and elevated echinocandin MIC values among Candida glabrata isolates from U.S. population-based surveillance. Antimicrob Agents Chemother. 2010;54(12):5042–7.
Fernandez-Silva F, Lackner M, Capilla J, et al. In vitro Antifungal susceptibility of Candida glabrata to caspofungin and the presence of FKS mutations correlate with treatment response in an immunocompromised murine model of invasive infection. Antimicrob Agents Chemother. 2014;58(7):3646–9.
Cleary JD, Garcia-Effron G, Chapman SW, Perlin DS. Reduced Candida glabrata susceptibility secondary to an FKS1 mutation developed during candidemia treatment. Antimicrob Agents Chemother. 2008;52(6):2263–5.
Castanheira M, Woosley LN, Diekema DJ, et al. Low prevalence of FKS1 hot spot 1 mutations in a worldwide collection of Candida strains. Antimicrob Agents Chemother. 2010;54(6):2655–9.
Shields RK, Nguyen MH, Press EG, et al. The presence of an FKS mutation rather than MIC is an independent risk factor for failure of echinocandin therapy among patients with invasive candidiasis due to Candida glabrata. Antimicrob Agents Chemother. 2012;56(9):4862–9.
Garcia-Effron G, Kontoyiannis DP, Lewis RE, Perlin DS, et al. Caspofungin-resistant Candida tropicalis strains causing breakthrough fungemia in patients at high risk for hematologic malignancies. Antimicrob Agents Chemother. 2008;52(11):4181–3.
Garcia-Effron G, Park S, Perlin DS. Correlating echinocandin MIC and kinetic inhibition of FKS1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. Antimicrob Agents Chemother. 2009;53(1):112–22.
Laverdiere M, Lalonde RG, Baril JG, et al. Progressive loss of echinocandin activity following prolonged use for treatment of Candida albicans oesophagitis. J Antimicrob Chemother. 2006;57(4):705–8.
Prigitano A, Esposito MC, Cogliati M, et al. Acquired echinocandin resistance in a Candida krusei blood isolate confirmed by mutations in the FKS1 gene. New Microbiol. 2014;37(2):237–40.
Costa-de-Oliveira S, Marcos Miranda I, Silva RM, et al. FKS2 mutations associated with decreased echinocandin susceptibility of Candida glabrata following anidulafungin therapy. Antimicrob Agents Chemother. 2011;55(3):1312–4.
Jensen RH, Justesen US, Rewes A, Perlin DS, Arendrup MC. Echinocandin failure case due to a previously unreported FKS1 mutation in Candida krusei. Antimicrob Agents Chemother. 2014;58(6):3550–2.
Arendrup MC, Perlin DS, Jensen RH, et al. Differential in vivo activities of anidulafungin, caspofungin, and micafungin against Candida glabrata isolates with and without FKS resistance mutations. Antimicrob Agents Chemother. 2012;56(5):2435–42.
Vermitsky JP, Edlind TD. Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a PDR1-like transcription factor. Antimicrob Agents Chemother. 2004;48(10):3773–81.
Acknowledgement
This work was supported by EraNet funding (Austrian Science Fund, Project ZFI006560/AspBIOmics).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
M. Lackner has received honoraria for invited talks by the pharmaceutical company Forest Pharmaceuticals. In the past 5 years, C. Lass-Flörl has received grant support from the Austrian Science Fund (FWF), MFF Tirol, Astellas Pharma, Gilead Sciences, Pfizer, Schering Plough, and Merck Sharp & Dohme. She has been an advisor/consultant to Gilead Sciences, Merck Sharp & Dohme, Pfizer, and Schering Plough. She has received travel/accommodation expenses from Gilead Sciences, Merck Sharp & Dohme, Pfizer, Astellas, and Schering Plough and has been paid for talks on behalf of Gilead Sciences, Merck Sharp & Dohme, Pfizer, Astellas, and Schering Plough. A. Martin-Vicente has no potential conflict of interest to state.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Current Management of Fungal Infections
Rights and permissions
About this article

Cite this article
Lackner, M., Martin-Vicente, A. & Lass-Flörl, C. Multidrug- and Cross-Resistant Candida: the Looming Threat. Curr Fungal Infect Rep 9, 23–36 (2015). https://doi.org/10.1007/s12281-014-0210-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12281-014-0210-1