
Abstract
Gefitinib, as the first-generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI), has achieved great advances in the treatment of non-small cell lung cancer (NSCLC), but drug resistance will inevitably occur. Therefore, exploring the resistance mechanism of gefitinib and developing new combination treatment strategies are of great importance. In our study, the results showed that selumetinib (AZD6244) synergistically inhibited the proliferation of NSCLC with gefitinib. Selumetinib also enhanced gefitinib-induced apoptosis and migration inhibition ability in gefitinib-resistant lung cancer cell lines. Subsequently, the negative regulation between MIG6 and STAT3 was observed and verified through the STRING database and western blotting assays. Sustained activation of STAT3 was significantly downregulated when co-treatment with selumetinib in gefitinib-resistant cells. However, the downregulation of p-STAT3, resulting from the combination of selumetinib and gefitinib was counteracted by the deletion of MIG6, suggesting that selumetinib enhanced gefitinib sensitivity by regulating MIG6/STAT3 in NSCLC. In contrast, p-STAT3 was further inhibited after treatment with gefitinib and selumetinib when MIG6 was overexpressed. Furthermore, the combined administration of selumetinib and gefitinib effectively promoted the sensitivity of lung cancer xenografts to gefitinib in vivo, and the tumor inhibition rate reached 81.49%, while the tumor inhibition rate of the gefitinib monotherapy group was only 31.95%. Overall, MIG6/STAT3 negative regulation plays an important role in the sustained activation of STAT3 and the resistance to EGFR-TKIs. Our study also suggests that EGFR-TKIs combined with MEK1/2 inhibitors, such as selumetinib, may be beneficial to those NSCLC patients who develop a primary or acquired resistance to EGFR-TKIs, providing theoretical support for combining TKIs and selumetinib in clinical cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer is still one of the types of malignant tumors with the highest incidence and mortality. The treatment of lung cancer is very challenging due to its difficulty in detection, ease of metastasis, and poor prognosis. The average five-year survival rate for lung cancer patients is approximately 15% (Sung et al. 2021). Therefore, it is urgent to discover effective drugs to treat lung cancer. In recent years, molecular targeted therapy has achieved significant efficacy in advanced non-small cell lung cancer (NSCLC) patients with driver genes. Currently, the most promising and representative example at the clinical level is targeted therapy of the epidermal growth factor receptor (EGFR) (Cooper et al. 2022). Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) are small-molecule EGFR inhibitors that can inhibit the activation of tyrosine kinase, and then block the EGFR signaling pathway by competitively binding EGFR to endogenous ligands, eventually producing a series of biological effects such as the proliferation and metastasis of tumor cells. Although three generations of EGFR inhibitors have been developed in recent years, including first-generation TKIs gefitinib and erlotinib, second-generation TKI afatinib, and third-generation TKI osimertinib, the occurrence of acquired resistance is inevitable, and the mechanisms of resistance are complex and incompletely understood (He et al. 2021).
Gefitinib, a first-generation EGFR-TKI, is the first small-molecule targeted drug for the treatment of advanced non-small cell lung cancer (NSCLC) that competitively binds to the ATP pocket of EGFR by simulating an adenine ring. Although gefitinib has achieved remarkable efficacy in the clinical treatment of lung cancer, drug resistance will inevitably occur (Sun et al. 2020). At the early stage of clinical treatment, the response rate of some NSCLC patients to gefitinib was disappointing, only approximately 4-23% (Köhler and Schuler 2013), which was caused by primary drug resistance. Most patients with partial EGFR mutations, including EGFR-L858R mutation and EGFR Del E746_A750 mutation, had better responses. These mutations were also called EGFR-sensitive mutations due to the increased binding force of EGFR and gefitinib. However, surprisingly, patients who initially responded to gefitinib also eventually developed secondary resistance to gefitinib after about 9-12 months (Pan et al. 2022). Therefore, the emergence of drug resistance is a great challenge for the clinical treatment of NSCLC, an important cause of clinical treatment failure of small molecule drugs, and also an urgent problem that needs to be solved at present.
The mechanism of drug resistance is a multigene, multilayer, and multidimensional complex process (Ramirez et al. 2016; Huang and Fu 2015). EGFR mutation, K-Ras mutation, and continued activation of STAT3 are the main mechanisms leading to primary EGFR-TKI resistance (Huang and Fu 2015). Abnormally activated STAT3 has been detected in a variety of tumors and is closely associated with poor survival and prognosis in tumor patients (Gu et al. 2020). Elevated STAT3 activity has been demonstrated in approximately 40–60% of non-small cell lung cancers (Harada et al. 2014). Changes in upstream STAT3 signaling, such as EGFR mutation or upregulation, Src overexpression, and abnormal activation of cytokines/gp130/JAK, can induce continued activation of STAT3 (Kandala and Srivastava 2012). The activity of STAT3 is regulated by upstream activated proteins and some endogenous negative regulatory proteins under normal physiological conditions. These negative regulatory proteins include the cytokine signaling inhibitor family, tyrosine phospholipases (PTPs), and the activated STAT3 inhibitor protein.
Previously, we showed that dual inhibition of FGFR and STAT3 is effective against gefitinib-resistant NSCLC (Song et al. 2021), indicating that combination therapy may be an effective strategy for overcoming gefitinib resistance. Selumetinib (AZD6244), is a potent, selective, non-ATP-competitive oral mitogen-activated protein kinase (MEK) 1/2 inhibitor (Yoon et al. 2012). Selumetinib is not presently approved for use in any clinical indication in humans, but its efficacy in combination with other chemotherapeutic agents for the treatment of tumors has been demonstrated in clinical studies (Hedayat et al. 2023; Carvajal et al. 2018). A phase III trial investigated the efficacy and safety of selumetinib plus docetaxel in patients harboring KRAS-mutant NSCLC patients (Das 2017). It was reported that the JAK2/STAT3 inhibitor AG490 synergistically increased the effects of AZD6244 on colon cancer in vitro and in vivo, providing a rationale for combining inhibitors of the JAK/STAT pathway and MEK inhibitors to reduce the potential impact of drug resistance (Jin et al. 2019). The combination of EGFR and MEK1/2 inhibitors showed synergistic effects in human gastric cancer cells and TNBC cell lines (Yoon et al. 2009; Maiello et al. 2015). The expression of ERRFI1(MIG6) was increased as a result of AZD6244 treatment in LoVo and SNU668 cell lines that contained wild-type EGFR and mutant KRAS. Treatment with a combination of an EGFR inhibitor and AZD6244 synergistically inhibited cell proliferation without activating AKT in AZD6244-resistant cells (Yoon et al. 2012).
In this study, to expand our understanding of the MEK inhibitor selumetinib plus the EGFR inhibitor gefitinib, we conducted in vitro and in vivo studies using three gefitinib-resistant lung cancer cell lines with EGFR mutation types consistent with those in clinical lung cancer patients. We demonstrate that selumetinib and gefitinib synergistically inhibit the proliferation of NSCLC, and enhance gefitinib-induced apoptosis and migration inhibition. Moreover, selumetinib overcomes gefitinib resistance by regulating MIG6/STAT3 in NSCLC. The results support the feasibility of combination therapy and the utility of selumetinib for NSCLC therapeutics.
Materials and methods
Chemicals and reagents
Gefitinib (S31076) was obtained from Shanghai Yuanye Bio-Technology Co., Ltd. Selumetinib (AZD6244, S125580) was purchased from Aladdin (Shanghai, China). DMSO (ST038) and RIPA lysis buffer (Lot#: P0013B) were purchased from Beyotime Biotechnology (Shanghai, China).
Cell culture
NCI-H1975 cells, A549 cells, and PC-9 cells were cultured in RPMI-1640 (Gibco, SH30809.01, Fisher Scientific, Hampton, NH, USA) and maintained in a medium supplemented with 10% FBS and 1% penicillin–streptomycin and propagated as monolayer cultures at 37 °C in a humidified 5% CO2 incubator. All the cells were kindly provided by Cell Bank, Chinese Academy of Sciences. Gefitinib-resistant PC9/GR cells were selected from a subculture that had acquired resistance to gefitinib using the concentration gradient method. PC-9 cells were exposed to 0.05 μM gefitinib and the medium was changed every 3 days until no dead cells were found. The concentration of gefitinib was increased gradually until the cells no longer died at 2 μM gefitinib. After about 30 days, the resistant sub-cloned cells were harvested.
Cell viability assay
A modified tetrazolium salt assay was used to measure the inhibition of cancer cell growth. Cancer cells were plated in a 96-well microtiter plate containing 100 μL of growth media per well and incubated for 24 h. Then, the cells were treated with gefitinib or selumetinib at the indicated concentrations for 72 h. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma–Aldrich, MO, USA) was added to each well of the 96-well plate. After 4 h of incubation, the formazan crystal was dissolved and spectrophotometrically quantified at a wavelength of 570 nm (Bio-Tek Instruments, Inc., Winooski, VT, USA). All the experimental data were obtained from six replicates, and all the experiments were repeated at least four times. The inhibition rate of each sample was calculated based on the A570 values as follows: % inhibition rate = (A570 nm control-treated cells/A570 nm control × 100%). The IC50 was defined as the concentration that led to 50% inhibition of cell viability.
Synergism and CI analyses: cells were seeded in quadruplicate in 96-well plates and treated with gefitinib or selumetinib alone or with the combination of gefitinib and selumetinib at the indicated doses. MTT assays were performed after 72 h of treatment. The synergistic effectiveness of the inhibitors was analyzed by using CompuSyn software (Comboyn Inc., Paramus, NJ, USA). CI < 1 indicates synergistic effects, CI > 1 indicates antagonistic effects, and CI = 1 indicates additive effects between the two drugs. The methodology behind the synergistic effect analysis was previously described (Chou 2010).
Cell clone formation assay
Cell clone formation ability was tested using plate clone formation assay. A549, NCI-H1975 cells, and PC9/GR cells were distributed in 6-well plates (500 cells/well). After 12 h of attachment, the cells were treated with selumetinib or gefitinib alone, or the two combinations for 48 h. Then, the medium was substituted with a drug-free medium every 2 days. The culture medium was removed after 14 days. The formed cell clones were fixed with methanol and stained with 0.1% crystal violet (G1063, Solarbio Science & Technology Co., Ltd, Beijing). The clone formation efficiency was calculated using Image J software (NIH, USA).
Flow cytometry analysis
Cells were seeded in 6-well plates and incubated for 24 h. Then, the cells were treated with selumetinib and gefitinib for an additional 48 h. Subsequently, cells were collected by centrifugation followed by resuspension with binding buffer. 100 µL of cell suspension was placed in flow cytometry tubes, and then 5 µL Annexin V/FITC (CA1020, Solarbio Science & Technology Co., Ltd, Beijing) was added and incubated for 5 min at room temperature in the dark. 5 µL PI was added for immediate detection by flow cytometry (Accuri™ C6 Plus, Becton, Dickinson and Company, NJ, America), and the data were analyzed with FlowJo software (FlowJo, Ashland, OR, USA).
Cell migration and invasion assay
For the cell scratch test, NCI-H1975 and A549 cells were seeded in 6-well plates (2 × 105 cells/well). After cell adherence, the formed cell monolayers were scratched with 100 μL pipette tips and washed with PBS to remove floating cells. Cells were incubated with gefitinib or selumetinib for 24 h. The migration distance was calculated using Image J software. Three replicates were performed at each concentration. For the transwell migration assay, cells (1 × 105 cells/well) were seeded in the upper compartment of the transwell chamber with serum-free conditioned medium (8 μm pore size, 3422, Corning, NY, USA). Then, a 0.6 mL culture medium with 10% FBS was added to the lower compartment. The plates were maintained for 24 h at 37 °C. After incubation, the filter was fixed with methanol and stained with 0.1% crystal violet (C0121, Beyotime Biotechnology, Shanghai, China). The cells on the upper surface of the filter were removed, and cells on the lower surface were photographed under a microscope at 100 magnifications followed by dissolution with 30% acetic acid. The solution was measured using a wavelength of 592 nm.
AO/EB fluorescence staining
AO/EB double staining is used to analyze the morphological characteristics of the apoptotic cells. EB (ethidium bromide) yields red–orange fluorescence, which can stain dead or apoptotic cells, while AO (acridine orange) can stain live cells with yellow-green fluorescence. NCI-H1975 and A549 cells (5 × 104 cells/well) were seeded at 48 well-plate and treated with gefitinib or selumetinib for 48 h at 37℃. After that, the mixed AO/EB solutions (R20292, Solarbio, Beijing, China) were added to cells and incubated for 5 min in the dark. Cell morphology was examined under a fluorescence microscope (IX71, Olympus, Japan).
Western blotting and antibodies
A549 cells were incubated with gefitinib or selumetinib at the indicated concentrations and for the indicated durations, washed with PBS twice, lysed on ice for 30 min in a loading buffer, and then boiled for approximately 10 min. The protein concentration was determined with a BCA reagent. Equal amounts of protein were electrophoresed in 6% to 10% SDS-PAGE gels, then the proteins were transferred to PVDF membranes (Pall, NY, USA), probed with antibodies, and detected by chemiluminescence (#32209, Thermo Fisher Scientific, MA, USA).
The antibodies to detect EGFR (#66455–1-Ig), STAT3 (#10253–2-AP), ERK 1/2 (#11257–1-AP), PARP1(#13371–1-AP), Cleaved Caspase 3 (#25128–1-AP), Caspase 8 (#13423–1-AP), Caspase 9 (#10380–1-AP), Bcl2 (#12789–1-AP), E-cadherin (#20874–1-AP), N-cadherin (#22018–1-AP), Vimentin (#10366–1-AP), MIG6 (#11630–1-AP), p-AKT (Ser473, #80455–1-RR), p-ERK (Thr202/Tyr204, #28733–1-AP) and GAPDH (#10,494–1-AP) were obtained from Proteintech Group, Inc, Wuhan, China. Antibodies against p-EGFR (Tyr1068, #3777S), and Cleaved Caspase9 (#9509) were obtained from Cell Signaling Technology (Boston, MA, USA). Cleaved-PARP was obtained from MultiSciences Biotech Co., Ltd (Hangzhou, China). Anti-Phospho-STAT3 (Tyr705) antibody (ET1603-40) was obtained from Hangzhou Huaan Biotechnology Co., Ltd. All the primary antibodies were diluted according to the instructions. Anti-rabbit and anti-mouse (1:5000) HRP-conjugated antibodies were used as the secondary antibodies.
siRNA and cDNA overexpression transfection
siRNA and cDNA overexpression transfection were conducted with the Lipofectamine® 3000 reagent according to the manufacturer’s instructions (L3000015, Thermo Fisher Scientific, MA, USA). Cells were transfected with a final concentration of 20 nmol/L siRNA for 4-6 h with the Lipofectamine® 3000 reagent, and then, the medium was replaced with a fresh growth medium. After transfection for 48 h, the cells were treated with the compounds or DMSO for 0.5 h, and cell lysate protein samples were used for Western blot analysis. The MIG6 siRNA gene sequences were as follows: 5′-CUACACUUUCUGAUUUCAA-3′. All of these siRNA molecules were obtained from Shanghai GenePharma Co., Ltd. MIG6 cDNA (Plasmid #82116) was purchased from Addgene (MA, USA).
Animal studies
All animal studies were conducted based on animal protocols approved by the Animal Care and Ethics Committee of Northwest University and in accordance with the National Health Guidelines on the Care and Use of Animals (NWU-AWC-20220903 M). Female BALB/c nude mice were obtained from GemPharmatech Co., Ltd. (Jiangsu, China). To assess the antitumor response of gefitinib and selumetinib, 1 × 107 A549 cells in 200 μL FBS-free medium were subcutaneously injected into the left axilla of the mice. Once the tumor volume reached 100–150 mm3, the mice were divided into four groups (6 mice per group): the control group was orally gavaged with the vehicle control [mixture of normal saline (87%), polyoxyethylenated castor oil (8%), ethyl alcohol (5%)]; single administration group was intraperitoneal injected with selumetinib (30 mg/kg) and gefitinib (30 mg/kg); in the combined administration group, gefitinib and selumetinib were administered sequentially by intraperitoneal injection at an interval of more than 1 h. All groups were administered every day for 21 days. The tumor volume and body weight were measured every 3 days. At the end of the experiments, the mice were sacrificed, and the primary tumors and some major organs including the heart, lung, liver, spleen, and kidney were collected and weighed. Calculate the organ index according to the following formula: organ index = (organ weight/mouse weight) × 100%.
Statistical analysis
The experimental data were analyzed with GraphPad Prism 8 software using one-way analysis of variance (ANOVA), and a p-value < 0.05 was considered statistically significant. The data are shown as the mean ± standard deviation (SD).
Results
Selumetinib and gefitinib synergistically inhibit the proliferation of NSCLC
To explore the antiproliferative effect of selumetinib combined with gefitinib in gefitinib-resistant NSCLC, we selected gefitinib primary resistant cell lines A549 (harboring EGFR wild type) and H1975 (harboring EGFR L858R/T790M mutant), as well as PC9/GR cells selected from a subculture that had acquired resistance to gefitinib. As shown in Fig. 1A, B the IC50 values of gefitinib on A549 cells and H1975 cells alone for 72 h were 14.94 μM and 10.07 μM, respectively. The IC50 values of selumetinib and gefitinib in the combined administration group on A549 cells and H1975 cells were 0.74 μM and 0.4 μM, respectively. In addition, PC9/GR cells exhibited a 110-fold increase in resistance to the growth-inhibitory effect of gefitinib compared with that in parent PC-9 cells as determined by MTT assay, and the resistant phenotype was stable for at least 6 months under drug-free conditions. Similarly, selumetinib potently enhanced the inhibitory effect of gefitinib on PC9/GR cells (Fig. 1C). Furthermore, according to the analysis of CompuSyn software, the combination of selumetinib and gefitinib synergistically inhibited the proliferation of A549 (Fig. 1D), H1975 (Fig. 1E) and PC9/GR cells (Fig. 1F) with CI values < 1. In addition, the synergistic effect of selumetinib and gefitinib was time-dependent. A549, H1975, and PC9/GR cell viability was examined following treatment with 5 µM gefitinib with or without the indicated selumetinib for 24, 48, and 72 h (Fig. 1G–I). The results of the MTT assay demonstrated that the combination of gefitinib and selumetinib increased the cell viability inhibition effect compared with that of gefitinib or selumetinib treatment alone, and the difference was statistically significant at 48 and 72 h (*p < 0.05, **p < 0.01). The clone formation assay also showed that gefitinib combined with selumetinib treatment significantly inhibited clone formation in A549, H1975, and PC9/GR cells compared with that of gefitinib treatment alone (*p < 0.05) (Fig. 1J). Clone formation efficiency is indicated in the histogram (Fig. 1K). In short, the results indicated that selumetinib and gefitinib synergistically inhibited the proliferation of NSCLC.

Selumetinib synergistically inhibited the proliferation of NSCLC with gefitinib. A–C MTT assay was used to examine the cell viability of A549, H1975, and PC9/GR cells treated with gefitinib (0, 1, 2, 5, 10, 20, 40, or 80 µM) with or without selumetinib (0, 1, 2, 5, 10, 20, 40 or 80 µM) for 72 h. D–F Gefitinib in combination with selumetinib induced the proliferation inhibition by the MTT assay (The CI value was calculated according to the CompuSyn software, n ≥ 3). The CI > 1 was antagonistic, CI = 1 was additive and CI < 1 was synergistic. G–I Cell viability of A549, H1975, and PC9/GR cells treated with 5 µM gefitinib and selumetinib for 24, 48, and 72 h. J Plate clone formation assay was used to examine the cell clone formation ability after treatment of gefitinib with or without ginsenoside CK in A549, H1975, and PC9/GR cells. (K) Clone formation efficiency was indicated in the histogram by Image J software. A statistically significant difference (*p < 0.05, or **p < 0.01) between the gefitinib/selumetinib combined group and the gefitinib group
Selumetinib enhanced gefitinib-induced apoptosis in gefitinib-resistant NSCLC
To evaluate the effects of selumetinib on apoptosis, AO/EB staining and flow cytometry were performed on A549 and H1975 cells. As illustrated in Fig. 2A, the AO/EB staining assay revealed that the number of cells emitting red fluorescence increased in the combined administration group in both A549 and H1975 cells, indicating that the number of apoptotic cells increased in the group co-induced by selumetinib, compared with that in the gefitinib group. Cell apoptosis triggered by gefitinib/selumetinib was determined using the Annexin V/PI apoptosis assay. The proportion of early and late apoptotic cells was markedly increased after cotreatment with selumetinib, while the rate of apoptotic cells increased from 7.18 ± 0.33% in the gefitinib group to 26.1 ± 1.27% of the cells cotreated with selumetinib (**p < 0.01) in A549 cells, and the rate of apoptotic cells increased from 7.27 ± 1.70% in the gefitinib group to 17.28 ± 2.28% of the cells cotreated with selumetinib (* p < 0.05) in H1975 cells (Fig. 2B, C). Subsequently, we explored the expression of important signaling proteins involved in apoptosis via western blotting assays (Fig. 2D, E). The combination of gefitinib and selumetinib increased the expression of cleaved caspase 9 and downregulated the expression of pro-caspase 9 and Bcl2, which were major members of the mitochondrial apoptotic pathway. Furthermore, as shown in Fig. 2D, the expression of caspase 8, a key member of the death receptor pathway, did not change significantly after cotreatment with selumetinib. However, co-treatment with selumetinib promoted the cleavage of pro-caspase 3 and pro-PARP1, which were the ultimate executors of apoptosis and its substrate protein. These results suggest that cotreatment with selumetinib may enhance apoptosis in gefitinib-resistant NSCLC cell lines mainly through the caspase-dependent mitochondrial pathway.

Selumetinib enhanced gefitinib-induced apoptosis in gefitinib-resistant NSCLC. A AO/EB staining was performed to assess apoptosis-induced morphological variation via fluorescence microscopy. Scale bars = 100 µm. B Flow cytometry was used to examine apoptosis in A549 cells treated with gefitinib (5 µM) with or without selumetinib (5 µM) for 48 h. *P < 0.05 vs. the gefitinib group. C Quantification plots were shown on the right. Statistics were exhibited as the mean ± SD of triplicate independent experiments; * p < 0.05 and ** p < 0.01 compared with the gefitinib group. D Protein expression levels of PARP1, Cleaved-PARP, Caspase 8, Caspase 9, Cleaved Caspase 3, Cleaved Caspase 9, and Bcl2 were determined by western blotting analysis in A549 cells treated with gefitinib (5 µM) with or without selumetinib (5 µM) for 48 h. GAPDH was used as a control protein. E Quantification plots were shown on the right
Selumetinib enhanced migration inhibition in gefitinib-resistant NSCLC
To test the antimigration effects of selumetinib on NSCLC cells, wound healing, and Transwell assays were conducted. The wound healing assay showed that the wound healing percentages of the combined group decreased evidently in both treated A549 and H1975 cells (Fig. 3A, B, * p < 0.05). Furthermore, the Transwell assay showed that the cell migration ability of the selumetinib cotreatment group showed an obvious reduction of 42.2% in A549 cells and 28.0% in H1975 cells (Fig. 3C, D, *p < 0.05, **p < 0.01), indicating the inhibitory effect of selumetinib on migration. Subsequently, we explored the expression of important signaling proteins involved in migration via western blotting assays (Fig. 3E, F). The combination of gefitinib and selumetinib significantly increased the protein expression levels of E-cadherin and decreased the protein expression levels of N-cadherin and Vimentin. These results demonstrated that the migration inhibition ability was enhanced after cotreatment with selumetinib in gefitinib-resistant A549 and H1975 cells.

Selumetinib enhanced migration inhibition in gefitinib-resistant NSCLC. A Wound healing assays were performed to investigate the migration ability alteration of A549 cells and H1975 cells after gefitinib treatment with or without selumetinib. Data were processed using ImageJ software. Quantification charts were listed on the right (B). C Transwell migration assays were performed to investigate the migration and invasion ability after gefitinib treatment with or without selumetinib. Quantification charts were listed on the right (D). Statistics were exhibited as the mean ± SD of triplicate independent experiments; * p < 0.05, ** p < 0.01. E Protein expression levels of E-Cadherin, N-Cadherin, and Vimentin were determined using western blotting analysis in A549 cells treated with gefitinib (5 µM) with or without selumetinib (5 µM) for 48 h. GAPDH was used as a control protein. F Quantification plots were shown on the right
Selumetinib could overcome gefitinib resistance by regulating MIG6/STAT3 in NSCLC
As previously described (Song et al. 2021), considering that sustained activation of STAT3 was an intrinsic mechanism underlying resistance to EGFR-TKIs, the inhibitory effect of the combination of gefitinib and selumetinib on STAT3 activation was examined. As shown in Fig. 4A, p-EGFR was significantly inhibited, but its downstream phosphorylation level of STAT3 was activated after exposure to gefitinib alone. When the cells were cotreated with selumetinib, p-STAT3, p-AKT, and p-ERK were significantly inhibited in a dose-dependent manner and were almost completely suppressed at high concentrations.

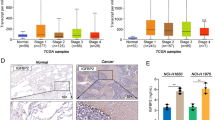
Selumetinib could overcome gefitinib resistance by regulating MIG6/STAT3 in NSCLC. A A549 cells were exposed to gefitinib (6.25, 12.5, or 25 µM) alone or co-treated with selumetinib (5 µM) for 24 h. Cells were harvested for western blotting analysis. Protein expression levels of p-EGFR, p-STAT3, p-AKT, and p-ERK were determined. B Protein interactions were analyzed using the STRING database. C MIG6 expression in breast cancer (BRCA), liver hepatocellular carcinoma (LIHC), lung squamous cell carcinoma (LUSC), and ovarian serous cystadenocarcinoma (OV) compared with the normal group based on GEPIA dataset. D A549 cells were exposed to selumetinib (2.5, 5, 10, or 20 µM) for 24 h. Cells were harvested for western blotting analysis. Protein expression levels of p-STAT3, MIG6, and p-ERK were determined. E A549 cells were transfected with 20 nM of control (NC), MIG6 siRNAs, or MIG6 cDNA for 48 h, then cells were treated with gefitinib (0, 1, 2, 5, 10, 20, 40 or 80 µM) with selumetinib (5 µM) for 48 h. Protein expression levels of MIG6 were determined using western blot analysis, and cell proliferation was detected by MTT assay. F A549 cells were interfered with siRNA MIG6 for 48 h, and treated with gefitinib (12.5 or 25 µM) in the presence of smeitinib (5 µM) for 24 h. Cells were harvested for western blotting analysis. G A549 cells were transfected with MIG6 cDNA for 48 h, then cells were treated with gefitinib (12.5 or 25 µM) in the presence of smeitinib (5 µM) for 24 h. Cells were harvested for western blotting analysis. Protein expression levels of p-EGFR and p-STAT3 were determined. GAPDH was used as a control protein
To further explore the mechanism by which selumetinib overcomes gefitinib resistance, proteins that might interact with STAT3 were analyzed, and a PPI network diagram was drawn using the STRING database (Fig. 4B). Mitogen-induced gene 6, MIG-6 (also known as ERRFI1) attracted our attention because it regulated receptor tyrosine kinase (RTK) signaling through negative feedback (Yoon et al. 2012). To confirm the expression of MIG6 in tumors, we searched and analyzed the data in the GEPIA database and found that the expression of MIG6 was lower in breast cancer (BRCA), liver hepatocellular carcinoma (LIHC), lung squamous cell carcinoma (LUSC), and ovarian serous cystadenocarcinoma (OV) tissues than in normal tissues (Fig. 4C). Therefore, the effect of selumetinib on MIG6 expression was examined. As shown in Fig. 4D, the expression of MIG6 was slightly elevated after selumetinib treatment, suggesting that MIG6 may play a certain regulatory role.
Next, to further investigate whether MIG6 was involved in EGFR-TKI resistance, the effects of gefitinib and selumetinib on cell proliferation and STAT3 activation were examined when MIG6 was knocked down or overexpressed in A549 cells. As shown in Fig. 4E, the effect of MIG6 on the proliferation rate of A549 cells after gefitinib and selumetinib cotreatment was analyzed using the MTT assay. The results showed that siMIG6 significantly decreased the gefitinib-mediated growth inhibition of A549 cells. However, transfection of A549 cells with the MIG6-overexpressing plasmid promoted gefitinib-mediated growth inhibition. Then, we further examined the effect of gefitinib on the activation of STAT3 when MIG6 was knocked down with siRNA in A549 cells (Fig. 4F). When MIG6 was not disturbed, p-STAT3 was strongly inhibited after gefitinib and selumetinib cotreatment. However, the decrease of p-STAT3 mediated by cotreatment with selumetinib and gefitinib in MIG6-intact A549 cells was blocked or much relieved when MIG6 expression was knocked down. Conversely, when MIG6 was overexpressed, p-STAT3 was further inhibited after treatment with gefitinib and selumetinib (Fig. 4G), suggesting that selumetinib enhanced gefitinib sensitivity by regulating MIG6/STAT3 in NSCLC.
Selumetinib promoted the sensitivity of lung cancer xenografts to gefitinib in vivo
Finally, to provide more evidence of the negative effect of selumetinib on tumor progression, xenograft mouse models were created with A549 cells. A schematic diagram of the animal experimental process is shown in Fig. 5A. The tumor size and weight were both decreased by selumetinib and gefitinib separately, but the reduction was more pronounced in the selumetinib and gefitinib combination groups (Fig. 5B, C). Tumor volumes were measured every 3 days during the experimental period, and the results demonstrated that selumetinib and gefitinib visibly inhibited solid tumor growth (Fig. 5D). The inhibition rates of selumetinib (30 mg/kg/d) and gefitinib (30 mg/kg/d) on tumor volume were 60.79% and 31.95%, respectively, but the inhibition rate of the combination group was 81.49%. After 21 days of administration, the average body weights of mice treated with gefitinib and selumetinib were 23.01 ± 0.48 g and 21.92 ± 0.61 g, respectively, which did not show an outstanding decrease compared to that of the normal group (21.75 ± 0.34 g). Meanwhile, the weight of mice in the combination group was 21.27 ± 0.40 g, which was similar to that of the normal group with no significance (Fig. 5E, p > 0.05).

Selumetinib promoted the sensitivity of lung cancer xenograft to gefitinib in vivo. A Schematic diagram of animal experimental process. B Representative pictures of A549-xenograft tumors of the control group, gefitinib-treated group, selumetinib-treated group, and gef + selumetinib combined group. C Tumor weight of xenograft mice in each group on the 21st day. D Tumor volume and E Body weight were measured every 3 days. F The organ indexes of mice including liver, spleen, kidney, lung, and heart were recorded. Statistics were exhibited as the mean ± SD (n = 6 in each group); *p < 0.05, **p < 0.01 compared with the gefitinib group, and #p < 0.05, ##p < 0.01 compared with the selumetinib group. G Western blotting of p-EGFR, p-STAT3, p-ERK in A549 xenograft tumor specimens. The results shown were representative of two independent experiments
Additionally, there were no statistically significant differences in organ indicators between each group, which demonstrated that the selumetinib and gefitinib combination group had no toxic side effects (Fig. 5F). Subsequently, the results of the immunoblotting assay in tumor tissues also showed that p-EGFR, p-STAT3, and p-ERK were more strongly inhibited in combination with selumetinib (Fig. 5G). Consequently, selumetinib effectively promoted the sensitivity of lung cancer xenografts to gefitinib in vivo.
Discussion
Lung cancer is driven by several oncogenes, including ALK, BRAF, EGFR, HER2, KRAS, MEK1, MET, NRAS, RET, and ROS1 (Roncarati et al. 2020). Mutation or amplification of these genes leads to the development of lung cancer, thus, small-molecule drugs targeting these oncogenes have provided a novel strategy for the treatment of lung cancer and have already achieved remarkable therapeutic results (Wang et al. 2021). EGFR overexpression is associated with survival rate, poor prognosis, and postoperative quality of life in NSCLC patients. Therefore, the development of small molecule inhibitors targeting EGFR is significant for the clinical treatment of NSCLC. Gefitinib, a first-generation EGFR-TKI, has achieved remarkable efficacy in the clinical treatment of advanced non-small cell lung cancer (NSCLC); However, drug resistance will inevitably occur. Therefore, exploring the resistance mechanism and overcoming strategies of gefitinib, as well as developing new combination treatment strategies have become the research hotspots in the field of lung cancer treatment.
In our study, the MEK1/2 inhibitor, selumetinib was selected to investigate the efficacy of combination with gefitinib in gefitinib-resistant NSCLC cells. Selumetinib (AZD6244), is a potent and selective adenosine triphosphate-uncompetitive inhibitor of MEK1/2 with anticancer activity both in vitro and in vivo. The drug is currently in phase III clinical development. However, selumetinib, as a single agent, failed to demonstrate clinical activity in different tumor types (Maiello et al. 2015). Therefore, we hypothesized that selumetinib and gefitinib had synergistic effects in lung cancer cells. The results of MTT assays illustrated that selumetinib, with gefitinib, synergistically inhibited the proliferation of NSCLC in primary resistant lung cancer cells A549 and H1975 cells, as well as in acquired resistant PC9/GR cells. Similar results were observed in the mouse xenograft model in vivo. The combined administration of selumetinib and gefitinib significantly inhibited the growth of solid tumors, and the tumor inhibition rate reached 81.49%. These results suggested that selumetinib could effectively enhance the inhibitory effect of gefitinib on NSCLC in vitro and in vivo.
Apoptosis, the most common form of cell death under physiological conditions, is crucial for the occurrence and development of tumors and drug resistance (Liu et al. 2017a, b). In this study, the results of AO/EB staining and flow cytometry showed an increased number of apoptotic A549 and H1975 cells after cotreatment with selumetinib. Subsequently, western blotting data showed that the expression levels of cleaved caspase 9, cleaved caspase 3, and cleaved PARP were significantly increased in cells after cotreatment with selumetinib. However, the expression levels of pro-caspase 9, pro-PARP1, and Bcl2 were decreased, indicating that cotreatment with selumetinib may enhance apoptosis in gefitinib-resistant cells mainly through the caspase-dependent mitochondrial pathway. Additionally, scratch assays and Transwell assays were performed to detect the tumor cell migration ability. Compared with those in the gefitinib monotherapy group, the proportion of wound healing and cell migration in the Transwell experiment in the selumetinib combined administration group was significantly reduced, indicating that selumetinib enhanced migration inhibition in gefitinib-resistant NSCLC.
STAT3, an important member of the signal transducer and activator of transcription (STAT) family, has dual functions in signal transduction and transcriptional regulation which regulate the growth, differentiation, apoptosis, and other physiological functions of cells (El-Tanani et al. 2022). Under normal physiological conditions, the activity of STAT3 is regulated by upstream activated proteins and some endogenous negative regulatory proteins. Once the regulation is lost, the overexpression of STAT3 will lead to a series of diseases. Studies have shown that persistent activation of STAT3 frequently occurs in non-small cell lung cancers, and is associated with survival, prognosis, and resistance (Zheng et al. 2021). It was reported that combined targeted inhibition of EGFR and STAT3 could enhance the efficacy of other EGFR-TKIs in lung cancer (Liu et al. 2017a, b). Our previous study also proved that some small molecule inhibitors could offset the sustained activation of STAT3 and enhance sensitivity in gefitinib-resistant cells (Song et al. 2021). In this study, the sustained activation of STAT3 was significantly inhibited after cotreatment with selumetinib, suggesting that the ability of selumetinib to overcome gefitinib resistance may be related to the regulation of STAT3.
Mitogen-induced gene 6 (MIG-6), also known as ErbB receptor feedback inhibitor 1 (ERRFI1), is considered an immediate early response gene that plays an important role in regulating the stress response, maintaining homeostasis, and functioning as a tumor suppressor in multiple kinds of human cancers (Liu et al. 2017a, b). A large number of studies have shown that MIG-6 is downregulated in many tumors including breast cancer, skin cancer, non-small cell lung cancer, ovarian cancer, pancreatic cancer, and endometrial cancer (Anastasi et al. 2005; Li et al. 2012; Cui et al. 2022). Loss of function in MIG6 is also associated with drug resistance (Boopathy et al. 2018). MIG-6 inhibits EGFR kinase activation by binding to the EGFR kinase domain and promoting the endocytogenic degradation of EGFR (Boopathy et al. 2018). Chang et al. (Chang et al. 2013) proved that the relative expression of MIG6 and EGFR is associated with resistance to EGFR kinase inhibitors. Recent studies have proven that abnormal activation of STAT3 and negative regulation of MIG6 are related to antitumor drug resistance (Yoo et al. 2018), but the mechanism underlying drug resistance remains unclear. In our study, the results of the GEPIA database illustrated that MIG6 was downregulated in many cancers compared with that in normal tissue. Yoon et al. (Yoon et al. 2012) reported that MIG6 was elevated after selumetinib treatment in LOVO cells containing wild-type EGFR and mutant KRAS. Meanwhile, the results showed that the expression of MIG6 was slightly elevated after selumetinib treatment in A549 cells harboring wild-type EGFR and mutant KRAS, suggesting that MIG6 may play a key regulatory role and that MIG6 may regulate the sensitivity of KRAS mutant cells and the MEK/ERK signaling pathway.
To further investigate the mechanism by which selumetinib overcomes gefitinib resistance, the coeffect of selumetinib on the activation of STAT3 was investigated when MIG6 was knocked down with siRNA or overexpressed with cDNA in A549 cells. The results showed that downregulation of p-STAT3 due to the combination of selumetinib and gefitinib was reversed by the deletion of MIG6, and p-STAT3 was further inhibited after cotreatment with selumetinib when MIG6 was overexpressed, suggesting that the negative regulatory axis of MIG6/STAT3 mediated the sensitization of selumetinib to gefitinib resistance in NSCLC. The above results indicate that the MIG6/STAT3 axis plays an important role in the sustained activation of STAT3 and the resistance to EGFR-TKIs. Our study also suggests that EGFR-TKIs combined with MEK1/2 inhibitors, such as selumetinib, may be beneficial to those patients with NSCLC who develop a primary or acquired resistance to EGFR-TKIs, also providing an effective strategy for combination treatment of clinical lung cancer.
Currently, the literature on NSCLC clinical trials of gefitinib and selumetinib in combination therapy is not available. However, selumetinib has shown promising results as a single agent or in combination with conventional chemotherapy and other targeted therapies both preclinically and clinically, in multiple cancers including pediatric low-grade glioma, non-small cell lung cancer, and melanoma, An initial phase II trial of selumetinib monotherapy for recurrent advanced NSCLC did not show a significant difference in PFS, but a subsequent phase II study of selumetinib combined with chemotherapy in KRAS wild-type or KRAS unknown tumors showed a higher response rate with the addition of selumetinib. In addition, in the phase 1 study of selumetinib combined with EGFR tyrosine kinase inhibitor erlotinib, patients with metastatic melanoma had an objective response (Campagne et al. 2021).
In the era of modern medicine, although many other treatment options, such as antibodies, nucleic acids, and cell and gene therapy, have emerged, small molecules are usually still the preferred option for many diseases (Beck et al. 2022). On the one hand, small-molecule drugs have precise targets and flexible delivery routes, on the other hand, small-molecule drugs also provide an alternative treatment option to single-targeted treatment options by combining chemotherapy with other drugs. As a result, the innovative use or combination of small-molecule drugs may be able to enhance clinical outcomes and improve patients' lives. In this study, we found that selumetinib could overcome gefitinib primary and acquired resistance by regulating MIG6/STAT3 in NSCLC, supporting the feasibility of combination therapy and the utility of selumetinib for NSCLC therapeutics.
References
Anastasi S, Sala G, Huiping C, Caprini E, Russo G, Iacovelli S, Lucini F, Ingvarsson S, Segatto O (2005) Loss of RALT/MIG-6 expression in ERBB2-amplified breast carcinomas enhances ErbB-2 oncogenic potency and favors resistance to Herceptin. Oncogene 24:4540–4548. https://doi.org/10.1038/sj.onc.1208658
Beck H, Härter M, Haß B, Schmeck C, Baerfacker L (2022) Small molecules and their impact in drug discovery: a perspective on the occasion of the 125th anniversary of the Bayer chemical research laboratory. Drug Discov Today 27:1560–1574. https://doi.org/10.1016/j.drudis.2022.02.015
Boopathy GTK, Lynn JLS, Wee S, Gunaratne J, Hong W (2018) Phosphorylation of Mig6 negatively regulates the ubiquitination and degradation of EGFR mutants in lung adenocarcinoma cell lines. Cell Signal 43:21–31. https://doi.org/10.1016/j.cellsig.2017.11.006
Campagne O, Yeo KK, Fangusaro J, Stewart CF (2021) Clinical pharmacokinetics and pharmacodynamics of selumetinib. Clin Pharmacokinet 60:283–303. https://doi.org/10.1007/s40262-020-00967-y
Carvajal RD, Piperno-Neumann S, Kapiteijn E, Chapman PB, Frank S, Joshua AM (2018) Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: a phase III, multicenter, randomized trial (SUMIT). J Clin Oncol 36:1232–1239. https://doi.org/10.1200/JCO.2017.74.1090
Chang XF, Izumchenko E, Solis LM, Kim MS, Chatterjee A, Ling SZ, Monitto CL, Harari PM, HidalgoM Goodman SN, Wistuba II, Bedi A, Sidransky D (2013) The relative expression of Mig6 and EGFR is associated with resistance to EGFR kinase inhibitors. PloS one 8:e68966. https://doi.org/10.1371/journal.pone.0068966
Chou TC (2010) Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res 70:440–446. https://doi.org/10.1158/0008-5472.CAN-09-1947
Cooper AJ, Sequist LV, Lin JJ (2022) Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat Rev Clin Oncol 19:499–514. https://doi.org/10.1038/s41571-022-00639-9
Cui YT, Kang Y, Zhang P, Wang YG, Yang ZY, Lu C, Zhang P (2022) Mig-6 could inhibit cell proliferation and induce apoptosis in esophageal squamous cell carcinoma. Thorac Cancer 13:54–60. https://doi.org/10.1111/1759-7714.14223
Das M (2017) Selumetinib does not improve survival outcomes in NSCLC. Lancet Oncol 18:e313. https://doi.org/10.1016/S1470-2045(17)30363-7
El-Tanani M, Al Khatib AO, Aladwan SM, Abuelhana A, McCarron PA, Tambuwala MM (2022) Importance of STAT3 signalling in cancer, metastasis and therapeutic interventions. Cell Signal 92:110275. https://doi.org/10.1016/j.cellsig.2022.110275
Gu Y, Mohammad IS, Liu Z (2020) Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors. Oncol Lett 19:2585–2594. https://doi.org/10.3892/ol.2020.11394
Harada D, Takigawa N, Kiura K (2014) The role of STAT3 in non-small cell lung cancer. Cancers (basel) 6:708–722. https://doi.org/10.3390/cancers6020708
He J, Huang Z, Han L, Gong Y, Xie C (2021) Mechanisms and management of 3rd generation EGFR TKI resistance in advanced non small cell lung cancer (review). Int J Oncol 59:90. https://doi.org/10.3892/ijo.2021.5270
Hedayat M, Jafari R, MajidiZolbanin N (2023) Selumetinib: a selective MEK1 inhibitor for solid tumor treatment. Clin Exp Med 23:229–244. https://doi.org/10.1007/s10238-021-00783-z
Huang L, Fu L (2015) Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B 5:390–401. https://doi.org/10.1016/j.apsb.2015.07.001
Jin J, Guo Q, Xie J, Jin D, Zhu Y (2019) Combination of MEK inhibitor and the JAK2-STAT3 pathway inhibition for the therapy of colon cancer. Pathol Oncol Res 25:769–775. https://doi.org/10.1159/000354627
Kandala PK, Srivastava SK (2012) Diindolylmethane suppresses ovarian cancer growth and potentiates the effect of cisplatin in tumor mouse model by targeting signal transducer and activator of transcription 3 (STAT3). BMC Med 10:9. https://doi.org/10.1186/1741-7015-10-9
Köhler J, Schuler M (2013) Afatinib, Erlotinib and gefitinib in the first-Line therapy of EGFR mutation-positive lung adenocarcinoma: a review. Oncol Res Treat 1(36):510–518. https://doi.org/10.1159/000354627
Li Z, Dong Q, Wang Y, Qu L, Qiu X, Wang E (2012) Downregulation of Mig-6 in nonsmall-cell lung cancer is associated with EGFR signaling. Mol Carcinog 51:522–534. https://doi.org/10.1002/mc.20815
Liu GB, Pei F, Yang FQ, Li LX, Amin AD, Liu SN, Buchan JR, Cho WC (2017a) Role of autophagy and apoptosis in non-small-cell lung cancer. Int J Mol Sci 18:367. https://doi.org/10.3390/ijms18020367
Liu J, Cho SN, Wu SP, Jin N, Moghaddam SJ, Gilbert JL, Wistuba I, DeMayo FJ (2017b) Mig-6 deficiency cooperates with oncogenic Kras to promote mouse lung tumorigenesis. Lung Cancer 112:47–56. https://doi.org/10.1016/j.lungcan.2017.08.001
Maiello MR, D’Alessio A, Bevilacqua S, Gallo M, Normanno N, De Luca A (2015) EGFR and MEK blockade in triple negative breast cancer cells. J Cell Biochem 116:2778–2785. https://doi.org/10.1002/jcb.25220
Pan ZZ, Wang K, Wang XN, Jia HR, Yang YQ, Duan YL, Huang LZ, Wu ZX, Zhang JY, Ding XS (2022) Cholesterol promotes EGFR-TKIs resistance in NSCLC by inducing EGFR/Src/Erk/SP1 signaling-mediated ERRα re-expression. Mol Cancer 21:77. https://doi.org/10.1186/s12943-022-01547-3
Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, Morinishi LS, Evans L, Ji WY, Hsu CH, Thurley K, Wei SG, Zhou AW, Koduru PR, Posner BA, Wu LF, Altschuler SJ (2016) Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun 7:1–8. https://doi.org/10.1038/ncomms
Roncarati R, Lupini L, Miotto E, Saccenti E, Mascetti S, Morandi L, Bassi C, Rasio D, Callegari E, Conti V, Rinaldi R, Lanza G, Gafà R, Papi A, Frassoldati A, Sabbioni S, S, Ravenna F, Casoni GL, Negrini M, (2020) Molecular testing on bronchial washings for the diagnosis and predictive assessment of lung cancer. Mol Oncol 14:2163–2175. https://doi.org/10.1002/1878-0261.12713
Song XP, Tang W, Peng H, Qi X, Li J (2021) FGFR leads to sustained activation of STAT3 to mediate resistance to EGFR-TKIs treatment. Invest New Drugs 39:1201–1212. https://doi.org/10.1007/s10637-021-01061-1
Sun C, Gao W, Liu J, Cheng H, Hao J (2020) FGL1 regulates acquired resistance to Gefitinib by inhibiting apoptosis in non-small cell lung cancer. Respir Res 21:210. https://doi.org/10.1186/s12931-020-01477-y
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71:209–249. https://doi.org/10.3322/caac.21660
Wang M, Herbst RS, Boshoff C (2021) Toward personalized treatment approaches for non-small-cell lung cancer. Nat Med 27:1345–1356. https://doi.org/10.1038/s41591-021-01450-2
Yoo JY, Yang WS, Lee JH, Kim BG, Broaddus RR, Lim JM, Kim TH, Jeong JW (2018) MIG-6 negatively regulates STAT3 phosphorylation in uterine epithelial cells. Oncogene 37:255–262. https://doi.org/10.1038/onc.2017.335
Yoon YK, Kim HP, Han SW, Hur HS, Oh DY, Im SA, Bang YJ, Kim TY (2009) Combination of EGFR and MEK1/2 inhibitor shows synergistic effects by suppressing EGFR/HER3-dependent AKT activation in human gastric cancer cells. Mol Cancer Ther 8(9):2526–2536. https://doi.org/10.1158/1535-7163
Yoon YK, Kim HP, Song SH, Han SW, Oh DY, Im SA, Bang YJ, Kim TY (2012) Down-regulation of mitogen-inducible gene 6, a negative regulator of EGFR, enhances resistance to MEK inhibition in KRAS mutant cancer cells. Cancer Lett 316:77–84. https://doi.org/10.1016/j.canlet.2011.10.023
Zheng QY, Dong H, Mo J, Zhang Y, Huang J, Ouyang SM, Shi S, Zhu K, Qu XM, Hu WH, Liu PQ, Wang YX, Zhang XL (2021) A novel STAT3 inhibitor W2014-S regresses human non-small cell lung cancer xenografts and sensitizes EGFR-TKI acquired resistance. Theranostics 11(2):824–840. https://doi.org/10.7150/thno.49600
Acknowledgements
This work was supported by the Natural Science Foundation of Shaanxi Province (2022JQ-112).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declared no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Song, X., Wang, L., Tang, W. et al. Selumetinib overcomes gefitinib primary and acquired resistance by regulating MIG6/STAT3 in NSCLC. Arch. Pharm. Res. 46, 924–938 (2023). https://doi.org/10.1007/s12272-023-01471-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-023-01471-0