
Abstract
Biological cell membranes are a natural barrier for living cells. In the last few decades, the cell membrane has been the main hurdle in the efficient delivery of bioactive and therapeutic agents. To increase the drug efficacy of these agents, additional mediators have been considered. Cell-penetrating peptides (CPPs), a series of oligopeptides composed of mostly hydrophobic and/or positively charged side chains, can increase the interaction with the cell membrane. CPP-based delivery platforms have shown great potential for the efficient and direct cytosol delivery of various cargos, including genes, proteins, and small molecule drugs. Bypassing endocytosis allows the CPP-based delivery systems greater defense against the degradation of protein-based drugs than other drug delivery systems. However, the delivery of CPPs exhibits intrinsically non-specific targeting, which limits their medical applications. To endow CPPs with specific targeting ability, the conjugation of pH-sensitive, enzyme-specific cleavable, and multiple targeting ligands has been reported. Optimization of the length and sequence of CPPs is still needed for various drugs of different sizes and surface charges. Toxicity issues in CPP-based delivery systems should be addressed carefully before clinical use.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many therapeutic agents have been developed and used to treat various diseases. However, the cell membrane that drugs must pass through to exert their therapeutic effect is a strong barrier that allows only specific substances to enter. Since the early 1980s when recombinant human insulin was first approved by the US Food and Drug Administration (FDA) for the treatment of diabetes mellitus, the interest in not only recombinant proteins but also biologically active molecules such as oligonucleotides and peptides has increased (Roberts et al. 2020; Muttenthaler et al. 2021). However, due to the hydrophilic nature of many bioactive molecules, they have difficulty passing through the cellular membrane. Therefore, many attempts have been made to overcome these limitations. In the late 1980s, it was reported that the transactivator of the transcription (TAT) protein of the HIV-1 virus penetrated the cell membrane in vitro (Green and Loewenstein 1988). Subsequently, in 1994, the homo-domain of Antennapedia, a Drosophila melanogaster homoprotein, was also reported to pass through cells (Derossi et al. 1994). Thirteen amino acid sequences corresponding to the residue 48–60 of TAT were found to play a key role in cellular uptake (Vivès et al. 1997). After that, peptides that pass through the cell membrane like TAT were commonly defined as cell-penetrating peptides (CPPs). CPPs are generally composed of 5–30 amino acids and pass-through cells and tissues by various mechanisms. The high permeability of CPP through the cell membrane even with companion drugs was a breakthrough that could overcome the limitations. Many published studies have reported the application of CPPs as drug delivery carriers to various diseases such as cancer, stroke, muscular dystrophy, bacterial infection, vaccine, etc. (Guidotti et al. 2017; Zhang et al. 2021; Buccini et al. 2021; Sadeghian et al. 2022).
CPPs can be classified by their origin, sequence, structure, function, etc. Various libraries of CPPs have different chemical and structural properties depending on the type and arrangement of amino acids and are categorized into cationic, anionic, amphipathic, and hydrophobic peptides. A highly positively charged cationic peptide is mainly composed of basic amino acids such as arginine and lysine. TAT, penetratin, and polyarginine are representative examples (Kurrikoff et al. 2016). Cationic CPPs have an electrostatic interaction with negatively charged carboxylic, phosphate, and sulfate groups of the cell membrane, and then enter the cell through various mechanisms.
Amphipathic CPPs have an arranged structure in which polar and nonpolar amino acids are ordered structurally (Hyun et al. 2018; Feger et al. 2020). Generally, lysine and arginine constitute the polar region, and valine, leucine, and alanine constitute the nonpolar region. More specifically, amphipathic CPPs are classified into primary, secondary, and proline-rich CPPs. Hydrophobic CPPs are typically composed of nonpolar amino acids and interact with the hydrophobic region of the cellular membrane and are represented by K-FGF, C105Y, and gH625 (Tréhin and Merkle 2004; Rhee and Davis 2006; Falanga et al. 2015). Although not completely understood, they may translocate by an energy-independent mechanism.
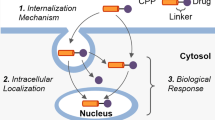
Various CPPs enter cells or deliver various cargos such as proteins, peptides, genes, and small drugs. However, it is very difficult to determine the cellular uptake mechanism of CPP. The penetrating mechanisms are very complex and diverse. Many reports indicate that cellular uptake mechanisms depended upon not only the type of cargo delivered but also various experimental conditions such as concentration, temperature, and pH. The processes of basic cellular uptake consist of steps that include interaction with the cell surface, the internalization process, and release to the target site. Many CPPs have a cationic character and the initial process begins through an electrostatic interaction with anionic groups of the cell membrane. In the next step, the internalization processes are very important and various mechanisms are reported to be involved, such as the carpet-like model (Xiao et al. 2018), transient pore model (Herce et al. 2009), and inverted micelle model (Kawamoto et al. 2011; Xu et al. 2019; Ruseska and Zimmer 2020) (Scheme 1). However, there are still several uncertain and controversial issues, and many studies have found that CPPs use various pathways rather than a single mechanism.

Schematic representation of the proposed internalization mechanisms of CPP-cargo
CPPs are highly effective as drug delivery tools and have potential for multiple uses. However, most CPPs have cationic properties and can be toxic to the target region as well as other organs and tissues. In addition, CPPs are easily deactivated due to protease attacks in living systems, resulting in low stability and short half-life (Munyendo et al. 2012; Reissmann 2014). For example, when various CPPs were administered to tumor-induced mice and observed 1 h later, only about 1% remained (Sarko et al. 2010).
Since CPPs were discovered, many related studies have been conducted for 30 years. In the early stage, the research mainly focused on identification of properties and mechanisms and enhancement of cellular uptake capacity (Xu et al. 2019). After the characteristics and mechanisms of CPP were revealed, studies focused on the stability, target selectivity, enhanced activity, increasing target types, and the possibility of clinical application (Heitz et al. 2009; Gallo et al. 2019). Many studies have been conducted using strategies such as cargo-CPP conjugated system, non-covalent complex type delivery system of various drugs or nucleic acids, active targeting strategy, development of specific signal activatable CPPs, and multifunctional system combining various features (Kurrikoff et al. 2016; Kim et al. 2021). These strategies were validated in vivo through animal models.
Recently, new combination strategies have been applied in the research of drug delivery systems to maximize delivery of many cargos. In this review, we introduce recent articles that have developed and studied CPPs for selective and targeted drug delivery of various kinds of cargo (Scheme 2).

Schematic illustration of cell-penetrating peptide (CPP)-based delivery of cargos and strategies for enhanced specificity
CPP-based targeting strategies
CPPs are very useful for increasing the cellular uptake of various cargos but generally have little cell specificity. In particular, a topical treatment or a direct injection system to the target site is quite applicable, but in other drug delivery cases such as intraperitoneal injection, intravenous injection, and oral administration, because the administration site and the target region do not match, drug accumulation at unexpected site increases, resulting in poor efficacy and many side effects. Therefore, rational strategies to overcome these limitations are needed for more specific biomedical applications and selective delivery. Many studies in progress are using specific environmental characteristics around the disease lesion such as pH conditions and overexpression of specific enzymes.
For example, acidic pH is the most common environment for tumors. In the tumor and disease-related tissues, the expression levels of disease-specific enzymes and membrane proteins are also significantly higher than in normal tissues. The differences in pH and expression level of biomolecules can be used as a key for enhancing the CPP-based drug delivery efficacy at the specific target sites. With these targeting strategies, CPP-mediated drug delivery can be used in various biomedical applications (Table 1).
pH-responsive change
One of the main strategies to increase the selectivity of the target site is to modify the CPP so that chemical or physical characteristics can be changed and delivered according to the environment of the target site. In general, the pH of a normal living body is 7.4, but in a specific inflammatory region of a lesion or cancer site, the pH becomes acidic (5.5–6.0). Controlling the cellular uptake efficiency of CPPs at a specific pH is a well-known method and is a very useful strategy to deliver the desired cargo selectively. Among the amino acids that constitute CPPs, histidine with the imidazole ring group is neutral at normal pH 7.4 but has a cationic charge at pH 6.0 or lower. Modified CPPs with histidine have been developed for pH-activatable drug delivery.
Malignant melanoma is a very aggressive and therapy-resistant cancer that is treated mainly by chemotherapy. However, there are many difficulties in using chemotherapy due to strong side effects, drug resistance, and poor malignant cell targeting. To complement this, studies on treatment methods that use combined chemotherapy are being actively conducted. Huang et al. (2020) tried to increase therapeutic effects against malignant melanoma by using a combination therapy of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and paclitaxel (PTX) (Fig. 1). They developed a targeting delivery system, TRAIL-[Lip-PTX]C18-TR, tailored to melanoma and an acidic tumor environment to increase the efficacy of the drug. First, PTX was encapsulated in a conventional liposome, and CPPs and TRAIL were introduced on the surface. CPPs were newly developed by connecting a pH-responsive peptide and stearic acid modified αvβ3 integrin binding CPP, TR (C(RGDfK)-AGYLLGHINLHHLAHL(Aib)HHIL- Lys-C18). Histidine-based pH-responsive C18-TR peptides selectively delivered liposomes to tumors and increased the release of PTX and TRAIL. In experiments using B16F10 melanoma cells in vitro, the IC50 of the TRAIL-[Lip-PTX]C18-TR treated group was 4.91-fold higher than the TRAIL-[Lip-PTX] treated group at pH 6.3. In addition, in vivo experiments using B16F10 melanoma-bearing C57 BL/6 mice found that TRAIL-[Lip-PTX]C18-TR treated mice had strong tumor reductions of 95.3% compared with the 67.7% of the group administered with TRAIL-[Lip-PTX].

Schematic illustration of the basic structure of TRAIL-[Lip-PTX]C18-TR and the mechanism of sequential tumor-site delivery. Reprinted with permission (Huang et al. 2020)
Artemisinin (ART) is used as an antimalarial drug but in addition to the treatment of malaria, ART is also used as a treatment for cancer and inflammatory diseases. However, ART has limited clinical applications due to its low solubility in water. Sun’s research group confirmed that ART had no effect at low micromolar concentrations and developed a pH-responsive targeted drug delivery system containing ART (Yu et al. 2021). A strategy of histidine-containing CPPs was used to increase the uptake efficiency under low acid conditions. First, ART was encapsulated in a liposome composed of cholesterol, SPC, and DSPE-PEG2000. Then the surface of the liposome was modified by (HE)10-G5-R6 CPPs (CHEHEHEHEHEHEHEHEHEHEGGGGRRRRRR) to develop an ART-loaded and surface-modified liposome (ART-Lip-HE-R6). By in vitro experiments with 4T1 cells, coumarin 6-loaded Lip-HE-R6 was mainly uptaken through clathrin-dependent endocytosis (62.49%). Lip-HE-R6 was confirmed to have a strong selective uptake at pH 6.5 rather than pH 7.4 compared to that of Lip-R6 without HE sequences. Even in tumor-bearing mice, DiR-loaded Lip-HE-R6 was tumor localized at a higher rate than DiR-loaded Lip-R6. In mice experiments in vivo, as well as being delivered at a high rate to the tumor location, the therapeutic effect was also higher than that of Lip-R6 which did not introduce HE, thereby overcoming the limitations of ART and demonstrating the possibilities of selective drug delivery.
Various CPPs and modified CPPs for targeted delivery generally operate at micromolar concentrations in vitro. On the other hand, LK CPP composed of lysine and leucine showed an effect at very low and tens of nanomolar concentrations. LK becomes a dimer connected by a disulfide bridge or helix loop helix structure. Both CPPs were confirmed to enter the cell at nanomolar concentrations. Nam et al. (2021) developed LH dimeric CPP (LH2, disulfide bridged dimer of LHHLCHLLHHLCHLAG) in which the lysine of the LK dimer was substituted with His (Fig. 2). LH2 was not only converted into a cationic charge at low pH but also underwent a structural change with increased helicity, showing pH responsiveness. As a result of examining the toxicity of various drugs in the form of conjugation and simple mixing with LK dimer in MDA-MB-231 cells, both showed higher cytotoxicity compared with the control group in weakly acidic conditions. In an MDA-MB-231 bearing mice model, when the LH2 dimer and LK2 dimer conjugated with Cyanine 5.5 were injected intravenously, LH2 showed a higher tumor localization rate as well as higher blood residual concentration. Above all, in a therapeutic effect experiment using tumor-bearing mice, both the conjugation of PTX and CPP (PTX-LH2(C)) and simple mixing (PTX-LH2(M)) showed a high antitumor effect even at low concentrations. This demonstrated the high possibility of selectively delivering with simple methods.

Schematic representation of pH-activatable penetration of cell-penetrating peptide, LH2, into the acidic tumor region. Reprinted with permission (Nam et al. 2021)
Enzyme-triggered cleavage
Another strategy for site-specific drug delivery is to target a substance overexpressed around a specific disease lesion. In particular, CPPs can be modified by introducing an overexpressed enzyme substrate and this can be a key to switching on/off cellular uptake of the CPPs or the release of the cargo, therefore, this could be a good strategy for targeted drug delivery. A large number of peptide sequences serve as substrates for overexpressed enzymes around disease lesions. Many studies have been conducted on CPPs fused to specific enzyme-cleavable peptides and applied to enzyme-responsive drug delivery carriers.
Tumor cells in deep tissue have a higher autophagy level than tumor cells in surface, and ATG4B, an autophagy-specific enzyme, is extensively expressed in deep tumor cells. The peptide fragment of the LC3 protein, GTFGF sequence, was a specific substrate of ATG4B, and was strongly sensitive to ATG4B. Wang et al. (2022) developed an autophagy-responsive CPP (GR9, GTFGFRRRRRRRRR) to target ATG4B, and PLGA-based autophagy-responsive multifunctional nanoparticles (PGNs) were combined with it. They synthesized enzyme-cleavable CPPs (DSPE-PEG2000-GR9) by conjugation with GTFGF, DSPE-PEG2000, and R9. PGNs were manufactured by mixing DSPE-PEG2000-GR9, PLGA polymer, docetaxel (DTX, a chemotherapy drug), and chloroquine (CQ), an autophagy inhibitor. PRN, nanoparticles using R9 instead of GR9, were also tested. For in vitro experiments, coumarin 6 loaded PGN stayed longer in B16F10 cells and also showed a higher uptake ratio in the B16F10 spheroid model. In a cytotoxicity test using CQ and DTX-loaded PGN and PRB, the PGN treated group had an IC50 value of 72.04 µg/mL, whereas the IC50 value of the PRN treated group was 118.85 µg/mL in 3D tumor spheroids. The PGN group also showed a higher therapeutic effect than other groups in therapeutic experiments in vivo.
Tumor associated neutrophil extracellular traps (NETs) are involved in both the progression and prognosis of various tumors. Inhibition of NET development can be an advantage in tumor inhibition. Matrix metalloproteinase 9 (MMP9) is a component of NET and has been studied as a substance for enzyme-cleavable targeting. Therefore, MMP9-cleavable CPP can be manufactured by adding MMP9-substrate peptides to CPP. By combining CPP with drugs or other functional materials, it is possible to ‘switch on’ cellular uptake and drug delivery under MMP9-rich conditions. Yin et al. (2021) developed an enzyme-cleavable targeted delivery system for the inhibition of malignant tumor growth designated as mP-NPs-DNase/PTX (Fig. 3). First, they prepared self-assembled PTX nanoparticles by conjugating PTX with a stearyl group via disulfide. Then, DNase I was conjugated to the MMP9 substrate peptide (PLGLAGGC) linked to TAT and used as a shell of PTX nanoparticles. In A549 cells, the intensity of coumarin-6, the encapsulated substance in the nanocarrier introduced with the MMP9 substrate sequence, was much higher than in other groups. The MMP9 substrate peptide (PLGLAGGC) linked CPP (TAT) acts as a navigator for selective delivery. Then, MMP9 degraded the peptides and DNase were released, following the degradation of the NETs. TAT exposure enabled PTX to enter the cell. In a 4T1 tumor mice model, the mouse group using the mP-NPs-DNase/PTX nanocarrier showed a higher inhibition in both tumor formation and lung metastasis.

Schematic representation of MMP9 responsive mP-NPs-DNase/PTX construction and NET regulation to enhance the inhibition of malignant tumor growth and distant metastasis. Reprinted with permission (Yin et al. 2021)
The most important mediator in preventing and treating secondary damage in traumatic brain injury (TBI) is the mitochondria. Cyclosporin A (CsA), a neutral cyclic polypeptide consisting of 11 amino acids, is the most promising neuroprotective therapeutic that maintains the integrity of mitochondrial function. It is difficult for CsA to enter the TBI region in sufficient amounts due to hydrophobicity and strong binding to plasma protein. In addition, high doses can lead to side effects such as neurotoxicity, nephrotoxicity, immunosuppression, and hepatotoxicity. Therefore, targeted delivery is required to bring about a neuroprotective effect. As a solution, Chen et al. (2020) developed CsA-loaded multifunctional nanocarriers, an MMP9-sensitive CPP-decorated reconstituted lipoprotein (CsA-MCRL). They developed a fusion peptide that combined an MMP9 activatable cell-penetrating peptide (MAP, RRRRRRRRR-PVGLIG-EGGEGGEGG) and an apolipoprotein mimic α-helix peptide (AC-FAEKFKEAVKDYFAKFWD) with a GSG linker. Then, the fusion peptide was bound to the surface of a lipoprotein biomimetic nanocarrier encapsulated with CsA. When TBI occurs, MMP9 expression increases, and the blood–brain barrier (BBB) opens. When DiR-labeled MCRL was applied to primary astrocytes and neurons with MMP9, both cellular uptake and localization on mitochondria were increased (0.84 of CsA-MCRLs, 0.46 of CsA-RLs). When cells were treated with oxygen-glucose derivatives and then treated with CsA-MCRL, it was shown that mitochondrial potential and neuron cell viability were restored compared to that of the CsA alone group. In addition, the intensity of DiR-labeled MCRL increased accumulation in the brain after intravenous injection into CCI mice.
Multiple targeting ligands
The active targeting ability of CPP conjugated drugs or vehicles is important in enhancing drug delivery efficacy in specific cells and reducing non-specific delivery. One of the most straightforward methods for cell-specific delivery is a direct conjugation of specific cell-targeting moieties along with CPP itself or CPP decorated vehicles. Obesity is excessive fat storage in the body, resulting from an impaired balance between energy intake and expenditure. There is no approved drug for obesity treatment that can increase energy spending. Brown adipose tissue (BAT) is a primary site of energy expenditure through thermogenesis. Transforming white adipose tissue (WAT) to BAT can be a promising strategy for obesity treatment. Hiradate et al. (2021) reported a white adipocyte-targeted CPP-mediated drug delivery system. They synthesized rosiglitazone (Rosi)-loaded nanoparticles (Rs-NPs) and functionalized the Rs-NPs with prohibitin-targeting peptides (CKGGRAKDC, PTNP) on the surface ligands of WAT and CPP moieties composed of octaarginine, R8. The dual target Rs-NPs (Rs-R8-PTNP) showed significantly higher intracellular delivery efficiency than CPP-free PTNP. Mature adipocyte-specific delivery of Rs-R8-PTNP was also confirmed by flow cytometry and confocal laser scanning microscopy (CLSM). Compared with a non-treated obese mouse model, Rs-R8-PTNP significantly reduced body weight (~ 7%) of the obese mouse model with no difference in food intake, and an upregulated fat browning marker (Ucp1) was confirmed by qRT-PCR.
Affibodies are small, engineered proteins (~ 6 kDa) composed of a few tens of peptides that can specifically target protein, imitating monoclonal antibodies (~ 150 kDa). To introduce cell-specific uptake ability, affibody conjugation on CPPs and CPP-functionalized drugs are attractive methods for efficient antitumor therapy. However, chemical conjugation methods might affect the 3D structure of the affibody and lower its binding affinity on target proteins. Chong et al. (2021) developed a fusion protein of HER2 affibody and alpha-helical CPP (LK-2-ZHER2) (Fig. 4). LK-2-ZHER2 showed a selective penetrating ability for HER2 positive SKBR-3 cells, which was 12 times more efficient than HER2 negative HCC-1937 cells. The CPP fusion protein does not require any additional chemical reaction that might affect the binding affinity of the HER2 affibody (Kd = 22 pM). They conjugated doxorubicin (DOX) with LK-2-ZHER2 (LK-2-ZHER2-DOX) and evaluated the drug efficacy under both cultured cell levels and a 3D spheroid cell system. Cell-specific drug efficacy of LK-2-ZHER2-DOX was confirmed by the cytotoxicity comparison between HER2 positive and negative cell lines. Penetration into the deep center of the 3D spheroid was also enabled by LK-2-ZHER2, whereas the HER2 affibody itself showed limited penetration in the peripheral region of the 3D spheroid. They also confirmed the LK-2-ZHER2 mediated drug delivery for selective targets in vivo. Using a tumor xenograft mouse model, a higher accumulation of Cy5.5 labeled LK-2-ZHER2 HER2 positive tumor over the HER2 negative tumor was confirmed by both an in vivo imaging system and CLSM.

LK-2-ZHER2 fusion protein as a selective cell-penetrating affibody for drug delivery in HER2 overexpressing cancer cells. Reprinted with permission (Chong et al. 2021)
Oral vaccination is one of the most efficient immunization methods for producing both mucosal IgA and serum IgG. Extensive intestine immune responses, such as recruiting M-cells through follicle-associated epithelium (FAE), can be initiated through the largest surface area of gut-associated lymphoid tissue. Despite the convenience of oral administration, several obstacles, which include low absorption rate and specificity, limit the number of developed oral vaccines for human use. Surwase et al. (2022) synthesized a chitosan-based tandem polymer conjugated with a mucus-penetrating polymer (low-molecular polyethylene glycol), CPPs, and integrin receptor targeting moieties (cyclic RGDfK peptide) for M-cell-specific delivery. They prepared vaccine microparticles (termed NiMOS) composed of alginic acids, ovalbumin (OVA), and cationic chitosan-based tandem polymers via emulsification and ionic gelation. The NiMOS showed efficient OVA loading at acidic pH (pH 1.2) and an OVA releasing property at neutral pH (pH 6.8). Efficient cellular uptake of FITC labeled OVA contained NiMOS was confirmed by CLSM and flow cytometry, and NiMOS showed marginal cytotoxicity on the human intestinal epithelial cell line. In addition, the NiMOS showed M-cell homing permeability in both cultured M-cell models that mimicked the FAE transportation in the intestine and mouse models. After the immunization of NiMOS, the production of serum IgG and mucosal IgA was significantly increased through induction by Th1 and Th2 cytokines from CD4+ and CD8+ T cells.
Application of the CPP-based delivery system
We summarized the various strategies that employ the specific targeting ability to the CPP-based cargo delivery system (Table 2). Based on these strategies, different types of cargo, such as drugs, genes, proteins, and nanoparticles can be delivered specifically for curing cancers and diseases with fewer off-target events. Here, we showed that such cargo in the CPP-mediated delivery platform can be used for specific biomedical applications, including anticancer and muscular dystrophy therapy.
Small molecule drugs
FDA approved small molecule drugs have been extensively used in cancer therapy for decades.The half life of small molecule drugs is usually too short to effectively treat cancer cells in vivo. Poor membrane permeability of FDA-approved small molecule drugs (e.g. oxaliplatin) limits their cancer treatment efficacy. In turn, an excessive dose of small molecule drugs is often required to treat cancer tissues. However, several small molecule drug candidates exhibit off-target cytotoxicity. The cytotoxicity could induce critical side effects, which is the main reason for clinical trial failure. Singh et al. (2021) developed octaarginine-oxaliplatin conjugates (CPP-Oxal) that enhanced intracellular delivery, which was mainly governed by macro-pinocytosis. To increase the half-life of oxaliplatin, they functionalized the CPP with dicarboxylic acids that can be conjugated with Pt(II) of oxaliplatin via bidentate chelation. CPP conjugation on the oxaliplatin resulted in not only increased intracellular delivery efficiency but also passive targeting ability in vivo. Dose-dependent cytotoxicity of CPP-Oxal was confirmed in three different cancer cell lines, whereas the physical mixture of CPP and oxaliplatin showed low antitumor activity under the same drug concentration range. Intracellular localization of CPP-Oxal was further studied after treatment with endocytosis inhibitors. Colocalization of CPP-Oxal and a mitotracker in cultured cells was also confirmed. In vivo antitumor activity of the CPP-Oxal conjugate showed an approximately two times higher tumor inhibition ratio than intact oxaliplatin in mice of the same body weight.
Cabazitaxel (CBT), a paclitaxel analog drug, has been used in anticancer therapy with an improved pharmacokinetic profile. With additional polymer formulation, the aqueous stability of CBT was increased. However, low cellular uptake of CBT remains an issue. Active targeting of small molecule drugs is essential to improving the efficacy of antitumor therapy. Park et al. (2021) synthesized cyclic CPP conjugated CBT (cCPP-CBT) via an ester bond that can be hydrolyzed by esterase. They also conjugated targeting peptides with cCPP-CBT (TP-cCPP-CBT) via a disulfide bond that can be labile under a reductive environment. With these environment-sensitive cleavable bonds, TP-cCPP-CBT can be fully activatable and function as intact CBT after intracellular delivery in a cell-specific manner. The prodrug, TP-cCPP-CBT, was successfully converted into CBT in human plasma for 24 h. The conversion process was confirmed under a slightly acidic pH (pH 6.5) and reductive environment (in the presence of glutathione). The cellular uptake of TP-cCPP-CBT in cells with the targeted ligand overexpressed was quantified and visualized by CLSM and flow cytometry.
Retinoblastoma (RB) is the most common cancer of the retina in infancy with brain and bone marrow metastases, causing life-threatening disease. Melphalan, which is a commonly used drug for RB, was delivered via multiple intravitreal injections by well-trained clinicians because of the fast elimination of melphalan in the eye. Even though topical instillation of drugs is usually favored, efficient delivery of small molecule drugs into the posterior ocular is hard to achieve without invasive injections. Jiang et al. (2022) synthesized ocular permeable peptide conjugated melphalan (89WP-Mel) and demonstrated its anticancer ability in a mouse model in vivo. The 89WP-Mel conjugate exhibited a targeted delivery through the sclera region and penetration across the posterior ocular barriers. They confirmed that the intracellular delivery of 89WP-Mel followed a clathrin-mediated endocytosis pathway. Penetration through the epithelial barrier, which was tested using in vitro models of ARPE-19 cell monolayer, was visualized in the case of dye-labeled 89WP. Topical instillation of 89WP-Mel efficiently inhibited the intraocular tumor proliferation in vivo using a mouse model. The effective tumor penetration ability of 89WP-Mel is comparable to the intravitreal injection of melphalan. CPP conjugation improved the pharmacokinetics of 89WP-Mel by enhancing (> 1000 times higher than intact melphalan) water solubility and penetration ability. Topical instillation of CPP conjugated small molecule drugs can be an attractive therapeutic method that replaces a complicated and invasive intravitreal injection.
Chronic rhinosinusitis with nasal polyps (CRSwNP) is a disease localized in the sinonasal cavity, which can develop into aspirin intolerance or asthma. Resveratrol (RSV) is a natural polyphenolic activator for SIRT1, relating to the downregulation of HIF1α. Downregulation of HIF1α induced the suppression of epithelial-to-mesenchymal transition (EMT), which is an important step for the formation of nasal polyps. Alpha-helix is one of the secondary structures in which the amino acids are stabilized in a spiral. Leucine and lysine-rich alpha-helical peptides (LK) are known to interact and penetrate the cell membrane even at low concentrations (10–100 nM). Kim et al. (2020) synthesized RSV-LK conjugates via a phenolic ester bond, which is designed to release free RSV after endocytosis (Fig. 5). The half-life of free RSV release from RSV-LK under esterase was 1.5 h. RSV-LK showed higher cellular uptake than the physical mixture of RSB/LK and RSV-TAT conjugates. After endocytosis, over 85% of RSV-LK was localized in the cytoplasm. In vitro assay for the RSV-LK-treated group showed a similar suppression effect on EMT markers with 20 times lower concentration compared to the free RSV-treated group. The in vivo inhibitory effect of RSV-LK on polyposis was comparable to a 10 times higher concentration of free RSV. The histochemical analysis confirmed that no significant inflammation markers were observed after RSV-LK treatment, whereas free RSV or free LK induced severe inflammation.

Schematic representation of the synthetic scheme of RSV-CPP conjugates efficiently delivering RSV-CPP conjugates with inhibitory effects on nasal polyp formation. Reprinted with permission (Kim et al. 2020)
Peptides
Peptide drugs are usually either too bulky or hydrophilic (with negative charges) to be delivered efficiently through the epithelial cell membrane. To increase intracellular delivery efficiency, CPPs have been conjugated with peptide drugs via chemical bond formation, electrostatic attraction, and hydrophobic interaction.
Insulin is one of the most common therapeutic peptides. It is hard to deliver through oral administration due to its high hydrophilicity and molecular weight. Amphipathic and cationic CPPs have been used as carrier proteins to enhance the intracellular delivery of hydrophilic therapeutic peptides. Diedrichsen et al. (2021) prepared CPP-insulin composites and studied their drug efficacy. The CPP-insulin composite was a few hundred nanometers in hydrodynamic size. For CPP-lipidated insulin composites, micron-sized aggregation composed of lipidated insulin was mainly formed with a low insulin recovery percentage (< 10%). They used three different CPPs, penetratin, shuffle, and penetramax, to prepare the CPP-insulin composites. Alpha-helix secondary structure of CPP-insulin composites was induced at a 10 times higher level than that of free CPPs. Circular dichroism confirmed that the alpha-helix degree of CPP-insulin after liposome treatment was about three times higher than without liposome, meaning the amphipathic CPP is stabilized by lipids. Cellular uptake of CPP-insulin was dramatically enhanced in the shuffle and penetramax cases. These shuffle and penetramax insulin composites showed high insulin bioavailability and regulated the blood glucose level in the normal range after administration. However, transient inflammation of connective tissues and epithelial cells in the intestine was observed, which requires further studies.
Another therapeutic peptide drug, glucagon-like peptide-2 (GLP-2), has been applied for treating depression. Despite the invasiveness, intracerebroventricular administration is the most common delivery method for GLP-2. To deliver protein drugs into the brain efficiently and non-invasively, the intranasal administration of GLP-2 has been studied. Akita et al. (2021) conjugated GLP-2 with CPP and penetration accelerating sequences (PAS-CPP-GLP-2, sequence: PAS(FFLIPKG)-CPP (RRRRRRRR)-spacer(GG)-GLP-2(HADGSFSDEMNTILDNLAARDFINWLIQTKITD)) (Fig. 6). PAS-CPP-GLP-2 was mainly delivered via micropinocytosis with CPP and escaped the endosomal structure by PAS. Colocalization of PAS-CPP-GLP-2 with early endosome was visualized by CLSM. Extracellular movement of PAS-CPP-GLP-2 via a transcellular route through the gap between the cells was also demonstrated using a microchamber device. Only PAS-CPP-GLP-2 had an antidepressive effect, whereas CPP-GLP-2 or PAS-GLP-2 did not exhibit a significant reduction in the depression score, which was quantified by counting periods of immobility.

Schematic representation of the intracellular delivery mechanism of GLP-2 peptide showing antidepression-like effect by intranasal bioactivity. Reprinted with permission (Akita et al. 2021)
Nucleic acids
Gene medicines including plasmid DNA (pDNA), an antisense oligonucleotide (ASO), siRNA, miRNA, and mRNA have been studied for a long time. Intrinsically, genes are not suitable for cellular uptake due to the electronic repulsion with an anionic cell surface. Therefore, additional methods such as viral vectors or non-viral carriers are essential. CPPs are useful in this field and some studies showed their potential for gene delivery without other reagents. McErlean et al. (2021) developed a linear CPP by rational design considering the role of amino acids including arginine, tryptophan, histidine, and cysteine, which are reported as advantageous for gene delivery (Fig. 7). They used the Protparam tool to compute the physicochemical property of CPP and Maestro 3D structure modeling software to predict its structure. During this process, they synthesized six sequences of CPP candidates and selected the sequence CHHHRRRWRRRHHC (CHAT) by comparing transfection efficiency in MCF7 and MDA-MB-231 cells. The cellular uptake pathway of CHAT was determined to be multiple, but caveolae and endosomal clathrin were dominant. After an intratumoral injection in a 4T1 breast cancer xenograft, CHAT complexed with luciferase pDNA showed significant luciferase expression that is comparable with the case of in vivo PEI, a commercial transfection agent. The authors also tried intravenous injection and observed bioluminescence in the liver, lung, kidney, and tumor. This result was expected because many CPPs have no specificity to target cells and targeting strategies would be helpful as mentioned above.

Gene delivery using cell-penetrating peptides (CPPs). Plasmid DNA delivery with 15 amino acid linear peptide and evaluation in 4T1 breast cancer xenograft. Reprinted with permission (McErlean et al. 2021)
Duchenne muscular dystrophy (DMD) is a genetic disorder due to the loss of dystrophin resulting in body-wide muscle degeneration. Exon skipping is a promising DMD therapy approved by the FDA, which uses phosphorodiamidate morpholino oligomer (PMO). To increase the efficiency of exon-skipping therapy, Yokota’s research group directly conjugated DG9 peptide to exon-skipping PMO (Lim et al. 2022). DG9 is a CPP that originated from the protein transduction domain in the human Hph-1 transcription factor. After retro-orbital injection into hDMDdel52; mdx mice, DG9-conjugated PMO showed a 2.2 to 12.3-fold and 14.4-fold higher skipping efficiency compared to unconjugated PMO in skeletal muscles and heart, respectively. After repeated injection of DG9-PMO once weekly for 3 weeks, 55 to 71% exon 51 skipping level and 2.8 to 3.9% dystrophin production were observed in skeletal muscles. There was no significant toxicity observed after the injection of DG9-PMO. In addition, the intramuscular injection of DG9-PMO in the tibialis anterior was also effective and showed dystrophin restoration demonstrating the potential of DG9-PMO for DMD therapy.
Similarly, Bersani et al. (2022) conjugated CPP to oligonucleotides and applied it to spinal muscular atrophy (SMA) therapy. SMA is a motor neuron disease and a representative genetic disease that results in infant death. The limited delivery of the currently used ASO in the rostral spinal and brain has reduced the therapeutic efficacy. The authors synthesized morpholino oligomer (MO) with increased survival motor neuron 1 (SMN) protein levels. They conjugated CPPs including TAT (YGRKKRRQRRRQ), r6, R6, and (RXRRBR)2XB (RXR) to the MOs and compared the amount of SMN proteins after injections in mice. Based on the results, they selected r6 and RXR peptides and applied them to symptomatic SMA mice. Interestingly, after intraperitoneal injection, RXR-MO and r6-MO conjugates were found in the central nervous system in a symptomatic phase with a completely closed BBB. In symptomatic SMA mice, RXR-MO and r6-MO conjugates showed improved median survivals of 41.4 and 23 days, respectively. These values are significantly higher than 17 days, in the case of naked MO without CPP. Pathological data showed that CPP-MOs ameliorate the degeneration of neuromuscular junctions more effectively compared to scrambled or naked MOs, demonstrating the usefulness of CPPs in gene therapy.
Nanoparticles
Nanoparticles (NPs) are regarded as promising carriers for drug and gene delivery (Koo et al. 2011). The wide human application of mRNA-containing lipid NP as a COVID-19 vaccine is a representative case (Hou et al. 2021). Most NP carriers require cellular uptake and intracellular release of their cargo including drugs and genes (Jo et al. 2020). To accelerate the cellular uptake of NPs, CPPs have been additionally decorated on the surface of NPs. For example, He et al. (2022) modified PLGA/PEI NPs complexed with the pDNA and KALA peptide (WEAKLAKALAKALAKHLAKALAKALKACEA), a CPP (Fig. 8). The resulting complex was about 200 nm in size and its zeta potential value is about + 15 mV. KALA peptide enabled fast cellular uptake and effective transfection with low cytotoxicity compared to PEI with cationic charges at high density resulting in plasma-membrane destabilization. The authors used a combination of KALA peptide-modified PLGA/PEI NPs containing pDNA and 3D alginate nanofibrous gene-activated matrix coated with polydopamine. In this combination, pDNA was slowly released from the matrix for 10 days, and HEK 293 T cells showed GFP expression by transfection over three weeks. For an in vivo test, the authors prepared full-thickness excisional wound models using SD rats. After the wound was covered with nanofiber scaffolds combined with KAKA-PLGA/PEI NPs containing VEGF-pDNA, nearly complete wound closure was observed after 21 days whereas 27.3 wound areas remained in the control group. Collagen deposition and rapid vascularization in tissue observed after Masson’s trichrome, CD31, and alpha-SMA staining showed effective therapy by KALA peptide-modified NPs and nanofiber scaffolds.

Nanoparticle (NP) delivery using cell-penetrating peptides (CPPs). KALA peptide-decorated PLGA/PNI NPs for delivery of the VEGF gene for wound healing in SD rats. Reprinted with permission (He et al. 2022)
Recently, Yang et al. (2022) introduced an intranasal siRNA delivery system for glioma therapy. They conjugated cholesterol to the N-terminal of the antimicrobial peptide DP7. The resulting CPP, DP7-C, enabled fast cellular uptake and could create a complex with siRNA. For bio-adhesive properties, the DP7-C/siRNA complex was coated with hyaluronic acid to form a core–shell nano-micelle structure. When compared with TAT and R9 peptides, DP7-C increased transfection efficiency in GL261 cells to over 80%. The biggest hurdle in drug delivery to glioma is the BBB, which blocks the entrance of drugs to the brain after oral or intravenous administration. Therefore, the authors used an intranasal route that is noninvasive and can bypass the BBB. After intranasal administration into mice, both HA/DP7-C/siRNA and DP7-C/siRNA showed a high accumulation of Cy5-labeled siRNA in the brain and the HA-coated one was the more efficient. In particular, the fluorescence signal in trigeminal nerves was higher than in olfactory bulbs revealing the main route of nose-to-brain transport. When HA/DP7-C/siRNA with VEGF or PLK1 sequence was treated on the GL261 tumor model, the median survival time was prolonged to 17 and 18 days, respectively. Body weights and pathological data showed no significant changes after treatment of HA/DP7-C/siRNA showing it was biocompatible.
Enhanced cellular uptake of NPs by CPP is also useful in cancer therapy. Liu et al. (2021) prepared DTX-loaded transfersomes modified with R8H3, a CPP containing 8 arginines and 3 histidines. The R8H3-transfersome had a faster cellular uptake and lower cell viability compared to those without R8H3 in B16F10 melanoma and 4T1 breast cancer cell lines due to the efficient delivery of DTX. For a more sustained release and supply of DTX, the transfersomes were encapsulated in oligopeptide hydrogel. The hydrogel was assembled by binding between Fmoc-Phe and Phe-Phe-Dopa. The authors prepared B16F10 tumor-bearing mice and surgically removed the primary tumor tissues. After the topical painting of the hydrogel mixed with R8H3-transfersome containing DTX onto the tumor area, the growth of recurrent tumors was efficiently suppressed. Importantly, the difference between groups with and without R8H3 was significant. In 4T1 tumor-bearing mice, the authors tested this system by intratumoral injection. They used luciferase-expressing 4T1 cells, which enabled the monitoring of tumor growth by luminescence imaging. In this model, the R8H3-transfersome showed near-complete tumor suppression due to the long-term drug supply from hydrogel and the efficient cellular uptake by R8H3 demonstrating the promising potential of CPPs.
Conclusion
In summary, we introduced the recent application of CPPs in drug and gene delivery. To improve the specificity of the CPP-based delivery system, researchers suggested rational strategies using pH-responsive structural change, enzyme-triggered cleavage, and multiple targeting ligands. Based on the advantages of CPPs and with the help of these strategies, CPPs have been applied in the delivery of various materials including small molecules, proteins, genes, and NPs. Many studies have demonstrated promising results in animal models and proved the usefulness of CPPs in wide applications in biomedical fields.
For human usage of CPP containing materials, the safety issue is maybe the biggest hurdle (Bobo et al. 2016). Most CPPs have arginine or lysine residues containing cationic charges, which can destabilize the cell membrane. Therefore, in human applications, the sequence of CPP should be considered carefully from the viewpoint of potential toxicity as well as the efficacy of cellular uptake. Cost is another important factor for commercialization. Sequences that are too long or complicated would increase the overall cost of massive production, which would be an obstacle to convincing investors (Cheng et al. 2012).
In contrast to other biological ligands, CPPs have intrinsic advantages. First, most of them enable the direct transfer of materials through membranes without an endo-lysosomal pathway. Most drugs need to move to the target organelles after cellular uptake and their entrapment in endosomes or lysosomes reduces their drug efficacy. This is the reason why endosomal escape has been a critical issue in drug and gene delivery research for a long time (Pittella et al. 2011). Particularly, in the case of fragile genes or proteins, they can be easily denatured or degraded in lysosomes with acidic pH and various enzymes (Biscans et al. 2022). In addition, CPPs can be synthesized by both solid-phase synthesis or recombinant DNA technology in living organisms. Researchers can select suitable methods according to the sequence and length of the CPPs. These intrinsic advantages of CPP and its fast cellular uptake make CPP an attractive component of drug and gene delivery, and we expect that the human application of CPP-based materials will increase in the near future.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Akita T, Kimura R, Akaguma S, Nagai M, Nakao Y, Tsugane M, Suzuki H, Oka JI, Yamashita C (2021) Usefulness of cell-penetrating peptides and penetration accelerating sequence for nose-to-brain delivery of glucagon-like peptide-2. J Control Release 335:575–583. https://doi.org/10.1016/j.jconrel.2021.06.007
Bersani M, Rizzuti M, Pagliari E, Garbellini M, Saccomanno D, Moulton HM, Bresolin N, Comi GP, Corti S, Nizzardo M (2022) Cell-penetrating peptide-conjugated Morpholino rescues SMA in a symptomatic preclinical model. Mol Ther 30:1288–1299. https://doi.org/10.1016/j.ymthe.2021.11.012
Biscans A, Ly S, Mchugh N, Cooper DA, Khvorova A (2022) Engineered ionizable lipid siRNA conjugates enhance endosomal escape but induce toxicity in vivo. J Control Release 349:831–843. https://doi.org/10.1016/j.jconrel.2022.07.041
Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR (2016) Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res 33:2373–2387. https://doi.org/10.1007/s11095-016-1958-5
Buccini DF, Cardoso MH, Franco OL (2021) Antimicrobial peptides and cell-penetrating peptides for treating intracellular bacterial infections. Front Cell Infect Microbiol. https://doi.org/10.3389/fcimb.2020.61.2931
Chen L, Song Q, Chen Y, Meng S, Zheng M, Huang J, Zhang Q, Jiang J, Feng J, Chen H, Jiang G, Gao X (2020) Tailored reconstituted lipoprotein for site-specific and mitochondria-targeted cyclosporine a delivery to treat traumatic brain injury. ACS Nano 14:6636–6648. https://doi.org/10.1021/acsnano.9b09186
Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A (2012) Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science 338:903–910. https://doi.org/10.1126/science.1226338
Chong S-E, Lee D, Oh JH, Kang S, Choi S, Nam SH, Yu J, Koo H, Lee Y (2021) A dimeric α-helical cell penetrating peptide mounted with an HER2-selective affibody. Biomater Sci 9:7826–7831. https://doi.org/10.1039/D1BM00819F
Derossi D, Joliot AH, Chassaing G, Prochiantz A (1994) The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem 269:10444–10450. https://doi.org/10.1016/S0021-9258(17)34080-2
Diedrichsen RG, Harloff-Helleberg S, Werner U, Besenius M, Leberer E, Kristensen M, Nielsen HM (2021) Revealing the importance of carrier-cargo association in delivery of insulin and lipidated insulin. J Control Release 338:8–21. https://doi.org/10.1016/j.jconrel.2021.07.030
Falanga A, Galdiero M, Galdiero S (2015) Membranotropic cell penetrating peptides: the outstanding journey. Int J Mol Sci 16:25323–25337. https://doi.org/10.3390/ijms161025323
Feger G, Angelov B, Angelova A (2020) Prediction of amphiphilic cell-penetrating peptide building blocks from protein-derived amino acid sequences for engineering of drug delivery nanoassemblies. J Phys Chem B 124:4069–4078. https://doi.org/10.1021/acs.jpcb.0c01618
Gallo M, Defaus S, Andreu D (2019) 1988–2018: Thirty years of drug smuggling at the nano scale. Challenges and opportunities of cell-penetrating peptides in biomedical research. Arch Biochem Biophys 661:74–86. https://doi.org/10.1016/j.abb.2018.11.010
Green M, Loewenstein PM (1988) Autonomous functional domains of chemically synthesized human immunodeficiency virus tat <em>trans</em>-activator protein. Cell 55:1179–1188. https://doi.org/10.1016/0092-8674(88)90262-0
Guidotti G, Brambilla L, Rossi D (2017) Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci 38:406–424. https://doi.org/10.1016/j.tips.2017.01.003
He S, Fang J, Zhong C, Ren F, Wang M (2022) Controlled pVEGF delivery via a gene-activated matrix comprised of a peptide-modified non-viral vector and a nanofibrous scaffold for skin wound healing. Acta Biomater 140:149–162. https://doi.org/10.1016/j.actbio.2021.11.037
Heitz F, Morris MC, Divita G (2009) Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol 157:195–206. https://doi.org/10.1111/j.1476-5381.2009.00057.x
Herce HD, Garcia AE, Litt J, Kane RS, Martin P, Enrique N, Rebolledo A, Milesi V (2009) Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys J 97:1917–1925. https://doi.org/10.1016/j.bpj.2009.05.066
Hiradate R, Khalil IA, Matsuda A, Sasaki M, Hida K, Harashima H (2021) A novel dual-targeted rosiglitazone-loaded nanoparticle for the prevention of diet-induced obesity via the browning of white adipose tissue. J Control Release 329:665–675. https://doi.org/10.1016/j.jconrel.2020.10.002
Hou X, Zaks T, Langer R, Dong Y (2021) Lipid nanoparticles for mRNA delivery. Nat Rev Mater. https://doi.org/10.1038/s41578-021-00358-0
Huang S, Zhang Y, Wang L, Liu W, Xiao L, Lin Q, Gong T, Sun X, He Q, Zhang Z, Zhang L (2020) Improved melanoma suppression with target-delivered TRAIL and Paclitaxel by a multifunctional nanocarrier. J Control Release 325:10–24. https://doi.org/10.1016/j.jconrel.2020.03.049
Hyun S, Lee Y, Jin SM, Cho J, Park J, Hyeon C, Kim K-S, Lee Y, Yu J (2018) Oligomer formation propensities of dimeric bundle peptides correlate with cell penetration abilities. ACS Cent Sci 4:885–893. https://doi.org/10.1021/acscentsci.8b00262
Jiang K, Fan X, Hu Y, Yao S, Liu Y, Zhan C, Lu W, Wei G (2022) Topical instillation of cell-penetrating peptide-conjugated melphalan blocks metastases of retinoblastoma. Biomaterials 284:121493. https://doi.org/10.1016/j.biomaterials.2022.121493
Jo MJ, Jin IS, Park C-W, Hwang BY, Chung YB, Kim J-S, Shin DH (2020) Revolutionizing technologies of nanomicelles for combinatorial anticancer drug delivery. Arch Pharmacal Res 43:100–109. https://doi.org/10.1007/s12272-020-01215-4
Kawamoto S, Takasu M, Miyakawa T, Morikawa R, Oda T, Futaki S, Nagao H (2011) Inverted micelle formation of cell-penetrating peptide studied by coarse-grained simulation: importance of attractive force between cell-penetrating peptides and lipid head group. J Chem Phys 134:095103. https://doi.org/10.1063/1.3555531
Kim GC, Cheon DH, Lee Y (2021) Challenge to overcome current limitations of cell-penetrating peptides. Biochim et Biophys Acta (BBA)—Prot Proteom 1869:140604. https://doi.org/10.1016/j.bbapap.2021.140604
Kim Y, Hwang S, Khalmuratova R, Kang S, Lee M, Song Y, Park J-W, Yu J, Shin H-W, Lee Y (2020) α-Helical cell-penetrating peptide-mediated nasal delivery of resveratrol for inhibition of epithelial-to-mesenchymal transition. J Control Release 317:181–194. https://doi.org/10.1016/j.jconrel.2019.11.034
Koo H, Huh MS, Sun I-C, Yuk SH, Choi K, Kim K, Kwon IC (2011) In vivo targeted delivery of nanoparticles for theranosis. Acc Chem Res 44:1018–1028. https://doi.org/10.1021/ar2000138
Kurrikoff K, Gestin M, Langel Þ (2016) Recent in vivo advances in cell-penetrating peptide-assisted drug delivery. Expert Opin Drug Deliv 13:373–387. https://doi.org/10.1517/17425247.2016.1125879
Lim KRQ, Woo S, Melo D, Huang Y, Dzierlega K, Shah MNA, Aslesh T, Roshmi RR, Echigoya Y, Maruyama R, Moulton HM, Yokota T (2022) Development of DG9 peptide-conjugated single- and multi-exon skipping therapies for the treatment of Duchenne muscular dystrophy. Proc Natl Acad Sci 119:e2112546119. https://doi.org/10.1073/pnas.2112546119
Liu C, Ma Y, Guo S, He B, Jiang T (2021) Topical delivery of chemotherapeutic drugs using nano-hybrid hydrogels to inhibit post-surgical tumour recurrence. Biomater Sci 9:4356–4363. https://doi.org/10.1039/D0BM01766C
Mcerlean EM, Ziminska M, Mccrudden CM, Mcbride JW, Loughran SP, Cole G, Mulholland EJ, Kett V, Buckley NE, Robson T, Dunne NJ, Mccarthy HO (2021) Rational design and characterisation of a linear cell penetrating peptide for non-viral gene delivery. J Control Release 330:1288–1299. https://doi.org/10.1016/j.jconrel.2020.11.037
Munyendo WL, Lv H, Benza-Ingoula H, Baraza LD, Zhou J (2012) Cell penetrating peptides in the delivery of biopharmaceuticals. Biomolecules 2:187–202
Muttenthaler M, King GF, Adams DJ, Alewood PF (2021) Trends in peptide drug discovery. Nat Rev Drug Discov 20:309–325. https://doi.org/10.1038/s41573-020-00135-8
Nam SH, Jang J, Cheon DH, Chong S-E, Ahn JH, Hyun S, Yu J, Lee Y (2021) pH-Activatable cell penetrating peptide dimers for potent delivery of anticancer drug to triple-negative breast cancer. J Control Release 330:898–906. https://doi.org/10.1016/j.jconrel.2020.10.063
Park SE, El-Sayed NS, Shamloo K, Lohan S, Kumar S, Sajid MI, Tiwari RK (2021) Targeted delivery of cabazitaxel using cyclic cell-penetrating peptide and biomarkers of extracellular matrix for prostate and breast cancer therapy. Bioconjug Chem 32:1898–1914. https://doi.org/10.1021/acs.bioconjchem.1c00319
Pittella F, Zhang M, Lee Y, Kim HJ, Tockary T, Osada K, Ishii T, Miyata K, Nishiyama N, Kataoka K (2011) Enhanced endosomal escape of siRNA-incorporating hybrid nanoparticles from calcium phosphate and PEG-block charge-conversional polymer for efficient gene knockdown with negligible cytotoxicity. Biomaterials 32:3106–3114. https://doi.org/10.1016/j.biomaterials.2010.12.057
Reissmann S (2014) Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci 20:760–784. https://doi.org/10.1002/psc.2672
Rhee M, Davis P (2006) Mechanism of uptake of C105Y, a novel cell-penetrating peptide*. J Biol Chem 281:1233–1240. https://doi.org/10.1074/jbc.M509813200
Roberts TC, Langer R, Wood MJA (2020) Advances in oligonucleotide drug delivery. Nat Rev Drug Discov 19:673–694. https://doi.org/10.1038/s41573-020-0075-7
Ruseska I, Zimmer A (2020) Internalization mechanisms of cell-penetrating peptides. Beilstein J Nanotechnol 11:101–123. https://doi.org/10.3762/bjnano.11.10
Sadeghian I, Heidari R, Sadeghian S, Raee MJ, Negahdaripour M (2022) Potential of cell-penetrating peptides (CPPs) in delivery of antiviral therapeutics and vaccines. European J Pharm Sci 169:106094. https://doi.org/10.1016/j.ejps.2021.106094
Sarko D, Beijer B, Garcia Boy R, Nothelfer E-M, Leotta K, Eisenhut M, Altmann A, Haberkorn U, Mier W (2010) The pharmacokinetics of cell-penetrating peptides. Mol Pharm 7:2224–2231. https://doi.org/10.1021/mp100223d
Singh T, Kang DH, Kim TW, Kong HJ, Ryu JS, Jeon S, Ahn TS, Jeong D, Baek MJ, Im J (2021) Intracellular delivery of oxaliplatin conjugate via cell penetrating peptide for the treatment of colorectal carcinoma in vitro and in vivo. Int J Pharm 606:120904. https://doi.org/10.1016/j.ijpharm.2021.120904
Surwase SS, Shahriar SMS, An JM, Ha J, Mirzaaghasi A, Bagheri B, Park J-H, Lee Y-K, Kim Y-C (2022) Engineered nanoparticles inside a microparticle oral system for enhanced mucosal and systemic immunity. ACS Appl Mater Interfaces 14:11124–11143. https://doi.org/10.1021/acsami.1c24982
Tréhin R, Merkle HP (2004) Chances and pitfalls of cell penetrating peptides for cellular drug delivery. Eur J Pharm Biopharm 58:209–223. https://doi.org/10.1016/j.ejpb.2004.02.018
Vivès E, Brodin P, Lebleu B (1997) A truncated HIV-1 tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus*. J Biol Chem 272:16010–16017. https://doi.org/10.1074/jbc.272.25.16010
Wang F, Xie D, Lai W, Zhou M, Wang J, Xu R, Huang J, Zhang R, Li G (2022) Autophagy responsive intra-intercellular delivery nanoparticles for effective deep solid tumor penetration. J Nanobiotechnol 20:300. https://doi.org/10.1186/s12951-022-01514-6
Xiao M, Jasensky J, Foster L, Kuroda K, Chen Z (2018) Monitoring antimicrobial mechanisms of surface-immobilized peptides in situ. Langmuir 34:2057–2062. https://doi.org/10.1021/acs.langmuir.7b03668
Xu J, Khan AR, Fu M, Wang R, Ji J, Zhai G (2019) Cell-penetrating peptide: a means of breaking through the physiological barriers of different tissues and organs. J Control Release 309:106–124. https://doi.org/10.1016/j.jconrel.2019.07.020
Yang Y, Zhang X, Wu S, Zhang R, Zhou B, Zhang X, Tang L, Tian Y, Men K, Yang L (2022) Enhanced nose-to-brain delivery of siRNA using hyaluronan-enveloped nanomicelles for glioma therapy. J Control Release 342:66–80. https://doi.org/10.1016/j.jconrel.2021.12.034
Yin H, Lu H, Xiong Y, Ye L, Teng C, Cao X, Li S, Sun S, Liu W, Lv W, Xin H (2021) Tumor-associated neutrophil extracellular traps regulating nanocarrier-enhanced inhibition of malignant tumor growth and distant metastasis. ACS Appl Mater Interfaces 13:59683–59694. https://doi.org/10.1021/acsami.1c18660
Yu Y, Zu C, He D, Li Y, Chen Q, Chen Q, Wang H, Wang R, Chaurasiya B, Zaro JL, Wang Y, Tu J, Sun C (2021) pH-dependent reversibly activatable cell-penetrating peptides improve the antitumor effect of artemisinin-loaded liposomes. J Colloid Interface Sci 586:391–403. https://doi.org/10.1016/j.jcis.2020.10.103
Zhang Y, Guo P, Ma Z, Lu P, Kebebe D, Liu Z (2021) Combination of cell-penetrating peptides with nanomaterials for the potential therapeutics of central nervous system disorders: a review. J Nanobiotechnol 19:255. https://doi.org/10.1186/s12951-021-01002-3
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (2021R1A4A3031875, 2022R1A2C1008699), the Korean Fund for Regenerative Medicine (KFRM) grant (21C0702L1) and Korea Health Technology R&D Project (HV22C0069) through the Korea Health Industry Development Institute (KHIDI) funded by the Korea government (the MSIT, the Ministry of Health & Welfare).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nam, S.H., Park, J. & Koo, H. Recent advances in selective and targeted drug/gene delivery systems using cell-penetrating peptides. Arch. Pharm. Res. 46, 18–34 (2023). https://doi.org/10.1007/s12272-022-01425-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-022-01425-y