
Abstract
Excessive alcohol consumption contributes to a broad clinical spectrum of liver diseases, from simple steatosis to end-stage hepatocellular carcinoma. The liver is the primary organ that metabolizes ingested alcohol and is exquisitely sensitive to alcohol intake. Alcohol metabolism is classified into two pathways: oxidative and non-oxidative alcohol metabolism. Both oxidative and non-oxidative alcohol metabolisms and their metabolites have toxic consequences for multiple organs, including the liver, adipose tissue, intestine, and pancreas. Although many studies have focused on the effects of oxidative alcohol metabolites on liver damage, the importance of non-oxidative alcohol metabolites in cellular damage has also been discovered. Furthermore, extrahepatic alcohol effects are crucial for providing additional information necessary for the progression of alcoholic liver disease. Therefore, studying the effects of alcohol-producing metabolites and interorgan crosstalk between the liver and peripheral organs that express ethanol-metabolizing enzymes will facilitate a comprehensive understanding of the pathogenesis of alcoholic liver disease. This review focuses on alcohol-metabolite-associated hepatotoxicity due to oxidative and non-oxidative alcohol metabolites and the role of interorgan crosstalk in alcoholic liver disease pathogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alcohol consumption occurs in most cultures, and alcohol abuse or alcoholism is found worldwide (Massey and Arteel 2012; Griswold et al. 2018). Excessive alcohol consumption leads to chronic liver disease and damages multiple organs, primarily brain, heart, gastrointestinal tract, and liver (Rehm et al. 2017; Stockwell and Zhao 2017; Evangelou et al. 2021). Aside from the detrimental effects of alcohol on personal health, excessive alcohol intake also has serious socioeconomic impacts at the population level (D'Angelo et al. 2022; Keyes 2022).
After drinking, a small amount of alcohol is quickly metabolized in the stomach, and most of it is absorbed by the gastrointestinal tract, particularly the stomach and upper small intestine (Jones 2019). Following absorption, the portal vein delivers the alcohol to the liver, which then contains a higher alcohol level than the peripheral blood. Because the liver is the primary site of ethanol metabolism, it sustains the greatest degree of damage from heavy drinking (Osna et al. 2017). The pathogenesis of alcoholic liver disease (ALD) has been predominantly classified into three categories: (1) liver damage due to alcohol and its metabolic by-products, such as acetaldehyde; (2) hepatic injury inducing damage-associated molecular patterns (DAMPs), which recruit innate and adaptive immune cells, perpetuating liver injury; and (3) alcohol consumption affecting gut microbiota and permeability, allowing bacterial compounds to enter the liver and stimulate immunological responses and liver injury (Dunn and Shah 2016; Han et al. 2020).
Excessive alcohol consumption causes a wide range of liver diseases, from simple steatosis (fatty liver) to end-stage liver cancer (hepatocellular carcinoma) (Seo and Jeong 2016). Simple steatosis occurs in more than 90% of problem drinkers who consume at least 16 g of alcohol each day (Osna et al. 2017). In a minority of heavy drinkers, alcoholic steatosis progresses to steatohepatitis. The late stages of ALD are characterized by the development of fibrosis, which is accompanied by excessive deposition of extracellular matrix proteins (Hyun et al. 2021). Despite many attempts over decades to ameliorate ALD progression, the only effective ALD treatment is alcohol abstinence. Liver transplantation is the only accepted standard of care for patients suffering from end-stage liver disease. However, for patients whose disease has progressed to hepatocellular carcinoma, 15% die while awaiting liver transplantation due to a shortage of grafts (Frazier et al. 2011; Husen et al. 2019).
Chronic alcohol consumption (15 or more drinks per week according to the guidelines of the National Institute on Alcohol Abuse and Alcoholism) can lead to a broad spectrum of liver diseases and has been a research focus for decades. However, studies on the mechanisms and detrimental effects of binge alcohol drinking (a heavy episodic alcohol intake) have not been studied intensively. Binge alcohol drinking is a pattern of drinking that typically brings a person’s blood alcohol concentration (BAC) to 0.08 g/dL or more (Majumdar and Tsochatzis 2022). Binge drinking-associated BAC is defined as the consumption of 5 or more drinks within a few hours (about 2 h) for men or 4 or more drinks for women (Fillmore and Jude 2011).
Binge drinking is linked to reduced resistance to microbial infection, suppression of innate immune responses, and increased intestinal permeability (Shukla et al. 2013; Szabo and Saha 2015) (Fig. 1). Interestingly, the administration of binge drinking to chronically ethanol-exposed mice dramatically amplifies liver injury (Ki et al. 2010; Bertola et al. 2013). Besides alcohol-induced liver damage, excessive alcohol exposure affects interorgan crosstalk, further contributing to ALD progression (Dasarathy and Brown 2017). Therefore, this review focuses on the pathophysiological aspects of alcohol-associated liver injury (hepatotoxicity by oxidative/non-oxidative ethanol metabolites) as well as interorgan communications in the progression of ALD.

A mechanistic distinction between the effects of acute (binge) and chronic (heavy) alcohol consumption. Acute alcohol intoxication (BAC \(\ge \) 0.08 g/dL in ~ 2 h) is a clinically harmful condition that follows the ingestion of large amounts of alcohol. Acute alcohol exposure, also known as “binge drinking,” can alter immunological and metabolic signaling and epigenetic pathways. It also exacerbates alcoholic liver disease in the chronically alcohol-exposed liver
Effects of alcohol on the liver
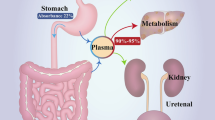
About 90% of alcohol is eliminated by the liver (only approximately 2–10% is removed by the kidneys and lungs) (Paton 2005; Jones 2019). Since alcohol is mostly metabolized by hepatocytes, the liver is a primary target for the detrimental effects of alcohol (Osna and Kharbanda 2016). Excessive alcohol consumption causes hepatic inflammation. The Kupffer cells, macrophages that reside in the liver, play a role in the innate immune response by producing various cytokines, eventually becoming involved in ALD pathogenesis (Kawaratani et al. 2013; Robinson et al. 2016). The production of several inflammatory cytokines (TNF-α, IFN-γ, and IL-17A) induces hepatic injury (Frank et al. 2020; Lee et al. 2020). In contrast, hepatoprotective and anti-inflammatory cytokines (e.g., IL-6 and IL-10) are also involved in ALD. IL-6 improves ALD by activating signal transducer and activator of transcription 3 (STAT3) and the subsequent induction of various hepatoprotective genes in hepatocytes (Giraldez et al. 2021). IL-10 inhibits alcoholic liver inflammation via the activation of STAT3 in the Kupffer cells and the subsequent inhibition of liver inflammation (Byun et al. 2013).
In addition to cytokine-associated liver injury, excessive alcohol intake accelerates liver inflammation by increasing the translocation of gut-derived endotoxins into the portal circulation, eventually activating the Kupffer cells. As a result, elevated levels of lipopolysaccharide (LPS) and proinflammatory cytokines are detected in serum. This finding suggests that bacterial toxins may be factors that accelerate ALD via LPS-Toll-like receptor 4 (TLR4) pathways (Seki et al. 2007; Kwong and Puri 2021). Furthermore, alcohol-induced intercellular communication between hepatic cells leads to ALD progression.
The liver consists of parenchymal cells (hepatocytes: > 70% of the total mass) and non-parenchymal cells (Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells, and hepatic lymphocytes: < 30% of the total mass) (Osna et al. 2022). Hepatic stellate cells and Kupffer cells play roles in ALD pathogenesis by interacting with hepatic immune cells (Yi and Jeong 2013). Signals exchanged between hepatic/non-parenchymal cells could affect the progression of liver disease, and this intercellular communication is achieved by multiple mechanisms, including direct cell-to-cell interactions, extracellular vesicles, chemokines, cytokines, and growth factors, all of which are recognized as mediators of intercellular communication (Osna et al. 2022). Therefore, a better understanding of the intercellular communication between hepatic and non-parenchymal cells in ALD pathophysiology will provide novel insights for use in the search for therapeutic approaches (Seo and Jeong 2016).
Drinking patterns can be classified into three categories according to drinking frequency over the preceding 6 month period and the amount of alcohol consumed: (1) continuous drinkers, (almost) daily alcohol consumption without binges; (2) frequent heavy drinkers, frequent alcohol consumption (more than 3 days per week) with frequent intoxication (more than once per week); and (3) episodic drinkers, less frequent, irregular alcohol consumption with longer sober periods (> 5 days) in between episodes and some binges (less than once per week) (Wetterling et al. 1999; Sarich et al. 2021). Alcohol consumption patterns are mixed and difficult to consider separately, though their effects are similar. For instance, acute alcohol intoxication not only leads to several metabolic alterations and hepatotoxicity, but is also associated with the development of ALD chronicity.
Several studies have demonstrated that chronic feeding plus a single ethanol feeding in mice induced more severe hepatic injury, inflammation, and liver steatosis than chronic alcohol feeding or single alcohol gavage alone (Bertola et al. 2013; Bertola 2020). These results imply that long-term alcohol drinking makes the liver more vulnerable to further liver injury after administration of high concentrations of alcohol (Torp et al. 2022). Additionally, a high-fat diet combined with binge alcohol administration causes acute hepatic inflammation and thus damage by increasing CXCL1 expression in the liver and promoting hepatic neutrophil infiltration (Hwang et al. 2020). Therefore, the combination of binge alcohol drinking and aspects of metabolic syndrome (such as obesity) are critical factors in determining the degree of ALD.
Alcohol metabolism and the involvement of alcohol metabolites in ALD
Oxidative alcohol metabolism
The liver, which has the highest concentration of alcohol-metabolizing enzymes, is primarily responsible for alcohol metabolism. The major pathway of oxidative alcohol metabolism involves alcohol dehydrogenase (ADH), an essential enzyme that converts ethanol to acetaldehyde, a highly reactive and toxic by-product that contributes to cytotoxicity. Additionally, acetaldehyde causes various forms of DNA damage, including DNA adducts, single- and/or double-stranded breaks, and point mutations (Mizumoto et al. 2017) (Fig. 2). Therefore, acetaldehyde has been classified as a possible human carcinogen (Group 2B) by the International Agency for Research on Cancer since 1987 (Kokkinakis et al. 2020). The ADH-catalyzed formation of acetaldehyde uses NAD+ as a cofactor and is responsible for the majority of alcohol oxidation in the liver (Zakhari 2006; Yang et al. 2022). Cytochrome P450 isozymes, including CYP2E1, 1A2, and 3A4, are predominantly present in the microsomes. CYP2E1 is the primary hepatic enzyme that catalyzes the oxidation of ethanol to acetaldehyde, and this enzyme is inducible by chronic alcohol consumption. However, the catalytic efficiency of CYP2E1 is considerably slower than that of ADH but has a tenfold higher capacity for binding ethanol (Harjumaki et al. 2021).

Schematic overview of oxidative and non-oxidative alcohol metabolism. Ingested alcohol is metabolized via two distinct pathways: the oxidative and non-oxidative pathways. Alcohol is oxidized into acetaldehyde by the oxidative alcohol-metabolizing enzymes ADH, CYP2E1, and catalase. Accumulated acetaldehyde, an intermediate metabolite in the process of oxidative alcohol metabolism, is a hepatotoxic compound. Alcohol conjugates with endogenous metabolites in the presence of several enzymes, leading to the production of fatty acid ethyl esters, phosphatidylcholine, ethyl glucuronide, and ethyl sulfate
Catalase, an enzyme found in peroxisomes, is also abundant in hepatocytes. This protein detoxifies hydrogen peroxide by converting it into water and oxygen (Osna et al. 2017). The catalase pathway has a relatively minor role during alcohol metabolism in the liver, but it has a significant ethanol-oxidizing function in the brain (Deitrich et al. 2006). The oxidative conversion of acetaldehyde to acetate, the final product of alcohol metabolism, by aldehyde dehydrogenase (ALDH) is the next stage in oxidative alcohol metabolism (Hyun et al. 2021). Most of the acetate generated during alcohol metabolism escapes the liver and is converted into acetyl-CoA by extrahepatic tissues.
Oxidative alcohol metabolite-mediated liver injury
Acetaldehyde is an intermediate metabolite of ethanol metabolism, and it induces a wide range of toxic, pharmacological, and behavioral responses (Guo and Ren 2010; Gao et al. 2019). Acetaldehyde is further metabolized into acetate by a mitochondrial, low-Km aldehyde dehydrogenase (ALDH2) enzyme (Jin et al. 2015; Jiang et al. 2020). Acetaldehyde stimulates the release of several signal molecules (e.g., epinephrine, norepinephrine, histamine, and bradykinin) and causes the cardiovascular symptoms of alcohol sensitivity, such as vasodilation and facial flushing, which are associated with abnormal heart rhythms and elevation of blood pressure (Quertemont and Didone 2006; Mustroph et al. 2019). Furthermore, acetaldehyde production promotes glutathione depletion, free radical-mediated toxicity, lipid peroxidation, and immune responses via the production of proinflammatory cytokines, all of which contribute to oxidative stress (Comporti et al. 2010; Seo et al. 2019).
Because aldehyde molecules are unstable, they react rapidly with cellular components to form adducts. These adducts can impair the functions of enzymes, DNA, structural proteins, and other macromolecules, thereby inhibiting cellular processes, eventually resulting in cytotoxicity. Acetaldehyde-derived adducts are generated in the progression of liver fibrosis in both alcoholic humans and murine models (Wang et al. 2020). ALDH2 is a key enzyme for acetaldehyde metabolism and is involved in the cellular mechanisms of alcohol-associated liver diseases. ALDH2 irreversibly metabolizes acetaldehyde to acetate in a redox reaction (Cederbaum 2012). Although the liver has long been thought to be the primary organ for acetaldehyde metabolism, the cumulative effect of ALDH2, which is found in various organs, is expected to contribute to systemic acetaldehyde clearance (Guillot et al. 2019). Additionally, a high concentration of acetaldehyde can change gut permeability and micro-equilibrium (increasing the abundance of Gram-negative bacteria), which causes direct hepatocyte damage (Malaguarnera et al. 2014). Owing to the toxic effects of acetaldehyde, the ALDH2*2 polymorphism is associated with a high risk of various cancers, such as hepatocellular carcinoma, gastric carcinoma, esophageal cancer, and colon cancer (Amanuma et al. 2015; Seo et al. 2019; Choi et al. 2021).
Non-oxidative alcohol metabolism
An intermediate product of oxidative alcohol metabolism, acetaldehyde, contributes to alcohol-induced liver injury. However, acetaldehyde-mediated cytotoxicity cannot account for damage to other organs, such as pancreas, heart, or brain, where oxidative alcohol metabolism is minimal or absent (Laposata and Lange 1986; Maenhout et al. 2013). Despite the presence of oxidative alcohol-metabolizing enzymes in the cardiovascular system, ethanol can directly harm the cardiovascular system or the hormonal modulation of heart function (Obad et al. 2018). Therefore, acetaldehyde-independent biochemical mediators (namely, non-oxidative ethanol metabolites or NOEMs) may be involved in the pathogenesis of alcohol-induced organ damage. Non-oxidative alcohol metabolism results in the enzymatic conjugation of ethanol with endogenous metabolites, such as glucuronic acid, sulfate, phospholipids, and fatty acids. The resultant metabolites are termed ethyl glucuronide (EtG), ethyl sulfate (EtS), phosphatidylethanol (PEth), and fatty acid ethyl ester (FAEE), respectively (Heier et al. 2016) (Fig. 2).
Non-oxidative alcohol metabolites are retained in body fluids and tissues for much longer periods than ethanol itself. Thus, they have been utilized as biomarkers for assessing ethanol intake in clinical and forensic settings (Heier et al. 2016). Moreover, accumulating evidence suggests that formation of non-oxidative alcohol metabolites interferes with cellular signaling pathways, disrupts organelle function, and contributes to ethanol toxicity in organs with limited oxidative capacity (Heier et al. 2016). Therefore, non-oxidative alcohol metabolism, in addition to the oxidative pathway, may also be an essential mechanism for understanding the pathophysiology of alcohol-mediated diseases in humans. However, NOEM-mediated hepatotoxicity has not yet been fully elucidated.
Non-oxidative alcohol metabolite-associated liver injury
FAEEs are the most well-known NOEMs proposed as indicators of alcohol-induced organ damage. FAEEs are a family of neutral lipids produced by the esterification of fatty acids with high concentrations of alcohol (Andresen-Streichert et al. 2018). Because FAEEs can be detected in the blood after ethanol ingestion, they have been proposed as markers of ethanol intake (Luginbuhl et al. 2016). Since organs that lack oxidative alcohol metabolism (pancreas, heart, or brain) exhibit cellular damage after alcohol intoxication, FAEEs and other NOEMs may play roles in producing alcohol-induced injury (Laposata and Lange 1986; Andresen-Streichert et al. 2018).
Notably, FAEEs circulate within low-density lipoprotein particles in human plasma after ethanol ingestion (Piano 2017). FAEEs are synthesized at high rates in the heart, providing a plausible link between alcohol ingestion and the subsequent development of alcohol-induced cardiomyopathy (Beckemeier and Bora 1998; Pfutzer et al. 2002). FAEE-associated cytotoxic effects can also be observed as acute pancreatitis via Ca2+-dependent mitochondrial inhibition, leading to a reduction in ATP, which impairs [Ca2+] homeostasis (Huang et al. 2014). Furthermore, the treatment of human hepatoblastoma cells with FAEEs causes significant morphological and biochemical alterations, suggesting that FAEEs play important roles in mediating ethanol-induced liver injury (Bhopale et al. 2006).
PEth is a phospholipid formed in the cell membrane in the presence of ethanol. In mammalian cells, PEth formation is catalyzed by phospholipase D (PLD), an enzyme that normally catalyzes phospholipid hydrolysis, leading to phosphatidic acid formation (Viel et al. 2012; Johansson et al. 2020). Phosphatidic acid is a lipid messenger involved in various cellular functions, including membrane trafficking, cytoskeletal remodeling, endocytosis, proliferation, and migration (Frohman 2015). A variety of tissues and cells contain PLD, with the highest activities reported in the lungs, adipose tissue, heart, and brain (Heier et al. 2016). PEth formation affects cellular signaling pathways by multiple mechanisms, including through competition with phosphatidic acid synthesis and functional disturbance of bio-membranes and membrane-associated proteins (Heier et al. 2016). Even 24 h after the last consumption of ethanol, PEth is still being produced in the neutrophils of alcoholics. This suggests that PEth could be a valuable marker of long-term ethanol ingestion (Aradottir et al. 2006; Lakso et al. 2019).
EtG and EtS have been used as biomarkers of recent ethanol intake and abstinence monitoring (Heier et al. 2016). The formation, pharmacokinetics, and elimination of EtG and EtS are < 0.1% of ingested ethanol (Wurst et al. 2015). EtG is formed by the transfer of a glucuronyl moiety from uridine 5′‐diphospho (UDP)‐glucuronic acid to ethanol. UDP-glucuronosyltransferases (UGTs), an enzyme family involved in phase II xenobiotic metabolism and glucuronidation of endogenous metabolites, catalyze this reaction (Rowland et al. 2013). Multiple organs, including adipose tissue, liver, brain, bone marrow, muscles, and hair, have detectable EtG levels (Schloegl et al. 2006; Wurst et al. 2015; Birkova et al. 2021). Blood EtG can be detected one hour after starting to consume ethanol, with peak concentrations occurring between 3.5 and 5.5 h (Halter et al. 2008; Karacaoğlu et al. 2019).
Like EtG, EtS is formed by the sulfonation of ethanol catalyzed by another class of phase II enzymes termed sulfotransferases (SULTs). EtS has been detected in the blood, urine, liver, kidneys, placenta, fetal tissues, and hair (Halter et al. 2008). Interestingly, inhibition of oxidative alcohol metabolism increases flux through the non-oxidative pathway, suggesting that the balance between oxidative and non-oxidative alcohol metabolisms may be important in determining the toxic effects of excessive alcohol consumption (Huang et al. 2014).
Interorgan crosstalk in ALD
Emerging evidence suggests that various organs, including the intestines, adipose tissue, and lungs, are affected by alcohol intoxication (Osna and Kharbanda 2016; Harris et al. 2019; Arteel 2020; Simon et al. 2022; White et al. 2022). Although the liver is responsible for most of the metabolism of ingested alcohol, other organs that express ethanol-metabolizing enzymes may be affected by oxidative or non-oxidative alcohol metabolites. Furthermore, interorgan crosstalk between the liver and peripheral organs is closely associated with the pathogenesis of ALD (Fig. 3).

The liver is the central organ affected by alcohol, but it is associated with peripheral organs. Ingested alcohol not only dysregulates hepatic function but also contributes to the pathogenesis of peripheral organs. Alcohol-associated liver diseases may be facilitated by interorgan crosstalk, which is essential for a comprehensive understanding of ALD progression
Gastrointestinal tract and ALD
The gastrointestinal tract participates in the digestion and absorption of nutrients, and its mucosal barrier protects the body from pathogens and extrinsic antigens. About 10% of ingested alcohol is absorbed through the stomach, and most of the remaining 90% is absorbed in the small intestine (Rajendram and Preedy 2005). Alcohol consumption disrupts the gut barrier, increases gut permeability, and induces bacterial translocation (Zhou and Zhong 2017). Studies have shown that alcohol-metabolizing enzymes located in the intestinal mucosa and gut microbiome also process acetaldehyde, an oxidative alcohol metabolite (Cederbaum 2012; Bishehsari et al. 2017). Additionally, it has been suggested that chronic alcohol consumption induces intestinal injury and is associated with the non-oxidative intestinal alcohol metabolite, PEth (Elamin et al. 2013; Heier et al. 2016).
Gut leakage is caused by excessive alcohol use, which leads to epithelial changes and blebbing of the lamina propria in the gut, allowing endotoxins to enter the portal and systemic circulations (Abdelmegeed et al. 2013). A study using chronically alcohol-fed animal models demonstrated the downregulation of tight junction proteins, such as occludin and zonula occludens-1, which are associated with gut permeability (Wang et al. 2014). Furthermore, alcohol-associated intestinal barrier dysfunction increases the translocation of pathogen-associated molecular patterns, including LPS, lipoteichoic acid, CpG DNA, flagellin, and β-glucan, into the portal and systemic circulations (Shi and Pamer 2011; Shim and Jeong 2020). Ligand-binding to TLRs activates downstream adaptor molecules, including nuclear factor-κB, interferon response factors, and mitogen-activated protein kinases, and initiates signaling cascades (Akira et al. 2006).
In addition to the provocation of TLR signaling pathways in the gut-liver axis, compelling evidence highlights alcohol consumption-induced compositional changes in intestinal microbiota in rodents and humans (Yan et al. 2011; Queipo-Ortuno et al. 2012; Ceccarelli et al. 2014; Engen et al. 2015). Notably, several studies have discovered that microbial dysbiosis, both quantitative and qualitative, are highly associated with alcohol consumption. Yan et al. (2011) reported that alcohol-fed mice showed bacterial overgrowth with a higher abundance of Bacteroidetes and Verrucomicrobia in comparison to mice fed isocaloric feed. Another study found altered intestinal microbiota compositions in sigmoidoscopies of alcoholic patients (Ciocan et al. 2018).
Additionally, compared with control patients, the abundance of potentially dangerous bacteria, such as Prevotellaceae, Enterobacteriaceae, Veillonellaceae, and Streptococcaceae, was significantly increased in patients with alcoholic cirrhosis (Chen et al. 2011). Research has shown that the prevalence of potentially pathogenic microbiomes in individuals with cirrhosis may be associated with hepatic disease prognoses (Liu et al. 2004; Woodhouse et al. 2018). Enteric dysbiosis leads to elevated levels of LPS, TLRs, NADPH oxidase homolog 4, and short-chain fatty acids, which activate the Kupffer cells and trigger liver inflammation and necrosis (Zeng et al. 2016; Zhang et al. 2019). Following alcohol intake, intestinal dysbiosis and bacterial overgrowth are commonly observed, resulting in intestinal barrier dysfunction, peripheral inflammation, and even liver injury (Kim et al. 2021). This highlights that dysregulation due to alcohol-induced intestinal barrier compromise and microbiota alterations are critical drivers of ALD progression.
Adipose tissue and ALD
Adipose tissue is primarily composed of adipocytes and the stromal vascular fraction. Adipose tissue has two functions: storing triglycerides as an energy source and regulating endocrine function by releasing various adipokines and cytokines (Guerreiro et al. 2022). Research suggests that alcohol is metabolized to acetaldehyde by either CYP2E1 or ADH in the adipose tissue. During chronic ethanol feeding CYP2E1 expression has been shown to increase in white adipose tissues (Zhang et al. 2015). Production of CYP2E1-dependent reactive oxygen species by adipocytes has been demonstrated in a chronic ethanol consumption rat model (Parker et al. 2018). ADH expression in adipose tissue is much lower than in the liver, and adipocyte ADH expression does not change with chronic ethanol consumption as it does in the liver. Nonetheless, chronic alcohol consumption significantly elevates ALDH activity in epididymal white adipose tissue and subcutaneous white adipose tissue (Zhang et al. 2015).
Several studies have demonstrated that chronic alcohol administration increases IL-6 expression in adipose tissues (He et al. 2015; Souza-Smith et al. 2017). In alcoholic individuals, increased IL-6 expression has been positively correlated with IL-18, osteopontin, α-smooth muscle actin, and semaphorins, emphasizing the importance of adipose tissue in the effects of ALD (Voican et al. 2015). Abundant evidence from animal experiments has demonstrated that chronic alcohol intake induces adipocyte lipolysis and the release of free fatty acids from white adipose tissue. This leads to ectopic fat accumulation within the liver, resulting in alcoholic fatty liver (Liangpunsakul et al. 2010; Zhong et al. 2012; Geisler and Renquist 2017). A study investigating lipolysis concluded that chronic alcohol administration decreased adipose tissue abundance and increased triglyceride degradation in male Wistar rats compared with pair-fed controls (Kang et al. 2007; Steiner and Lang 2017).
In addition to lipolysis, Sebastian et al. (2011) examined the effects of chronic alcohol administration (Lieber-DeCarli liquid diet for 25 days) on mice and discovered that long-term alcohol administration increased adipocyte death and inflammation via a CYP2E1/Bid/C1q-dependent pathway. Not surprisingly, chronic alcohol administration upregulates proinflammatory cytokine proliferation in adipose tissues (Fulham et al. 2019). A recent study has also demonstrated that adipocyte death can trigger infiltration and lipolysis of adipose tissue macrophages and subsequent liver damage through CCR2+ macrophage activation, suggesting that lipolysis-related lipotoxicity contributes to adipocyte death-associated liver injury (Kim et al. 2019).
Lungs and ALD
Studies on pulmonary ethanol metabolism suggest that ingested alcohol reaches the respiratory system via the bronchial circulation and is metabolized via oxidative and non-oxidative alcohol metabolism (Liang et al. 2012; Kaphalia and Calhoun 2013). Like the liver, the lungs of patients with alcoholism may be more susceptible to inflammation-related damage. Chronic alcohol consumption considerably changes the permeability of the alveolar epithelium (Massey et al. 2015a; Sadikot et al. 2019). Both ADH and CYP2E1 generate acetaldehyde, which is rapidly absorbed through the lungs (Kaphalia and Calhoun 2013). Acetaldehyde causes oxidative stress, lowers pulmonary macrophage phagocytic activity, and damages the nasal epithelium (Wyatt et al. 2012; Hoyt et al. 2017). Interestingly, several studies have focused on FAEE formation, one of the end products of non-oxidative alcohol metabolism in alcohol-induced pulmonary injury (Manautou and Carlson 1991; Manautou et al. 1992). Recently, Kaphalia et al. (2019) demonstrated exposure of alveolar macrophages to ethanol increased FAEE synthesis, endoplasmic reticulum stress, and oxidative stress. It was further demonstrated that ethanol-induced oxidative stress may occur through the formation of FAEEs.
Chronic alcohol abuse is a risk factor for acute respiratory distress syndrome (ARDS). Alcohol intake affects host systemic immunity and makes them more susceptible to lung infection (Liang et al. 2012). Studies have indicated that alcohol misuse increases the risk of lung injury secondary to pulmonary infection (Simet and Sisson 2015). Alcohol consumption disrupts mucus-facilitated clearance of bacterial pathogens from the upper respiratory tract (Wyatt et al. 2004; Chen et al. 2022), making alcoholic patients more susceptible to ARDS (Moss et al. 1996; Meza et al. 2022). Chronic ethanol administration in mice exacerbated LPS-induced pulmonary damage, resulting in the production of TNF-α responsive chemokines, macrophage inflammatory protein-2, and keratinocyte chemoattractant in the bronchoalveolar lavage fluid (Massey et al. 2015b). Similarly, a study using an acute-on-chronic binge ethanol-fed mice model also demonstrated neutrophil infiltration associated with functional changes in the central airways (Poole et al. 2019).
The lung-liver axis concept is based on clinical data from patients with a history of alcohol consumption who had increased risks of ARDS, death from ARDS, and hepatopulmonary syndrome (Afshar et al. 2014; Yang et al. 2019). Importantly, the death rate for ARDS patients with end-stage hepatic failure is approximately 100%, demonstrating that lung defense systems are strongly associated with hepatic parenchymal detoxification (Herrero et al. 2020). Additionally, crosstalk between the lungs and liver in ALD may be bidirectional. Mechanical ventilation generates lung-derived mediators (lung perfusates) and leads to inflammatory chemokine production in the sinusoidal endothelial cells of the liver (Markovic et al. 2009; Jaecklin et al. 2011). Another study discovered that acute and chronic alcohol consumption reduces protective antioxidants, oxidizes reduced glutathione, and suppresses innate and adaptive immunity in the lungs (Kaphalia and Calhoun 2013; Yeligar et al. 2016).
Alcohol damages the hepatic and pulmonary tissues through similar modes of action, which accelerates the progression of alcoholic liver and pulmonary diseases. Both the liver and lungs have resident macrophages that play significant roles in orchestrating inflammatory responses, including the induction of proinflammatory cytokines. Tissue injury and organ dysfunction are also exacerbated by tissue remodeling, which results in increased deposition of extracellular matrix components, such as fibronectin and collagen, and increased oxidative stress (Massey et al. 2015a). Therefore, the understanding of interdependent mechanisms in ALD progression is a very crucial factor to search for and set new therapeutic targets.
Conclusions and perspectives
Excessive alcohol consumption leads to a broad clinical-histological spectrum, from simple steatosis to cirrhosis and hepatocellular carcinoma. For decades, most related research has focused on pathophysiological changes associated with chronic alcohol exposure, whereas the importance of acute alcohol abuse has been underestimated. Based on previous studies, binge alcohol drinking not only leads to acute hepatotoxicity, but also promotes chronic alcoholic liver damage (Bertola et al. 2013; Molina and Nelson 2018). Research has highlighted the synergistic effects of long- and short-term alcohol exposure on ALD progression. Because the liver is the primary site of alcohol metabolism, alcoholic liver damage is mainly caused by alcohol metabolites. Almost 90% of the alcohol consumed is oxidatively metabolized into acetaldehyde by three hepatic enzymatic pathways, including the liver ADH pathway (90%), the microsomal ethanol-oxidizing system (8–10%), and the catalase pathway (< 2%) (Jiang et al. 2020).
In addition to acetaldehyde-mediated liver injury, non-oxidative alcohol metabolism occurs in the organs most injured by alcohol abuse. Several non-oxidative routes of ethanol metabolism have been described previously; these result in the enzymatic conjugation of ethanol to endogenous metabolites. FAEEs are esterification products of ethanol and fatty acids and have been implicated as important mediators of ethanol-induced cytotoxicity, including those affecting hepatocytes (Alhomsi et al. 2006). These metabolites represent alternative biomarkers since they can be detected several hours or days after ethanol exposure (Dinis-Oliveira 2016).
Excessive alcohol consumption mainly affects the liver, the primary organ of its metabolism. However, it also significantly affects other organs that contain alcohol-metabolizing enzymes. These extrahepatic organs (gastrointestinal tract, adipose tissue, and lungs) can also be damaged by oxidative or non-oxidative metabolites, and interorgan crosstalk between the liver and extrahepatic organs contributes to ALD development. Therefore, it is expected that understanding (1) the hepatic damage caused by the various alcohol-mediated metabolites, and (2) the crosstalk between liver and extrahepatic organs will provide a novel insight into the underlying mechanisms behind alcohol-associated liver injury and its therapeutic targets.
References
Abdelmegeed MA, Banerjee A, Jang S, Yoo SH, Yun JW, Gonzalez FJ, Keshavarzian A, Song BJ (2013) CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic Biol Med 65:1238–1245. https://doi.org/10.1016/j.freeradbiomed.2013.09.009
Afshar M, Smith GS, Terrin ML, Barrett M, Lissauer ME, Mansoor S, Jeudy J, Netzer G (2014) Blood alcohol content, injury severity, and adult respiratory distress syndrome. J Trauma Acute Care Surg 76:1447–1455. https://doi.org/10.1097/TA.0000000000000238
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801. https://doi.org/10.1016/j.cell.2006.02.015
Alhomsi K, Cluette-Brown JE, Laposata M (2006) Fatty acid ethyl esters in human mononuclear cells: production by endogenous synthesis greatly exceeds the uptake of preformed ethyl esters. Alcohol Clin Exp Res 30:560–566. https://doi.org/10.1111/j.1530-0277.2006.00062.x
Amanuma Y, Ohashi S, Itatani Y, Tsurumaki M, Matsuda S, Kikuchi O, Nakai Y, Miyamoto S, Oyama T, Kawamoto T, Whelan KA, Nakagawa H, Chiba T, Matsuda T, Muto M (2015) Protective role of ALDH2 against acetaldehyde-derived DNA damage in oesophageal squamous epithelium. Sci Rep 5:14142. https://doi.org/10.1038/srep14142
Andresen-Streichert H, Muller A, Glahn A, Skopp G, Sterneck M (2018) Alcohol biomarkers in clinical and forensic contexts. Dtsch Arztebl Int 115:309–315. https://doi.org/10.3238/arztebl.2018.0309
Aradottir S, Asanovska G, Gjerss S, Hansson P, Alling C (2006) PHosphatidylethanol (PEth) concentrations in blood are correlated to reported alcohol intake in alcohol-dependent patients. Alcohol Alcoholi 41:431–437. https://doi.org/10.1093/alcalc/agl027
Arteel GE (2020) Liver–lung axes in alcohol-related liver disease. Clin Mol Hepatol 26:670–676. https://doi.org/10.3350/cmh.2020.0174
Beckemeier ME, Bora PS (1998) Fatty acid ethyl esters: potentially toxic products of myocardial ethanol metabolism. J Mol Cell Cardiol 30:2487–2494. https://doi.org/10.1006/jmcc.1998.0812
Bertola A (2020) Mouse model of alcoholic steatohepatitis. Methods Mol Biol 2164:145–157. https://doi.org/10.1007/978-1-0716-0704-6_15
Bertola A, Mathews S, Ki SH, Wang H, Gao B (2013) Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 8:627–637. https://doi.org/10.1038/nprot.2013.032
Bhopale KK, Wu H, Boor PJ, Popov VL, Ansari GA, Kaphalia BS (2006) Metabolic basis of ethanol-induced hepatic and pancreatic injury in hepatic alcohol dehydrogenase deficient deer mice. Alcohol 39:179–188. https://doi.org/10.1016/j.alcohol.2006.09.005
Birkova A, Hubkova B, Cizmarova B, Bolerazska B (2021) Current view on the mechanisms of alcohol-mediated toxicity. Int J Mol Sci. https://doi.org/10.3390/ijms22189686
Bishehsari F, Magno E, Swanson G, Desai V, Voigt RM, Forsyth CB, Keshavarzian A (2017) Alcohol and gut-derived inflammation. Alcohol Res 38:163–171
Byun JS, Suh YG, Yi HS, Lee YS, Jeong WI (2013) Activation of toll-like receptor 3 attenuates alcoholic liver injury by stimulating kupffer cells and stellate cells to produce interleukin-10 in mice. J Hepatol 58:342–349. https://doi.org/10.1016/j.jhep.2012.09.016
Ceccarelli S, Nobili V, Alisi A (2014) Toll-like receptor-mediated signaling cascade as a regulator of the inflammation network during alcoholic liver disease. World J Gastroenterol 20:16443. https://doi.org/10.3748/wjg.v20.i44.16443
Cederbaum AI (2012) Alcohol metabolism. Clin Liver Dis 16:667–685. https://doi.org/10.1016/j.cld.2012.08.002
Chen C-H, Wang W-L, Hsu M-H, Mochly-Rosen D (2022) Alcohol consumption, ALDH2 polymorphism as risk factors for upper aerodigestive tract cancer progression and prognosis. Life 12:348. https://doi.org/10.3390/life12030348
Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, Wang Y, Zhu B, Li L (2011) Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54:562–572. https://doi.org/10.1002/hep.24423
Choi CK, Shin MH, Cho SH, Kim HY, Zheng W, Long J, Kweon SS (2021) Association between ALDH2 and ADH1B polymorphisms and the risk for colorectal cancer in Koreans. Cancer Res Treat 53:754–762. https://doi.org/10.4143/crt.2020.478
Ciocan D, Rebours V, Voican CS, Wrzosek L, Puchois V, Cassard A-M, Perlemuter G (2018) Characterization of intestinal microbiota in alcoholic patients with and without alcoholic hepatitis or chronic alcoholic pancreatitis. Sci Rep 8:1–12. https://doi.org/10.1038/s41598-018-23146-3
Comporti M, Signorini C, Leoncini S, Gardi C, Ciccoli L, Giardini A, Vecchio D, Arezzini B (2010) Ethanol-induced oxidative stress: basic knowledge. Genes Nutr 5:101–109. https://doi.org/10.1007/s12263-009-0159-9
D’angelo A, Petrella C, Greco A, Ralli M, Vitali M, Giovagnoli R, De Persis S, Fiore M, Ceccanti M, Messina MP (2022) Acute alcohol intoxication: a clinical overview. Clin Ter 173:280–291. https://doi.org/10.7417/CT.2022.2432
Dasarathy S, Brown JM (2017) Alcoholic liver disease on the rise: interorgan cross talk driving liver injury. Alcohol Clin Exp Res 41:880–882. https://doi.org/10.1111/acer.13370
Deitrich R, Zimatkin S, Pronko S (2006) Oxidation of ethanol in the brain and its consequences. Alcohol Res Health 29:266
Dinis-Oliveira RJ (2016) Oxidative and non-oxidative metabolomics of ethanol. Curr Drug Metab 17:327–335. https://doi.org/10.2174/1389200217666160125113806
Dunn W, Shah VH (2016) Pathogenesis of alcoholic liver disease. Clin Liver Dis 20:445–456. https://doi.org/10.1016/j.cld.2016.02.004
Elamin EE, Masclee AA, Dekker J, Jonkers DM (2013) Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr Rev 71:483–499. https://doi.org/10.1111/nure.12027
Engen PA, Green SJ, Voigt RM, Forsyth CB, Keshavarzian A (2015) The gastrointestinal microbiome: alcohol effects on the composition of intestinal microbiota. Alcohol Res 37:223–236
Evangelou E, Suzuki H, Bai W, Pazoki R, Gao H, Matthews PM, Elliott P (2021) Alcohol consumption in the general population is associated with structural changes in multiple organ systems. Elife. https://doi.org/10.7554/eLife.65325
Fillmore MT, Jude R (2011) Defining “binge” drinking as five drinks per occasion or drinking to a. 08% BAC: which is more sensitive to risk? Am J Addict 20:468–475. https://doi.org/10.1111/j.1521-0391.2011.00156.x
Frank K, Abeynaike S, Nikzad R, Patel RR, Roberts AJ, Roberto M, Paust S (2020) Alcohol dependence promotes systemic IFN-gamma and IL-17 responses in mice. PLoS ONE 15:e0239246. https://doi.org/10.1371/journal.pone.0239246
Frazier TH, Stocker AM, Kershner NA, Marsano LS, Mcclain CJ (2011) Treatment of alcoholic liver disease. Therap Adv Gastroenterol 4:63–81. https://doi.org/10.1177/1756283X10378925
Frohman MA (2015) The phospholipase D superfamily as therapeutic targets. Trends Pharmacol Sci 36:137–144. https://doi.org/10.1016/j.tips.2015.01.001
Fulham MA, Ratna A, Gerstein RM, Kurt-Jones EA, Mandrekar P (2019) Alcohol-induced adipose tissue macrophage phenotypic switching is independent of myeloid toll-like receptor 4 expression. Am J Physiol Cell Physiol 317:C687–C700. https://doi.org/10.1152/ajpcell.00276.2017
Gao Y, Zhou Z, Ren T, Kim SJ, He Y, Seo W, Guillot A, Ding Y, Wu R, Shao S, Wang X, Zhang H, Wang W, Feng D, Xu M, Han E, Zhong W, Zhou Z, Pacher P, Niu J, Gao B (2019) Alcohol inhibits T-cell glucose metabolism and hepatitis in ALDH2-deficient mice and humans: roles of acetaldehyde and glucocorticoids. Gut 68:1311–1322. https://doi.org/10.1136/gutjnl-2018-316221
Geisler CE, Renquist BJ (2017) Hepatic lipid accumulation: cause and consequence of dysregulated glucoregulatory hormones. J Endocrinol 234:R1–R21. https://doi.org/10.1530/JOE-16-0513
Giraldez MD, Carneros D, Garbers C, Rose-John S, Bustos M (2021) New insights into IL-6 family cytokines in metabolism, hepatology and gastroenterology. Nat Rev Gastroenterol Hepatol 18:787–803. https://doi.org/10.1038/s41575-021-00473-x
Griswold MG, Fullman N, Hawley C, Arian N, Zimsen SR, Tymeson HD, Venkateswaran V, Tapp AD, Forouzanfar MH, Salama JS (2018) Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the global burden of disease study 2016. The Lancet 392:1015–1035. https://doi.org/10.1016/S0140-6736(18)31310-2
Guerreiro VA, Carvalho D, Freitas P (2022) Obesity, adipose tissue, and inflammation answered in questions. J Obes 2022:2252516. https://doi.org/10.1155/2022/2252516
Guillot A, Ren T, Jourdan T, Pawlosky RJ, Han E, Kim SJ, Zhang L, Koob GF, Gao B (2019) Targeting liver aldehyde dehydrogenase-2 prevents heavy but not moderate alcohol drinking. Proc Natl Acad Sci USA 116:25974–25981. https://doi.org/10.1073/pnas.1908137116
Guo R, Ren J (2010) Alcohol and acetaldehyde in public health: from marvel to menace. Int J Environ Res Public Health 7:1285–1301. https://doi.org/10.3390/ijerph7041285
Halter CC, Dresen S, Auwaerter V, Wurst FM, Weinmann W (2008) Kinetics in serum and urinary excretion of ethyl sulfate and ethyl glucuronide after medium dose ethanol intake. Int J Legal Med 122:123–128. https://doi.org/10.1007/s00414-007-0180-8
Han H, Desert R, Das S, Song Z, Athavale D, Ge X, Nieto N (2020) Danger signals in liver injury and restoration of homeostasis. J Hepatol 73:933–951. https://doi.org/10.1016/j.jhep.2020.04.033
Harjumaki R, Pridgeon CS, Ingelman-Sundberg M (2021) CYP2E1 in Alcoholic and non-alcoholic liver injury. roles of ROS, reactive intermediates and lipid overload. Int J Mol Sci. https://doi.org/10.3390/ijms22158221
Harris B, Mcalister A, Willoughby T, Sivaraman V (2019) Alcohol-dependent pulmonary inflammation: a role for HMGB-1. Alcohol 80:45–52. https://doi.org/10.1016/j.alcohol.2018.09.008
He Z, Li M, Zheng D, Chen Q, Liu W, Feng L (2015) Adipose tissue hypoxia and low-grade inflammation: a possible mechanism for ethanol-related glucose intolerance? Br J Nutr 113:1355–1364. https://doi.org/10.1017/S000711451500077X
Heier C, Xie H, Zimmermann R (2016) Nonoxidative ethanol metabolism in humans-from biomarkers to bioactive lipids. IUBMB Life 68:916–923. https://doi.org/10.1002/iub.1569
Herrero R, Sanchez G, Asensio I, Lopez E, Ferruelo A, Vaquero J, Moreno L, De Lorenzo A, Banares R, Lorente JA (2020) Liver-lung interactions in acute respiratory distress syndrome. Intensive Care Med Exp 8:48. https://doi.org/10.1186/s40635-020-00337-9
Hoyt LR, Randall MJ, Ather JL, Depuccio DP, Landry CC, Qian X, Janssen-Heininger YM, Van Der Vliet A, Dixon AE, Amiel E, Poynter ME (2017) Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol 12:883–896. https://doi.org/10.1016/j.redox.2017.04.020
Huang W, Booth DM, Cane MC, Chvanov M, Javed MA, Elliott VL, Armstrong JA, Dingsdale H, Cash N, Li Y, Greenhalf W, Mukherjee R, Kaphalia BS, Jaffar M, Petersen OH, Tepikin AV, Sutton R, Criddle DN (2014) Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 63:1313–1324. https://doi.org/10.1136/gutjnl-2012-304058
Husen P, Hornung J, Benko T, Klein C, Willuweit K, Buechter M, Saner FH, Paul A, Treckmann JW, Hoyer DP (2019) Risk Factors for high mortality on the liver transplant waiting list in times of organ shortage: a single-center analysis. Ann Transplant 24:242–251. https://doi.org/10.12659/AOT.914246
Hwang S, Ren T, Gao B (2020) Obesity and binge alcohol intake are deadly combination to induce steatohepatitis: a model of high-fat diet and binge ethanol intake. Clin Mol Hepatol 26:586–594. https://doi.org/10.3350/cmh.2020.0100
Hyun J, Han J, Lee C, Yoon M, Jung Y (2021) Pathophysiological aspects of alcohol metabolism in the liver. Int J Mol Sci. https://doi.org/10.3390/ijms22115717
Jaecklin T, Engelberts D, Otulakowski G, O’brodovich H, Post M, Kavanagh BP (2011) Lung-derived soluble mediators are pathogenic in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 300:L648–L658. https://doi.org/10.1152/ajplung.00305.2010
Jiang Y, Zhang T, Kusumanchi P, Han S, Yang Z, Liangpunsakul S (2020) Alcohol metabolizing enzymes, microsomal ethanol oxidizing system, cytochrome P450 2E1, catalase, and aldehyde dehydrogenase in alcohol-associated liver disease. Biomedicines. https://doi.org/10.3390/biomedicines8030050
Jin S, Chen J, Chen L, Histen G, Lin Z, Gross S, Hixon J, Chen Y, Kung C, Chen Y, Fu Y, Lu Y, Lin H, Cai X, Yang H, Cairns RA, Dorsch M, Su SM, Biller S, Mak TW, Cang Y (2015) ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc Natl Acad Sci USA 112:9088–9093. https://doi.org/10.1073/pnas.1510757112
Johansson K, Johansson L, Pennlert J, Soderberg S, Jansson JH, Lind MM (2020) Phosphatidylethanol levels, as a marker of alcohol consumption, are associated with risk of intracerebral hemorrhage. Stroke 51:2148–2152. https://doi.org/10.1161/STROKEAHA.120.029630
Jones AW (2019) Alcohol, its absorption, distribution, metabolism, and excretion in the body and pharmacokinetic calculations. Wiley Interdiscip Rev 1:e1340. https://doi.org/10.1002/wfs2.1340
Kang L, Chen X, Sebastian BM, Pratt BT, Bederman IR, Alexander JC, Previs SF, Nagy LE (2007) Chronic ethanol and triglyceride turnover in white adipose tissue in rats: inhibition of the anti-lipolytic action of insulin after chronic ethanol contributes to increased triglyceride degradation. J Biol Chem 282:28465–28473. https://doi.org/10.1074/jbc.M705503200
Kaphalia L, Calhoun WJ (2013) Alcoholic lung injury: metabolic, biochemical and immunological aspects. Toxicol Lett 222:171–179. https://doi.org/10.1016/j.toxlet.2013.07.016
Kaphalia L, Srinivasan MP, Kakumanu RD, Kaphalia BS, Calhoun WJ (2019) Ethanol exposure impairs AMPK signaling and phagocytosis in human alveolar macrophages: role of ethanol metabolism. Alcohol Clin Exp Res 43:1682–1694. https://doi.org/10.1111/acer.14131
Karacaoğlu E, Odabaşı AB, Akçan R, Tümer AR, Lale A, Kocadağlı T, Gökmen V (2019) Time dependent change of ethanol consumption biomarkers, ethyl glucuronide and ethyl sulphate, after single dose ethanol intake. Turkish J Biochem 44:379–387. https://doi.org/10.1515/tjb-2018-0173
Kawaratani H, Tsujimoto T, Douhara A, Takaya H, Moriya K, Namisaki T, Noguchi R, Yoshiji H, Fujimoto M, Fukui H (2013) The effect of inflammatory cytokines in alcoholic liver disease. Mediators Inflamm 2013:495156. https://doi.org/10.1155/2013/495156
Keyes KM (2022) Age, period, and cohort effects in alcohol use in the United States in the 20th and 21st centuries: implications for the coming decades. Alcohol Res. https://doi.org/10.35946/arcr.v42.1.02
Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, Gao B (2010) Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology 52:1291–1300. https://doi.org/10.1002/hep.23837
Kim DH, Sim Y, Hwang JH, Kwun IS, Lim JH, Kim J, Kim JI, Baek MC, Akbar M, Seo W, Kim DK, Song BJ, Cho YE (2021) Ellagic acid prevents binge alcohol-induced leaky gut and liver injury through inhibiting gut dysbiosis and oxidative stress. Antioxidants (basel). https://doi.org/10.3390/antiox10091386
Kim SJ, Feng D, Guillot A, Dai S, Liu F, Hwang S, Parker R, Seo W, He Y, Godlewski G, Jeong WI, Lin Y, Qin X, Kunos G, Gao B (2019) Adipocyte death preferentially induces liver injury and inflammation through the activation of chemokine (C-C Motif) receptor 2-positive macrophages and lipolysis. Hepatology 69:1965–1982. https://doi.org/10.1002/hep.30525
Kokkinakis M, Tsakiris I, Tzatzarakis M, Vakonaki E, Alegakis A, Papachristou S, Karzi V, Kokkinaki A, Goumenou M, Kallionakis M, Kalogeraki A (2020) Carcinogenic, ethanol, acetaldehyde and noncarcinogenic higher alcohols, esters, and methanol compounds found in traditional alcoholic beverages. Risk Assess Approach Toxicol Rep 7:1057–1065. https://doi.org/10.1016/j.toxrep.2020.08.017
Kwong EK, Puri P (2021) Gut microbiome changes in Nonalcoholic fatty liver disease & alcoholic liver disease. Transl Gastroenterol Hepatol 6:3. https://doi.org/10.21037/tgh.2020.02.18
Lakso HA, Wuolikainen A, Sundkvist A, Johansson I, Marklund SL (2019) Long-term stability of the alcohol consumption biomarker phosphatidylethanol in erythrocytes at − 80 degrees C. Clin Mass Spectrom 11:37–41. https://doi.org/10.1016/j.clinms.2018.12.002
Laposata EA, Lange LG (1986) Presence of nonoxidative ethanol metabolism in human organs commonly damaged by ethanol abuse. Science 231:497–499. https://doi.org/10.1126/science.3941913
Lee JH, Shim YR, Seo W, Kim MH, Choi WM, Kim HH, Kim YE, Yang K, Ryu T, Jeong JM, Choi HG, Eun HS, Kim SH, Mun H, Yoon JH, Jeong WI (2020) Mitochondrial double-stranded RNA in exosome promotes interleukin-17 production through toll-like receptor 3 in alcohol-associated liver injury. Hepatology 72:609–625. https://doi.org/10.1002/hep.31041
Liang Y, Yeligar SM, Brown LA (2012) Chronic-alcohol-abuse-induced oxidative stress in the development of acute respiratory distress syndrome. Sci World J 2012:740308. https://doi.org/10.1100/2012/740308
Liangpunsakul S, Crabb DW, Qi R (2010) Relationship among alcohol intake, body fat, and physical activity: a population-based study. Ann Epidemiol 20:670–675. https://doi.org/10.1016/j.annepidem.2010.05.014
Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Riordan SM (2004) Synbiotic modulation of gut flora: effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 39:1441–1449. https://doi.org/10.1002/hep.20194
Luginbuhl M, Schrock A, Konig S, Schurch S, Weinmann W (2016) Determination of fatty acid ethyl esters in dried blood spots by LC-MS/MS as markers for ethanol intake: application in a drinking study. Anal Bioanal Chem 408:3503–3509. https://doi.org/10.1007/s00216-016-9426-y
Maenhout TM, De Buyzere ML, Delanghe JR (2013) Non-oxidative ethanol metabolites as a measure of alcohol intake. Clin Chim Acta 415:322–329. https://doi.org/10.1016/j.cca.2012.11.014
Majumdar A, Tsochatzis EA (2022) Binge drinking and fibrogenesis: more than just a hangover. Liver Int 42:6–8. https://doi.org/10.1111/liv.15119
Malaguarnera G, Giordano M, Nunnari G, Bertino G, Malaguarnera M (2014) Gut microbiota in alcoholic liver disease: pathogenetic role and therapeutic perspectives. World J Gastroenterol 20:16639–16648. https://doi.org/10.3748/wjg.v20.i44.16639
Manautou JE, Buss NJ, Carlson GP (1992) Oxidative and non-oxidative metabolism of ethanol by the rabbit lung. Toxicol Lett 62:93–99. https://doi.org/10.1016/0378-4274(92)90082-u
Manautou JE, Carlson GP (1991) Ethanol-induced fatty acid ethyl ester formation in vivo and in vitro in rat lung. Toxicology 70:303–312. https://doi.org/10.1016/0300-483x(91)90005-l
Markovic N, Mccaig LA, Stephen J, Mizuguchi S, Veldhuizen RA, Lewis JF, Cepinskas G (2009) Mediators released from LPS-challenged lungs induce inflammatory responses in liver vascular endothelial cells and neutrophilic leukocytes. Am J Physiol Gastrointest Liver Physiol 297:G1066–G1076. https://doi.org/10.1152/ajpgi.00278.2009
Massey VL, Arteel GE (2012) Acute alcohol-induced liver injury front physiol. Front Physiol 3:193. https://doi.org/10.3389/fphys.2012.00193
Massey VL, Beier JI, Ritzenthaler JD, Roman J, Arteel GE (2015a) Potential role of the gut/liver/lung axis in alcohol-induced tissue pathology. Biomolecules 5:2477–2503. https://doi.org/10.3390/biom5042477
Massey VL, Poole LG, Siow DL, Torres E, Warner NL, Schmidt RH, Ritzenthaler JD, Roman J, Arteel GE (2015b) Chronic alcohol exposure enhances lipopolysaccharide-induced lung injury in mice: potential role of systemic tumor necrosis factor-alpha. Alcohol Clin Exp Res 39:1978–1988. https://doi.org/10.1111/acer.12855
Meza V, Arnold J, Diaz LA, Ayala Valverde M, Idalsoaga F, Ayares G, Devuni D, Arab JP (2022) Alcohol consumption: medical implications, the liver and beyond. Alcohol Alcohol 57:283–291. https://doi.org/10.1093/alcalc/agac013
Mizumoto A, Ohashi S, Hirohashi K, Amanuma Y, Matsuda T, Muto M (2017) Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int J Mol Sci. https://doi.org/10.3390/ijms18091943
Molina PE, Nelson S (2018) Binge drinking’s effects on the body. Alcohol Res 39:99–109
Moss M, Bucher B, Moore FA, Moore EE, Parsons PE (1996) The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA 275:50–54. https://doi.org/10.1001/jama.1996.03530250054027
Mustroph J, Lebek S, Maier LS, Neef S (2019) Mechanisms of cardiac ethanol toxicity and novel treatment options. Pharmacol Ther 197:1–10. https://doi.org/10.1016/j.pharmthera.2018.12.006
Obad A, Peeran A, Little JI, Haddad GE, Tarzami ST (2018) Alcohol-mediated organ damages: heart and brain. Front Pharmacol 9:81. https://doi.org/10.3389/fphar.2018.00081
Osna NA, Donohue TM Jr, Kharbanda KK (2017) Alcoholic liver disease: pathogenesis and current management. Alcohol Res 38:147–161
Osna NA, Eguchi A, Feldstein AE, Tsukamoto H, Dagur RS, Ganesan M, New-Aaron M, Arumugam MK, Chava S, Ribeiro M, Szabo G, Mueller S, Wang S, Chen C, Weinman SA, Kharbanda KK (2022) Cell-to-cell communications in alcohol-associated liver disease. Front Physiol 13:831004. https://doi.org/10.3389/fphys.2022.831004
Osna NA, Kharbanda KK (2016) Multi-organ alcohol-related damage: mechanisms and treatment. Biomolecules. https://doi.org/10.3390/biom6020020
Parker R, Kim SJ, Gao B (2018) Alcohol, adipose tissue and liver disease: mechanistic links and clinical considerations. Nat Rev Gastroenterol Hepatol 15:50–59. https://doi.org/10.1038/nrgastro.2017.116
Paton A (2005) Alcohol in the body. BMJ 330:85–87. https://doi.org/10.1136/bmj.330.7482.85
Pfutzer RH, Tadic SD, Li HS, Thompson BS, Zhang JY, Ford ME, Eagon PK, Whitcomb DC (2002) Pancreatic cholesterol esterase, ES-10, and fatty acid ethyl ester synthase III gene expression are increased in the pancreas and liver but not in the brain or heart with long-term ethanol feeding in rats. Pancreas 25:101–106. https://doi.org/10.1097/00006676-200207000-00021
Piano MR (2017) Alcohol’s effects on the cardiovascular system. Alcohol Res 38:219–241
Poole LG, Beier JI, Torres-Gonzales E, Schlueter CF, Hudson SV, Artis A, Warner NL, Nguyen-Ho CT, Dolin CE, Ritzenthaler JD, Hoyle GW, Roman J, Arteel GE (2019) Chronic + binge alcohol exposure promotes inflammation and alters airway mechanics in the lung. Alcohol 80:53–63. https://doi.org/10.1016/j.alcohol.2018.10.008
Queipo-Ortuno MI, Boto-Ordonez M, Murri M, Gomez-Zumaquero JM, Clemente-Postigo M, Estruch R, Cardona Diaz F, Andres-Lacueva C, Tinahones FJ (2012) Influence of red wine polyphenols and ethanol on the gut microbiota ecology and biochemical biomarkers. Am J Clin Nutr 95:1323–1334. https://doi.org/10.3945/ajcn.111.027847
Quertemont E, Didone V (2006) Role of acetaldehyde in mediating the pharmacological and behavioral effects of alcohol. Alcohol Res Health 29:258–265
Rajendram R, Preedy VR (2005) Effect of alcohol consumption on the gut. Dig Dis 23:214–221. https://doi.org/10.1159/000090168
Rehm J, Gmel GE Sr, Gmel G, Hasan OSM, Imtiaz S, Popova S, Probst C, Roerecke M, Room R, Samokhvalov AV, Shield KD, Shuper PA (2017) The relationship between different dimensions of alcohol use and the burden of disease-an update. Addiction 112:968–1001. https://doi.org/10.1111/add.13757
Robinson MW, Harmon C, O’farrelly C, (2016) Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol 13:267–276. https://doi.org/10.1038/cmi.2016.3
Rowland A, Miners JO, Mackenzie PI (2013) The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 45:1121–1132. https://doi.org/10.1016/j.biocel.2013.02.019
Sadikot RT, Bedi B, Li J, Yeligar SM (2019) Alcohol-induced mitochondrial DNA damage promotes injurious crosstalk between alveolar epithelial cells and alveolar macrophages. Alcohol 80:65–72. https://doi.org/10.1016/j.alcohol.2018.08.006
Sarich P, Canfell K, Egger S, Banks E, Joshy G, Grogan P, Weber MF (2021) Alcohol consumption, drinking patterns and cancer incidence in an Australian cohort of 226,162 participants aged 45 years and over. Br J Cancer 124:513–523. https://doi.org/10.1038/s41416-020-01101-2
Schloegl H, Rost T, Schmidt W, Wurst FM, Weinmann W (2006) Distribution of ethyl glucuronide in rib bone marrow, other tissues and body liquids as proof of alcohol consumption before death. Forensic Sci Int 156:213–218. https://doi.org/10.1016/j.forsciint.2005.03.024
Sebastian BM, Roychowdhury S, Tang H, Hillian AD, Feldstein AE, Stahl GL, Takahashi K, Nagy LE (2011) Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J Biol Chem 286:35989–35997. https://doi.org/10.1074/jbc.M111.254201
Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF (2007) TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13:1324–1332. https://doi.org/10.1038/nm1663
Seo W, Gao Y, He Y, Sun J, Xu H, Feng D, Park SH, Cho YE, Guillot A, Ren T, Wu R, Wang J, Kim SJ, Hwang S, Liangpunsakul S, Yang Y, Niu J, Gao B (2019) ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J Hepatol 71:1000–1011. https://doi.org/10.1016/j.jhep.2019.06.018
Seo W, Jeong WI (2016) Hepatic non-parenchymal cells: master regulators of alcoholic liver disease? World J Gastroenterol 22:1348–1356. https://doi.org/10.3748/wjg.v22.i4.1348
Shi C, Pamer EG (2011) Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11:762–774. https://doi.org/10.1038/nri3070
Shim YR, Jeong WI (2020) Recent advances of sterile inflammation and inter-organ cross-talk in alcoholic liver disease. Exp Mol Med 52:772–780. https://doi.org/10.1038/s12276-020-0438-5
Shukla SD, Pruett SB, Szabo G, Arteel GE (2013) Binge ethanol and liver: new molecular developments. Alcohol Clin Exp Res 37:550–557. https://doi.org/10.1111/acer.12011
Simet SM, Sisson JH (2015) Alcohol’s effects on lung health and immunity. Alcohol Res 37:199–208
Simon L, Souza-Smith FM, Molina PE (2022) Alcohol-associated tissue injury: current views on pathophysiological mechanisms. Annu Rev Physiol 84:87–112. https://doi.org/10.1146/annurev-physiol-060821-014008
Souza-Smith FM, Ford SM Jr, Simon L, Molina PE (2017) Repeated binge-like alcohol intoxication: depot-specific adipose tissue immuno-metabolic dysregulation. Shock 48:243–250. https://doi.org/10.1097/SHK.0000000000000843
Steiner JL, Lang CH (2017) Alcohol adipose tissue and lipid dysregulation. Biomolecules. https://doi.org/10.3390/biom7010016
Stockwell T, Zhao J (2017) Alcohol’s contribution to cancer is underestimated for exactly the same reason that its contribution to cardioprotection is overestimated. Addiction 112:230–232. https://doi.org/10.1111/add.13627
Szabo G, Saha B (2015) Alcohol’s effect on host defense. Alcohol Res 37:159–170
Torp N, Israelsen M, Nielsen MJ, Astrand CP, Juhl P, Johansen S, Hansen CD, Madsen B, Villesen IF, Leeming DJ, Thiele M, Hansen T, Karsdal M, Krag A (2022) Binge drinking induces an acute burst of markers of hepatic fibrogenesis (PRO-C3). Liver Int 42:92–101. https://doi.org/10.1111/liv.15120
Viel G, Boscolo-Berto R, Cecchetto G, Fais P, Nalesso A, Ferrara SD (2012) Phosphatidylethanol in blood as a marker of chronic alcohol use: a systematic review and meta-analysis. Int J Mol Sci 13:14788–14812. https://doi.org/10.3390/ijms131114788
Voican CS, Njike-Nakseu M, Boujedidi H, Barri-Ova N, Bouchet-Delbos L, Agostini H, Maitre S, Prevot S, Cassard-Doulcier AM, Naveau S, Perlemuter G (2015) Alcohol withdrawal alleviates adipose tissue inflammation in patients with alcoholic liver disease. Liver Int 35:967–978. https://doi.org/10.1111/liv.12575
Wang W, Wang C, Xu H, Gao Y (2020) Aldehyde dehydrogenase, liver disease and cancer. Int J Biol Sci 16:921–934. https://doi.org/10.7150/ijbs.42300
Wang Y, Tong J, Chang B, Wang BF, Zhang D, Wang BY (2014) Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol Med Rep 9:2352–2356. https://doi.org/10.3892/mmr.2014.2126
Wetterling T, Veltrup C, Driessen M, John U (1999) Drinking pattern and alcohol-related medical disorders. Alcohol Alcohol 34:330–336. https://doi.org/10.1093/alcalc/34.3.330
White BA, Ramos GP, Kane S (2022) The impact of alcohol in inflammatory bowel diseases. Inflamm Bowel Dis 28:466–473. https://doi.org/10.1093/ibd/izab089
Woodhouse C, Patel V, Singanayagam A, Shawcross D (2018) the gut microbiome as a therapeutic target in the pathogenesis and treatment of chronic liver disease. Aliment Pharmacol Ther 47:192–202. https://doi.org/10.1111/apt.14397
Wurst FM, Thon N, Yegles M, Schruck A, Preuss UW, Weinmann W (2015) Ethanol metabolites: their role in the assessment of alcohol intake. Alcohol Clin Exp Res 39:2060–2072. https://doi.org/10.1111/acer.12851
Wyatt TA, Gentry-Nielsen MJ, Pavlik JA, Sisson JH (2004) Desensitization of PKA-stimulated ciliary beat frequency in an ethanol-fed rat model of cigarette smoke exposure. Alcohol-Clin Exp Res 28:998–1004. https://doi.org/10.1097/01.Alc.0000130805.75641.F4
Wyatt TA, Kharbanda KK, Mccaskill ML, Tuma DJ, Yanov D, Devasure J, Sisson JH (2012) Malondialdehyde-acetaldehyde-adducted protein inhalation causes lung injury. Alcohol 46:51–59. https://doi.org/10.1016/j.alcohol.2011.09.001
Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA, Schnabl B (2011) Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53:96–105. https://doi.org/10.1002/hep.24018
Yang P, Formanek P, Scaglione S, Afshar M (2019) Risk factors and outcomes of acute respiratory distress syndrome in critically ill patients with cirrhosis. Hepatol Res 49:335–343. https://doi.org/10.1111/hepr.13240
Yang YM, Cho YE, Hwang S (2022) Crosstalk between oxidative stress and inflammatory liver injury in the pathogenesis of alcoholic liver disease. Int J Mol Sci 23:774. https://doi.org/10.3390/ijms23020774
Yeligar SM, Chen MM, Kovacs EJ, Sisson JH, Burnham EL, Brown LA (2016) Alcohol and lung injury and immunity. Alcohol 55:51–59. https://doi.org/10.1016/j.alcohol.2016.08.005
Yi HS, Jeong WI (2013) Interaction of hepatic stellate cells with diverse types of immune cells: foe or friend? J Gastroenterol Hepatol 28(Suppl 1):99–104. https://doi.org/10.1111/jgh.12017
Zakhari S (2006) Overview: how is alcohol metabolized by the body? Alcohol Res Health 29:245–254
Zeng T, Zhang CL, Xiao M, Yang R, Xie KQ (2016) Critical roles of kupffer cells in the pathogenesis of alcoholic liver disease: from basic science to clinical trials. Front Immunol 7:538. https://doi.org/10.3389/fimmu.2016.00538
Zhang WL, Zhong W, Sun XH, Sun Q, Tan XB, Li Q, Sun XG, Zhou ZX (2015) Visceral white adipose tissue is susceptible to alcohol-induced lipodystrophy in rats: role of acetaldehyde. Alcohol-Clin Exp Res 39:416–423. https://doi.org/10.1111/acer.12646
Zhang ZY, Tang HS, Chen P, Xie H, Tao YG (2019) Demystifying the manipulation of host immunity, metabolism, and extraintestinal tumors by the gut microbiome. Signal Transduct Tar. https://doi.org/10.1038/s41392-019-0074-5
Zhong W, Zhao Y, Tang Y, Wei X, Shi X, Sun W, Sun X, Yin X, Sun X, Kim S, Mcclain CJ, Zhang X, Zhou Z (2012) Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am J Pathol 180:998–1007. https://doi.org/10.1016/j.ajpath.2011.11.017
Zhou Z, Zhong W (2017) Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res 1:197–207. https://doi.org/10.1016/j.livres.2017.12.004
Acknowledgements
This work was supported by the National Research Foundation of Korea (Grant Numbers: 2014M3A9D5A01073556, 2018R1A5A2025286, 2021R1F1A1062136, and 2022R1C1C1008912).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Park, S.H., Lee, YS., Sim, J. et al. Alcoholic liver disease: a new insight into the pathogenesis of liver disease. Arch. Pharm. Res. 45, 447–459 (2022). https://doi.org/10.1007/s12272-022-01392-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-022-01392-4