
Abstract
The intestinal mucosa is continuously exposed to a large number of commensal or pathogenic microbiota and foreign food antigens. The intestinal epithelium forms a dynamic physicochemical barrier to maintain immune homeostasis. To efficiently absorb nutrients from food, the epithelium in the small intestine has thin, permeable layers spread over a vast surface area. Epithelial cells are renewed from the crypt toward the villi, accompanying epithelial cell death and shedding, to control bacterial colonization. Tight junction and adherens junction proteins provide epithelial cell–cell integrity. Microbial signals are recognized by epithelial cells via toll-like receptors. Environmental signals from short-chain fatty acids derived from commensal microbiota metabolites, aryl hydrocarbon receptors, and hypoxia-induced factors fortify gut barrier function. Here we summarize recent findings regarding various environmental factors for gut barrier function. Further, we discuss the role of gut barriers in the pathogenesis of human intestinal disease and the challenges of therapeutic strategies targeting gut barrier restoration.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
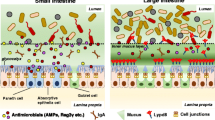
A well-structured network of epithelial and stromal cells in the gut facilitates efficient and selective nutrient absorption while providing a physical barrier against noxious agents. The intestine is covered by a single layer of epithelial cells which are differentiated from pluripotent intestinal epithelial stem cells (IESCs) at the base of the crypt (Santos et al. 2018). The small intestine and colon differ in the gross organ structure and composition/distribution of intestinal epithelial cells (IECs) (Fig. 1). In the small intestine, thin and long villi increase the mucosal surface area for efficient nutrient absorption. In contrast, villi are absent in the colon, but extended crypts efficiently absorb water and metabolic products produced by the microbiome (Allaire et al. 2019).

The difference in epithelial cell composition between the small intestine and colon. Gross features of the small intestine (a) and colon (b). There are several differences between the two. First, thin and long villi extend from the luminal surface area for nutrient absorption in the small intestine but are absent in the colon. Second, antimicrobial peptide (AMP) producing Paneth cells are in the small intestinal crypt but not the colon. Instead, Reg4+ deep crypt secretory cells in the colon play the same role as the Paneth cells. Third, the colon has a more abundant bilayer mucus than the small intestine due to a larger number of goblet cells. Therefore, the small intestine absorbs nutrients better than the colon. The small intestine and colon both have Lgr5+ stem, TA, enteroendocrine, and tuft cells. Lgr5+ stem cells can self-renew and differentiate into TA cells, which move from the crypt to the villi and can differentiate into tuft, enteroendocrine, and goblet cells
The small intestine has fewer goblet cells than the colon, but both Paneth cells and M cells are located here. In particular, Peyer’s patches and Paneth cells are mostly found in the ileum and are closely associated with a high bacterial density (Ramanan and Cadwell 2016). As there are no Paneth cells in the colon, the expression of antimicrobial molecules is lower here than in the small intestine (Cunliffe and Mahida 2004). However, there are more goblet cells in the colon than in the small intestine and the colon has a thick double mucus layer, firm inner layer, and loose outer layer. As the inner layer is anchored to the intestinal epithelium, which has polymerized mucin 2 (MUC2), microorganisms cannot easily invade the intestinal epithelium. Therefore, there are no microorganisms in the inner mucus layer (Johansson et al. 2008). The inner mucus layer is converted to the outer layer by a MUC2 proteolytic process by the host or bacteria. As a result, the loosened mucus of the outer layer contains microorganisms (Atuma et al. 2001; Johansson et al. 2008).
Regarding physicochemical barriers, the gut mucosa has multiple layers to maintain tissue homeostasis (Fig. 2) (Allaire et al. 2019). The outer layer is a microbial ecosystem that competes for and represses pathogens. The next layer is a mucous barrier containing antimicrobial peptides produced by secretory epithelial cells. The third layer is a monolayer epithelium tightly interconnected with tight and adherens junctions, including claudins, occludin, zonula occludens (ZO), and E-cadherin. The final barrier is an immunological surveillance system that controls for and combats external invaders. These multi-layered gut barriers sustain sterile conditions in most organs within the body. In the low-oxygen microenvironment of the gut, fermented short-chain fatty acid (SCFA) metabolites from dietary fibers or other signals help epithelial cells fortify barrier functions in the epithelium. Here we discuss the crucial environmental sensors maintaining the gut barrier, focusing on regulation of the epithelial barrier and its relationship with inflammatory intestinal diseases.

Disrupted gut barriers induce inflammation and an IBD outbreak. There are three physicochemical barriers in the gut other than the commensal microbiome that protect against pathogens in the gut. First, antimicrobial peptides produced by Paneth cells can disrupt microbial cell walls and membranes by forming pores that induce cell lysis. Second, the mucus layer produced by goblet cells can prevent pathogen invasion. The third barrier is a physical barrier composed of epithelial cells connected by tight and adherens junctions. Gut barrier integrity failures increase gut permeability and bacterial translocation, which lead to an inflammatory response induced by innate immune cells. IBD is induced if these responses lead to chronicity. In the lumen, short-chain fatty acids (SCFAs) produced by anaerobes also contribute to the intestinal barrier integrity
Gut epithelium
The repertoire of intestinal epithelial cells
The most prominent subtypes of intestinal epithelial cells are enterocytes, which absorb water and nutrients (Kong et al. 2018; Serra et al. 2019). Secretory subtypes of IECs include enteroendocrine cells, goblet cells, and Paneth cells (Table 1). Enteroendocrine cells regulate digestive function and the immune and nervous systems via hormone secretion and are divided into at least eight subsets according to their secretory hormones. For example, enterochromaffin cells secrete 5-hydroxy-tryptamine (5-HT, also called serotonin), D cells secrete somatostatin (SST), G cells secrete gastrin, I cells secrete cholecystokinin (CCK), and K cells secrete gastric inhibitory peptide (GIP). Enterochromaffin-like cells secrete histamine, L cells secrete glucagon-like peptide (GLP) and peptide YY (PYY), Mo cells secrete motilin, N cells secrete neurotensin (NTS), and S cells secrete secretin (SCT) (Helander and Fandriks 2012; Gribble and Reimann 2016). Goblet cells secrete mucins and Paneth cells secrete antimicrobial molecules to form physical and biochemical barriers (Ma et al. 2018; Lueschow and McElroy 2020). Goblet cells secrete glycosylated mucins into the intestinal lumen, forming a mucus layer through the disulfide bond between glycosylated mucins (Fu et al. 2011).
The most abundant MUC2 plays an important role in mucus layer organization on the epithelial surface of the colon but also binds to the glycan receptors of dendritic cells to maintain gut homeostasis and induce anti-inflammatory signaling (Heazlewood et al. 2008; Shan et al. 2013). Mucin secretion by goblet cells can be regulated by gut microbes or their metabolites (SCFAs or cytokines) (Shimotoyodome et al. 2000; Fallon et al. 2002). Goblet cell-derived products such as intestinal trefoil factor (ITF) and resistin-like molecule-β (RELM-β) also regulate intestinal physical barriers. ITF regulates tight junctions and cell apoptosis and promotes epithelial repair. RELM-β controls TH2-mediated responses (Artis et al. 2004; Aihara et al. 2017). Antimicrobial peptides (AMPs) secreted by IECs are small and basic amino acid-rich proteins. Epithelial cells produce AMPs, such as regenerating islet-derived protein IIIγ (REGIIIγ), in the small intestine, and β-defensin and cathelicidin in the colon (Hase et al. 2002; Cunliffe and Mahida 2004; Vaishnava et al. 2008, 2011).
Paneth cells in the crypts of the small intestine secrete lysozymes and a variety of AMPs, such as α-defensin, cathelicidin, REGIIIγ, and sPLA2 (Bevins and Salzman 2011). Defensins and cathelicidin interact with the negatively charged microbial cell membrane to form a pore-like structure, causing cell membrane disruption (Lai and Gallo 2009). REGIIIγ binds to the cell wall peptidoglycans of gram-positive bacteria and catalyzes the formation of pores, inducing bacterial cell lysis (Cash et al. 2006; Mukherjee et al. 2014). Paneth cells are also an important cellular niche for Lgr5+ stem cells via molecules such as Wnt3, EGF, and Notch ligands (Sato et al. 2011). However, there are no typical Paneth cells located in the colon. Instead, Reg4+ deep crypt secretory cells function as the colon equivalent of Paneth cells (Sasaki et al. 2016). Microfold cells (M cells) found in the follicle-associated epithelium specialize in the uptake of luminal antigens and delivery to antigen-presenting cells (APCs) (Mabbott et al. 2013). Cup cells are wine-like cells, accounting for 6 % of ileum epithelial cells, but their function is unknown (Madara 1982). Tuft cells (taste-chemosensory epithelial cells) secrete cytokines to initiate an immune response to parasites (Gerbe et al. 2016; Howitt et al. 2016; Middelhoff et al. 2017).
Homeostasis of intestinal epithelium from stem cells
Intestinal epithelial cells are some of the most proliferative cells in the body. To maintain the integrity and homeostasis of the intestinal barrier against pathogen and xenobiotic attack, epithelial cells constantly regenerate from the crypt region’s intestinal stem cells (ISC) (Fig. 3a) (Volk and Lacy 2017). Self-renewal and differentiation of intestinal epithelial cells are associated with the leucine-rich repeat-containing G-protein coupled receptor (GPR) 5 (LGR5). LGR5 is expressed in various body tissues and is a member of the Wnt signaling pathway which plays an essential role in recovering intestinal epithelium (Hsu et al. 1998; Cordero and Sansom 2012). LGR5 is a representative marker of ISCs, especially in adult tissue, and cancers, as differentiation of the epithelial cells is mediated by multipotent LGR5+ ISCs (Barker et al. 2007; Beumer and Clevers 2016). ISCs divide to proliferate at the bottom of the crypt, and their daughter cells climb to the upper villi, much like a conveyer belt. These rapidly dividing daughter cells, called transit-amplifying (TA) cells, then differentiate into an absorptive progenitor or a secretory progenitor (Hsu et al. 2014).

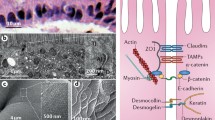
Dynamic homeostasis of the gut epithelium. a Proliferation and differentiation of Lgr5+ stem cells into TA cells simultaneously occur in the intestinal crypt. TA cells are differentiated into secretory and absorptive epithelial lineages during migration from the crypt to villi. When these differentiated cells reach the villi tips, apoptosis occurs via cell signaling. b There are various junctions in the paracellular spaces between adjacent cells. The tight junction is composed of claudin, occludin, and ZO-1, and the adherens junction is composed of E-cadherin. There are also desmosomes, gap junctions, and hemidesmosomes in the paracellular space that are involved in transporting nutrients and forming the physical intestinal barrier. c TA cells derived from Lgr5+ stem cells can differentiate into secretory or absorptive epithelial lineages. Math1 activation by Wnt/β-catenin signaling, ATOH1, and DLL1 can induce TA cell differentiation into secretory epithelial progenitors. HES 1 activation by Notch signaling can induce TA cell differentiation into absorptive epithelial progenitors
TA cell plasticity and differentiation are strictly controlled by three major signaling pathways: Wnt, Notch, and bone morphogenetic protein (BMP) (Fig. 3c). Notch-HES1 signaling promotes the TA cells’ absorptive lineage (Demitrack and Samuelson 2016; Kim and Jang 2020) post-differentiation to an absorptive enterocyte (Gui et al. 2017). The Wnt-Math1 signaling pathway leads the TA cells to differentiate into a secretory lineage (Yang et al. 2001; Gui et al. 2017). ATOH1 and DLL1 coordinate to form a secretory progenitor (van Es et al. 2012; Tomic et al. 2018). The differentiation process then forms the enteroendocrine cells, goblet cells, and Paneth cells, which are regulated by NEUROG3, GHI1, SPDEF, SOX9, and EPHB3. BMP is critical for balancing Wnt-driven homeostatic proliferation (Medema and Vermeulen 2011). At the crypt domain, Wnt concentration is more dominant than BMP although both Wnt and BMP signals are important for axis between self-renewal and differentiation. In contrast, at the villi domain, BMP concentration is more dominant than Wnt which mediates proliferation versus differentiation (Spit et al. 2018). The strict, rapid differentiation and proliferation of epithelial cells promote intestinal homeostasis. Impaired or damaged cells can be removed or replaced by newly differentiated cells (Gu et al. 2011; Bischoff et al. 2014). Increased epithelial proliferation removes parasitized or infected epithelial cells (MacDonald 1992), while crowding of epithelial cells extrudes the villi, preventing excessive epithelial cell accumulation and tumor formation (Eisenhoffer et al. 2012).
Intestinal organoid is one of useful in vitro models for patient specific investigation of the intestinal epithelium hereafter the first establishment in 2009 for self-organizing ‘mini-guts’ (Sato et al. 2009). Single Lgr5+ ISCs or isolated crypt containing ISCs are seeded into a supporting Matrigel matrix which provides stem cell niche. The isolated ISCs have the ability to survive, proliferate, self-organize into 3-dimensional structures in vitro. They require Wnt/Notch for self-renewal and differentiation (Angus et al. 2019). Wnt in the small intestine can be provided by mesenchymal cells and Paneth cells. Unlike intestinal organoid originated from small intestine, colonic organoid does not contain Wnt-producing Paneth cell, which therefore require more exogenous Wnt supplement (Takahashi and Shiraishi 2020). Patient-derived gut organoids can be utilized for investigating infectious disease, whole-genome sequencing, drug screening and regenerative medicine.
Epithelial cell junctions
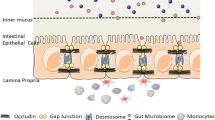
Cell-cell junctions are a well-organized, structural continuum of the extracellular connection between adjacent cells. They are composed of different cytoskeleton elements (Fig. 3b). These junctions maintain homeostasis by regulating tissue integrity and ion, solute, and microbe diffusion across the tissue.
Tight junction
Mammals have tight junctions in the apex of the lateral plasma membranes between adjacent cells. Tight junctions surround each cell form a proteinaceous film that regulates ion and solute diffusion via a paracellular pathway. Tight junctions maintain the division of apical and basolateral membrane proteins and lipids (Zihni et al. 2016). Tight junctions are composed of transmembrane protein families, including claudin, occludin, and the peripheral membrane adaptor protein ZO (Van Itallie and Anderson 2014; Lee et al. 2018a). In human studies, claudin 1, 2, 3, 4, 5, 7, 8, 12, and 15 are expressed in the small intestine (Lu et al. 2013). Claudin and occludin form homotypic complexes between cells. ZO-1, 2, and 3 connect occludin and claudin to the actin cytoskeleton, which maintains the tight junction formation. The mutual assembly of tight junction proteins forms different pore-sized networks, mediating the differential diffusion of ions and solutes (Zihni et al. 2016). Since the permeability of ions of different sizes and charges is determined according to the amino acids in the claudins, diffusion varies with the expressed claudin type (Van Itallie and Anderson 2006). While the precise mechanism of action remains elusive, occludin regulates tight junction stability, permeability, and barrier function through phosphorylation and a ZO-1 interaction (Lee et al. 2018a).
Adherens junction
Adherens junctions are protein complexes that are usually more basal than the tight junction (Guo et al. 2007). The adherens junction plays a role in cell-cell adhesion, actin cytoskeleton regulation, cell signaling, and gene transcription (Takeichi 2014). Cadherin, such as E-cadherin, is the main type of transmembrane protein comprising the adherens junction. The cadherins connect to adjacent cadherins in a calcium-dependent manner. These cadherins indirectly bind α-catenin via β-catenin, and in turn α-catenin links to the actin cytoskeleton (Knudsen et al. 1995). P120-catenin linked with the cadherin ternary complex is associated with the cadherin juxtamembrane domain, which suppresses cadherin endocytosis (Davis et al. 2003). E-cadherin adhesion can be intensified through the protein vinculin’s link to a force-dependent conformation of α-catenin (le Duc et al. 2010). Nectins bind to afadins which are involved in Ca2+-independent cellular adhesion (Takai and Nakanishi 2003). These nectin-based adhesions make the first cell-cell junction, which then recruit the cadherin-catenin complex to form the adherens junction (Tachibana et al. 2000; Honda et al. 2003).
Environmental sensors for gut barriers
Epithelial cells are the primary cellular determinant of the epithelial barrier function in the gut. Epithelial cells express toll-like receptors (TLRs), which are essential for the recognition of conserved microbial factors. There are several environmental sensors involved in maintaining the gut epithelial barrier, such as hypoxia-induced factor (HIF), aryl hydrocarbon receptor (AhR), and SCFAs. Here, we will focus on the cellular mechanism and relationship of these environmental sensor signals for gut barrier integrity.
Short-chain fatty acids (SCFAs)
SCFAs are microbiota-derived metabolites, such as acetate, propionate, butyrate, and valerate, which are produced from dietary fibers through the fermentation of anaerobic flora (Fig. 4). SCFAs are passively diffused from the lumen to the cell and then transported into the cells via carrier proteins, such as proton-coupled monocarboxylate transporter 1 (MCT1) and sodium-coupled monocarboxylate transporter 1 (SMCT1) (Sivaprakasam et al. 2017; Parada Venegas et al. 2019). These are agonists for the G-protein coupled receptors (GPR) 41/FFAR3, GPR43/FFAR2, and GPR109A/NIACR1, leading to various immune responses in IECs, dendritic cells, and macrophages (Priyadarshini et al. 2018). Butyrate can facilitate differentiation of IL-10 producing Tregs and reduce the pro-inflammatory cytokine TNF-α from macrophages, which promotes inflammation by recruiting neutrophils in the gut (Vinolo et al. 2009; Lee and Hase 2014). SCFAs enhance antimicrobial peptides such as REGIIIγ and β-defensin via activation of mTOR and STAT3 in the epithelium barrier. (Zhao et al. 2018; Chen and Vitetta 2020). In ISCs of colon crypt, butyrate can suppress cell proliferation by inhibiting histone deacetylases (HDAC) and enhancing promoter activity for the negative cell-cycle regulator FoxO3. Fully differentiated colonocytes metabolize and reduce butyrate levels through Acyl-CoA dehydrogenase (ACADS) (Kaiko et al. 2016; Xiao et al. 2018). However, lactate accelerates stem cell proliferation dependent on GPR81 (Lee et al. 2018b). Butyrate can promote TGF-β expression through HDAC inhibition and SP1 (Martin-Gallausiaux et al. 2018). In association with other environmental signals, butyrate can stabilize HIF as O2 consumption for its metabolic process causes cellular hypoxic conditions (Kelly et al. 2015). These findings suggest that crosstalk between microbiota-derived SCFAs and intestinal epithelial HIF augments gut barrier function.

The role of SCFAs in gut barrier function. SCFAs produced by the microbiota after anaerobic fermentation of dietary fiber can be recognized by G-protein coupled receptors such as GPR41, 43, 81, and 109 A. When butyrate is recognized by GPR43, STAT3 expression is induced by the mTOR pathway and STAT3 can induce REGIIIγ and β-defensin expression. This can induce SP1 expression, the transcription factor for TGF-β expression in colonocytes that inhibits ISC proliferation by HDAC inhibition and FoxO3 expression. Lactate, which is recognized by GPR81, can induce ISC proliferation. Mitochondrial oxygen consumption during butyrate metabolism can indirectly stabilize HIF-α
Toll-like receptor (TLR)
TLRs are representative pattern recognition receptors (PRR) with a transmembrane protein form that can recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (Kawasaki and Kawai 2014). TLRs are identified from TLR1 to TLR13, with TLR1 to TLR10 found in human cells. Each TLR is combined in a heterodimer or homodimer that can recognize various ligands according to its combination. If TLR ligands are recognized, two major types of adaptor proteins, such as Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) and myeloid differentiation factor 88 (MyD88), can mediate downstream signals (Kamdar et al. 2018). NF-κB is a transcription factor that expresses pro-inflammatory cytokines such as IL-1, IL-6, IL-12, and TNF-α (Mukherjee et al. 2016).
In both Crohn’s disease and ulcerative colitis (UC) patients, expression of TLR1, TLR2, TLR6, TLR8, and TLR9 remains unchanged, while expression of TLR4 and TLR5 is increased compared to healthy controls (Kordjazy et al. 2018). In immunocompetent cells, TLR signaling generally induces innate immune responses. In chronic inflammation, it mediates intestinal barrier breakdown via inflammatory mediators, such as TNF-α (Peterson et al. 2010). However, activation of TLRs by the commensal microflora controls intestinal epithelial homeostasis and protects against injury (Rakoff-Nahoum et al. 2004). In intestinal epithelial cells, TLR2 stimulation efficiently preserves ZO-1-associated barrier integrity against stress-induced damage, which is controlled by PI3K/AKT and conventional protein kinase C (PKC) isoforms via MyD88 (Cario 2008). TLR1-deficient mice have increased permeability and reduced transmucosal resistance followed by increased bacterial translocation to systemic organs (Kamdar et al. 2018). These findings suggest that TLR1–TLR2 signaling sustains epithelial integrity through the tightening of intercellular junctions. Conversely, TLR4 activation induces enhanced barrier permeability and leaky gut through upregulation of myosin light chain kinase (MLCK), which mediates the opening of tight junctions by promoting actin-myosin filament contraction(Nighot et al. 2017). In indirect association with other environmental sensors, TLR3 and TLR4 activation can upregulate HIF-1α gene expression at the mRNA level via the NF-κB pathway (Han et al. 2016). LPS induces TLR4 signaling, which drives ferritin-mediated iron sequestration and results in deprivation of an essential PHD cofactor, free iron, followed by HIF-1α stabilization (Siegert et al. 2015). These findings demonstrate that different TLR signals are associated with the differential control of the intercellular barrier integrity by enhancing or disrupting intestinal epithelial barrier junction molecules. This depends on the type of microbe.
Aryl hydrocarbon receptor (AhR)
AhR is a ligand-dependent intracellular transcription factor (Rothhammer and Quintana 2019). AhR is usually combined with the heat shock protein 90 (HSP90) dimer and X-associated protein 2 (XAP2) in the cytoplasm (Fig. 5). AhR contains two regions, the Per-Arnt-Sim (PAS) domain and the basic Helix/Loop/Helix (bHLH) domain. The PAS domain is located in AhR’s C-terminal, which plays a role in maintaining the cytosolic AhR complex in the absence of ligands. The bHLH domain is in AhR’s N-terminal, which contains a nuclear localization sequence (NLS) dependent on AhR ligands and a nuclear export sequence (NES) (Hao and Whitelaw 2013). Once AhR ligands such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), 6-Formyl indolo[3,2-b]carbazole (FICZ), and 2-(1ʹH-indole-3ʹ-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) are bound to the AhR-HSP90 dimer-XAP2 complex, AhR NLS is exposed and the ligand-AhR complex is transported into the nucleus (Ikuta et al. 1998; Ehrlich et al. 2018). In the nucleus, the AhR-ligand complex that has dissociated from the HSP 90 dimer-XAP2 combines with the AhR nuclear translocator (ARNT, also known as HIF-1β). The AhR-ARNT complex is recruited to the xenobiotic response element (XRE) and acts as a transcription factor for the expression of molecules such as cytochrome P450 family-1 subfamily-A polypeptide-1 (CYP1A1), CYP1B1, and AhR repressor (AhRR) (Zhu et al. 2019). CYP1A1 is a representative metabolizing enzyme in the cytoplasm that can reduce AhR signaling via ligand consumption (Schiering et al. 2017). AhRR also competes with the AhR-ligand complex for ARNT. When ARNT is combined with AhRR, exposure to AhR NES mediates the relocation of the AhR-ligand complex from the nucleus to the cytoplasm. It is then finally degraded by proteosomes in the cytoplasm (Ikuta et al. 1998; Rothhammer and Quintana 2019).

The role of AhR signaling in gut barrier function. Ligands such as TCDD, FICZ, and ITE are essential for AhR activation. When ligands bind to the AhR/Hsp90/XAP2 complex, it moves from the cytoplasm to the nucleus and binds to ARNT. This complex acts as the transcription factor for XRE, which is involved in CYP1A1 and AhRR expression. If the AhR-ARNT complex is not formed by AhRR, AhR moves from the nucleus to the cytoplasm and it is degraded by the proteasome. CYP1A1 can also metabolize the AhR ligand, inactivating AhR signaling. AhR activation can induce tight junction component expression and goblet cell differentiation and inhibit Lgr5+ stem cell proliferation by regulating Wnt/β-catenin signaling
AhR is expressed in epithelial and immune cells in the gut and plays a role in the intestinal barrier’s homeostatic and inflammatory conditions. AhR promotes anti-inflammatory DC and induces Th17 differentiation and Treg stabilization. In a 2, 4, 6-trinitrobenzenesulphonic acid (TNBS) colitis model, activation of AhR by FICZ reduced inflammatory cytokines and induced IL-22 expression in DCs and CD4+ T cells, which triggers AMP production and reinforces the mucus barrier (Monteleone et al. 2011). In dextran sulfate sodium (DSS)-induced colitis, AhR activation by FICZ enhanced the expression of tight junction proteins such as ZO-1, claudin-1, and occludin to reduce barrier permeability (Yu et al. 2018). AhR-deficient IECs abnormally function in the Wnt/β-catenin and ubiquitin E3 ligase signaling pathways (Metidji et al. 2018). AhR controls IEC self-renewal by limiting ISC proliferation and promoting its differentiation. AhR in IECs enhances IL-10 receptor expression to boost their responsiveness to IL-10 and enhance epithelial barrier function (Lanis et al. 2017). In the IECs of a Crohn’s disease patient, miRNA-124 induced pro-inflammatory transcriptional programs by targeting AhR (Zhao et al. 2016). In contrast with AhR’s barrier protection, dietary and microbial oxazoles activate indoleamine 2, 3-dioxygenase 1(IDO-1) to generate tryptophan metabolites, which inhibited IL-10 production and induced intestinal inflammation (Iyer et al. 2018). SCFAs, especially butyrate, enhance the AhR pathway and AhR-dependent genes in IECs, which suggests that butyrate may be a potential AhR ligand (Marinelli et al. 2019). In association with other environmental sensors, AhR competes with HIF-1α to interact with ARNT (HIF-1β) (Chan et al. 1999). This integration of HIF-1α and AhR might lead to interference between the two signaling pathways in a variety of cellular responses. AhR promotes HIF-1α degradation in Tr1 cells (Mascanfroni et al. 2015). Research must examine the crosstalk between HIF-1α and AhR in IECs and its effect on gut barrier function.
Hypoxia-inducible factor (HIF)
HIF is an oxygen-sensitive transcription factor and a cellular survival mechanism for hypoxic stress that is related to cellular metabolism, the intestinal barrier, erythropoiesis, and angiogenesis (Glover and Colgan 2011). Receptor tyrosine kinase (RTK) in the plasma membrane recognizes growth factors and activates the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of the rapamycin (mTOR) signaling pathway. Activated mTOR can induce production of the HIF-α molecule as a transcription factor (Guo et al. 2015). These factors consist of three oxygen-sensitive α-subunits of HIF molecules called HIF-1α, HIF-2α, and HIF-3α and a conserved β-subunit, HIF-1β, also known as aryl hydrocarbon receptor nuclear translocator (ARNT) (Rankin and Giaccia 2008).
In normoxia, HIF-α is hydroxylated at the proline or asparagine residues by prolyl hydroxylase (PHD) and asparaginyl hydroxylase factor inhibiting HIF (FIH) (Fig. 6). Hydroxylated HIF-α in the cytoplasm is ubiquitinated by the von Hippel-Lindau protein (pVHL), leading to their proteasomal degradation. In hypoxia, HIF-α accumulates in stable condition and translocates from the cytoplasm to the nucleus, where it binds to HIF-1β and 300-kilodalton coactivator protein (p300)/CREB binding protein (CBP). This complex acts as a transcription factor for hypoxia response element (HRE), expressing genes that allow for adaptation or survival under hypoxic conditions (Cavadas et al. 2013). HIF’s hypoxia-induced responses can induce or regulate inflammation according to different cells in the gut. Hypoxia may represent an environmental cause for inflammatory bowel disease (IBD) pathogenesis. Both HIF-1α and HIF-2α are found in high levels in IECs in active UC or Crohn’s disease patients (Xue et al. 2013).

The role of HIF signaling in gut barrier function. In normoxia, proline or asparagine hydroxylation occurs in HIF-α by PHD or FIH. pVHL is also conjugated into hydroxylated HIF-α, leading to ubiquitination and proteasomal degradation. In hypoxia, HIF-α are stabilized and accumulate without hydroxylation. They move from the cytoplasm to the nucleus and bind to p300/CBP and HIF-β, forming a heterodimer. This acts as the transcription factor for HRE, with the gene expression depending on subtype. HIF-1α induces the expression of genes involved in enhancing intestinal barrier function, such as β-defensin, MUC3, and ITF. HIF-2α induces pro-proliferative and pro-inflammatory gene expression, which decreases barrier integrity in the IBD model
Hypoxia promotes IECs to produce TNF, leading to an increase in epithelial barrier permeability (Taylor et al. 1998). HIF signals in the innate immune cells, including neutrophils, macrophages, and dendritic cells, enhance pro-inflammatory cytokine production (Bosco et al. 2011). In contrast, hypoxia-exposed IECs in the physiological gut lumen induce barrier-preservative factors to reduce the inflammatory burden of ITF, MUC3, and CD73 expression. Even transmigrating neutrophils rapidly deplete microenvironmental oxygen, which leads to the stabilization of HIF molecules in the gut epithelium (Campbell et al. 2014). In experimental animal models of oxazolone and TNBS-induced colitis, HIF-1α isoform expression was beneficial in ameliorating inflammation via induction of barrier-protective genes (Karhausen et al. 2004). In experimental murine DSS-induced colitis, HIF-2α augmented intestinal inflammation via increased inflammatory responses (Shah et al. 2008). The PHDs consist of three isoforms, PHD1, PHD2, and PHD3, which mediate diverse functions in immunocompetent and non-immune cells. Deficiency of PHD1 or PHD3 protects intestinal epithelial barrier integrity in mice (Tambuwala et al. 2010; Chen et al. 2015). These results suggest differential effects of HIF isoforms in gut homeostasis.
Relation with human IBD and therapeutic strategies
Gut barriers in IBD
Defects in the gut barrier are associated with a broad range of human diseases, such as IBD, and extra-intestinal diseases, such as non-alcoholic fatty liver disease and neurologic brain diseases (Fig. 2) (Vancamelbeke and Vermeire 2017). IBD is a chronic inflammatory disease in the gastrointestinal tract comprising uncreative colitis and Crohn’s diseases. Possible causes include combined host genetic alterations and environmental factors. IBD is thought to be closely related to immune function alterations caused by commensal microbiota (Zhang and Li 2014). In a healthy adult gut, Firmicutes, Bacteroidetes, Proteobacteria, and Actinomycetes co-exist in balance. Patients with IBD, especially UC patients, have decreased Firmicutes and Bacteroidetes and increased Proteobacteria and Actinomycetes (Machiels et al. 2014). This dysbiosis disrupts the balanced microbial composition and accumulates bacterial toxins such as LPS. IBD patients have higher intestinal permeability than healthy groups (Michielan and D’Inca 2015). Occludin expression systemically decreases at the mRNA and protein levels in patients with uncreative colitis and Crohn’s diseases. Expression of tight junction and adherens junction components such as ZO-1, claudin, E-cadherin, and β-catenin decreases in epithelial cells in inflammatory regions. This is mainly caused by circulating pro-inflammatory cytokines, such as IFN-γ, TNF-α, and IL-13 (Fries et al. 2013). TNF-α increases epithelial permeability through alterations in tight junction function, structure, and dynamics upon infection (Capaldo and Nusrat 2015).
Gut barrier fortification as a target of IBD therapeutics
Current novel IBD drugs have mainly focused on controlling inflammation, as this is the most important disease symptom (Neurath 2017). Recently, biologics to block key mediators of pathogenic inflammation, such as TNFs or integrin, have been increasingly used in IBD patients. TNF-α blockers can reduce the excessive inflammatory response and gut permeability (Suenaert et al. 2002). In addition, therapies that block cytokine signaling have been developed, such as Janus kinases (JAKs) inhibitors and tofacitinib (Danese et al. 2016). Fecal microbiota transplant involves feces from healthy donors being transplanted into IBD patients. The changes in intestinal bacterial composition are expected to have beneficial effects in IBD patients (Lopez and Grinspan 2016). However, novel fundamental modulators need to be further investigated.
One current IBD therapeutic strategy is to restore the gut barrier function (Fig. 7). The protective role of HIF-1α during gut inflammation has led to the investigation of PHD inhibitors as a potential therapeutic strategy. Pan-PHD inhibitors, such as dimethyloxalylglycine (DMOG) and FG-4497, can reduce the symptoms of experimental murine colitis (Cummins et al. 2008; Robinson et al. 2008). Oral administration of AKB-4924 or TRC160334 protects against murine colitis and reduces systemic off-target effects in extra-intestinal organs (Gupta et al. 2014; Marks et al. 2015). Several pan-PHD inhibitors are currently being examined for the treatment of various diseases, including IBD (Marks et al. 2015). Further, local treatment of AKB-4924 is currently under phase I clinical trials (NCT02914262).

Overview of IBD and therapeutic approaches. IBD, a chronic inflammatory disease in the lower gastrointestinal tract, has complex genetic and environmental reasons. On a cellular level, decreased mucus, dysbiosis in the lumen, infection, and a severe inflammatory response via innate immune cells are related to IBD pathogenesis. Recent research regarding the role of gut environmental sensors in reducing inflammation via fortification of the gut barrier has shed light on the development of novel IBD therapeutics
Based on the beneficial effect of SCFA on the gut barrier, Phase 2 clinical trials with GLPG0974, a GPR43-specific antagonist, in individuals with mild-to-moderate UC did not change clinical outcomes over a short period (Bolognini et al. 2016).While SCFAs have presented beneficial effects in experimental systems of intestinal inflammation, clinical effects remain controversial (Galvez et al. 2005).
Oral treatment with the microbial metabolite Urolithin A (UroA) derived from polyphenolics in berries and pomegranate fruits and its analog UAS03 significantly enhanced gut barrier function and inhibited gut inflammation, suggesting a potential therapeutic application for the IBD treatment (Singh et al. 2019). AhR ligands such as indigo from plants can promote mucosal healing by inducing IL-22 production from type 3 innate lymphocytes cells (ILC3) (Zelante et al. 2013). In a prospective, randomized, double-blind, placebo-controlled trial, indigo naturalis effectively induced a clinical response in UC patients. However, its safety has not been established because of potential adverse effects, including pulmonary arterial hypertension (Naganuma et al. 2018). TLR2-p treatment significantly reduced colitis-associated conditions, suggesting that the TLR2 signaling pathways are promising therapeutic targets (Laroui et al. 2012). TLR9 activation improved mucosal healing and symptomatic remission in UC patients (Atreya et al. 2016). Palmitoylethanolammide (PEA) improves the acute phase of the intestinal inflammation that occurs in UC through enteric glia/toll-like receptor 4-dependent PPAR-α activation (Esposito et al. 2014). MLCK is a potential therapeutic target as epithelial MLCK–dependent barrier dysfunction following TLR signaling mediates intestinal inflammation (Clayburgh et al. 2005). Divertin blocks MLCK1 recruitment without inhibiting enzymatic function, which corrects barrier dysfunction and prevents intestinal inflammation (Graham et al. 2019).
Concluding remarks
Biological barrier homeostasis is critical for protecting against infection. In the gut mucosa, barrier integrity is critical since the gut has a unique environment consisting of nutritional and microbial factors. Barrier function breakdown initiates local inflammation in luminal products followed by the onset of intestinal inflammatory disease. Research indicates that epithelial cells utilize gut environmental sensors to maintain these barriers. Currently, novel therapeutic targets for barrier modulation via environmental sensors have been studied in animal models and human trials. Barrier and anti-inflammatory modulators are a promising therapeutic option for treating IBD.
References
Aihara E, Engevik KA, Montrose MH (2017) Trefoil factor peptides and gastrointestinal function. Annu Rev Physiol 79:357–380. https://doi.org/10.1146/annurev-physiol-021115-105447
Allaire JM, Crowley SM, Law HT, Chang SY, Ko HJ, Vallance BA (2019) The intestinal epithelium: central coordinator of mucosal immunity: (Trends in Immunology 39, 677–696, 2018). Trends Immunol 40(2):174. https://doi.org/10.1016/j.it.2018.12.0082018.12.008
Angus HCK, Butt AG, Schultz M, Kemp RA (2019) Intestinal organoids as a tool for inflammatory bowel disease research. Front Med (Lausanne) 6:334. https://doi.org/10.3389/fmed.2019.00334
Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, Schad GA, Scott P, Wu GD (2004) RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci USA 101(37):13596–13600. https://doi.org/10.1073/pnas.0404034101
Atreya R, Bloom S, Scaldaferri F, Gerardi V, Admyre C, Karlsson A, Knittel T, Kowalski J, Lukas M, Lofberg R, Nancey S, Petryka R, Rydzewska G, Schnabel R, Seidler U, Neurath MF, Hawkey C (2016) Clinical effects of a topically applied toll-like receptor 9 agonist in active moderate-to-severe ulcerative colitis. J Crohns Colitis 10(11):1294–1302. https://doi.org/10.1093/ecco-jcc/jjw103
Atuma C, Strugala V, Allen A, Holm L (2001) The adherent gastrointestinal mucus gel layer: thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol 280(5):G922–G929. https://doi.org/10.1152/ajpgi.2001.280.5.G922
Barker N, Van Es JH, Kuipers J, Kujala P, Van Den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H (2007) Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449(7165):1003–1007. https://doi.org/10.1038/nature06196
Beumer J, Clevers H (2016) Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Development 143(20):3639–3649. https://doi.org/10.1242/dev.133132
Bevins CL, Salzman NH (2011) Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9(5):356–368. https://doi.org/10.1038/nrmicro2546
Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, Wells JM (2014) Intestinal permeability–a new target for disease prevention and therapy. BMC Gastroenterol 14:189. https://doi.org/10.1186/s12876-014-0189-7
Bolognini D, Tobin AB, Milligan G, Moss CE (2016) The pharmacology and function of receptors for short-chain fatty acids. Mol Pharmacol 89(3):388–398. https://doi.org/10.1124/mol.115.102301
Bosco MC, Pierobon D, Blengio F, Raggi F, Vanni C, Gattorno M, Eva A, Novelli F, Cappello P, Giovarelli M, Varesio L (2011) Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood 117(9):2625–2639. https://doi.org/10.1182/blood-2010-06-292136
Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, Mcnamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, Ehrentraut SF, Curtis VF, Burgess A, Garvey JF, Sorensen A, Nemenoff R, Jedlicka P, Taylor CT, Kominsky DJ, Colgan SP (2014) Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40(1):66–77. https://doi.org/10.1016/j.immuni.2013.11.020
Capaldo CT, Nusrat A (2015) Claudin switching: physiological plasticity of the Tight Junction. Semin Cell Dev Biol 42:22–29. https://doi.org/10.1016/j.semcdb.2015.04.003
Cario E (2008) Barrier-protective function of intestinal epithelial Toll-like receptor 2. Mucosal Immunol 1(Suppl 1):S62–S66. https://doi.org/10.1038/mi.2008.472008.47
Cash HL, Whitham CV, Behrendt CL, Hooper LV (2006) Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313(5790):1126–1130. https://doi.org/10.1126/science.1127119
Cavadas MA, Nguyen LK, Cheong A (2013) Hypoxia-inducible factor (HIF) network: insights from mathematical models. Cell Commun Signal 11(1):42. https://doi.org/10.1186/1478-811X-11-42
Chan WK, Yao G, Gu YZ, Bradfield CA (1999) Cross-talk between the aryl hydrocarbon receptor and hypoxia inducible factor signaling pathways. Demonstration of competition and compensation. J Biol Chem 274(17):12115–12123. https://doi.org/10.1074/jbc.274.17.12115
Chen J, Vitetta L (2020) The role of butyrate in attenuating pathobiont-induced hyperinflammation. Immune Netw 20(2):e15. https://doi.org/10.4110/in.2020.20.e15
Chen Y, Zhang HS, Fong GH, Xi QL, Wu GH, Bai CG, Ling ZQ, Fan L, Xu YM, Qin YQ, Yuan TL, Sun H, Fang J (2015) PHD3 stabilizes the tight junction protein occludin and protects intestinal epithelial barrier function. J Biol Chem 290(33):20580–20589. https://doi.org/10.1074/jbc.M115.653584
Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR (2005) Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest 115(10):2702–2715. https://doi.org/10.1172/JCI24970
Cordero JB, Sansom OJ (2012) Wnt signalling and its role in stem cell-driven intestinal regeneration and hyperplasia. Acta Physiol (Oxf) 204(1):137–143. https://doi.org/10.1111/j.1748-1716.2011.02288.x
Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT (2008) The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology 134(1):156–165. https://doi.org/10.1053/j.gastro.2007.10.012
Cunliffe RN, Mahida YR (2004) Expression and regulation of antimicrobial peptides in the gastrointestinal tract. J Leukoc Biol 75(1):49–58. https://doi.org/10.1189/jlb.0503249
Danese S, Grisham M, Hodge J, Telliez JB (2016) JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol 310(3):G155–G162. https://doi.org/10.1152/ajpgi.00311.2015
Davis MA, Ireton RC, Reynolds AB (2003) A core function for p120-catenin in cadherin turnover. J Cell Biol 163(3):525–534. https://doi.org/10.1083/jcb.200307111
Demitrack ES, Samuelson LC (2016) Notch regulation of gastrointestinal stem cells. J Physiol 594(17):4791–4803. https://doi.org/10.1113/JP271667
Ehrlich AK, Pennington JM, Bisson WH, Kolluri SK, Kerkvliet NI (2018) TCDD, FICZ, and other high affinity AhR ligands dose-dependently determine the fate of CD4 + T cell differentiation. Toxicol Sci 161(2):310–320. https://doi.org/10.1093/toxsci/kfx215
Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J (2012) Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 484(7395):546–549. https://doi.org/10.1038/nature10999
Esposito G, Capoccia E, Turco F, Palumbo I, Lu J, Steardo A, Cuomo R, Sarnelli G, Steardo L (2014) Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut 63(8):1300–1312. https://doi.org/10.1136/gutjnl-2013-305005
Fallon PG, Jolin HE, Smith P, Emson CL, Townsend MJ, Fallon R, Smith P, Mckenzie AN (2002) IL-4 induces characteristic Th2 responses even in the combined absence of IL-5, IL-9, and IL-13. Immunity 17(1):7–17. https://doi.org/10.1016/s1074-7613(02)00332-1
Fries W, Belvedere A, Vetrano S (2013) Sealing the broken barrier in IBD: intestinal permeability, epithelial cells and junctions. Curr Drug Targets 14(12):1460–1470. https://doi.org/10.2174/1389450111314120011
Fu J, Wei B, Wen T, Johansson ME, Liu X, Bradford E, Thomsson KA, Mcgee S, Mansour L, Tong M, Mcdaniel JM, Sferra TJ, Turner JR, Chen H, Hansson GC, Braun J, Xia L (2011) Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest 121(4):1657–1666. https://doi.org/10.1172/JCI45538
Galvez J, Rodriguez-Cabezas ME, Zarzuelo A (2005) Effects of dietary fiber on inflammatory bowel disease. Mol Nutr Food Res 49(6):601–608. https://doi.org/10.1002/mnfr.200500013
Gerbe F, Sidot E, Smyth DJ, Ohmoto M, Matsumoto I, Dardalhon V, Cesses P, Garnier L, Pouzolles M, Brulin B, Bruschi M, Harcus Y, Zimmermann VS, Taylor N, Maizels RM, Jay P (2016) Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529(7585):226–230. https://doi.org/10.1038/nature16527
Glover LE, Colgan SP (2011) Hypoxia and metabolic factors that influence inflammatory bowel disease pathogenesis. Gastroenterology 140(6):1748–1755. https://doi.org/10.1053/j.gastro.2011.01.056
Graham WV, He W, Marchiando AM, Zha J, Singh G, Li HS, Biswas A, Ong M, Jiang ZH, Choi W, Zuccola H, Wang Y, Griffith J, Wu J, Rosenberg HJ, Wang Y, Snapper SB, Ostrov D, Meredith SC, Miller LW, Turner JR (2019) Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat Med 25(4):690–700. https://doi.org/10.1038/s41591-019-0393-7
Gribble FM, Reimann F (2016) Enteroendocrine cells: chemosensors in the intestinal epithelium. Annu Rev Physiol 78:277–299. https://doi.org/10.1146/annurev-physiol-021115-105439
Gu Y, Forostyan T, Sabbadini R, Rosenblatt J (2011) Epithelial cell extrusion requires the sphingosine-1-phosphate receptor 2 pathway. J Cell Biol 193(4):667–676. https://doi.org/10.1083/jcb.201010075
Gui X, Meng Z, Mcconnell YJ, Liu S, Falck VG, Mack LA, Temple WJ (2017) Differing expression profiles of Notch/enterocyte and Wnt/secretory lineage signallings are associated with morphological diversity of appendiceal tumours. J Clin Pathol 70(1):40–50. https://doi.org/10.1136/jclinpath-2016-203645
Guo H, German P, Bai S, Barnes S, Guo W, Qi X, Lou H, Liang J, Jonasch E, Mills GB, Ding Z (2015) The PI3K/AKT pathway and renal cell carcinoma. J Genet Genom 42(7):343–353. https://doi.org/10.1016/j.jgg.2015.03.003
Guo R, Sakamoto H, Sugiura S, Ogawa M (2007) Endothelial cell motility is compatible with junctional integrity. J Cell Physiol 211(2):327–335. https://doi.org/10.1002/jcp.20937
Gupta R, Chaudhary AR, Shah BN, Jadhav AV, Zambad SP, Gupta RC, Deshpande S, Chauthaiwale V, Dutt C (2014) Therapeutic treatment with a novel hypoxia-inducible factor hydroxylase inhibitor (TRC160334) ameliorates murine colitis. Clin Exp Gastroenterol 7:13–23. https://doi.org/10.2147/CEG.S51923
Han S, Xu W, Wang Z, Qi X, Wang Y, Ni Y, Shen H, Hu Q, Han W (2016) Crosstalk between the HIF-1 and Toll-like receptor/nuclear factor-kappaB pathways in the oral squamous cell carcinoma microenvironment. Oncotarget 7(25):37773–37789. https://doi.org/10.18632/oncotarget.9329
Hao N, Whitelaw ML (2013) The emerging roles of AhR in physiology and immunity. Biochem Pharmacol 86(5):561–570. https://doi.org/10.1016/j.bcp.2013.07.004
Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF (2002) Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun 70(2):953–963. https://doi.org/10.1128/iai.70.2.953-963.2002
Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, Adams R, Kato M, Nelms KA, Hong NA, Florin TH, Goodnow CC, Mcguckin MA (2008) Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 5(3):e54. https://doi.org/10.1371/journal.pmed.0050054
Helander HF, Fandriks L (2012) The enteroendocrine “letter cells”—time for a new nomenclature? Scand J Gastroenterol 47(1):3–12. https://doi.org/10.3109/00365521.2011.638391
Honda T, Shimizu K, Kawakatsu T, Yasumi M, Shingai T, Fukuhara A, Ozaki-Kuroda K, Irie K, Nakanishi H, Takai Y (2003) Antagonistic and agonistic effects of an extracellular fragment of nectin on formation of E-cadherin-based cell-cell adhesion. Genes Cells 8(1):51–63. https://doi.org/10.1046/j.1365-2443.2003.00616.x
Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, Artis D, Garrett WS (2016) Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351(6279):1329–1333. https://doi.org/10.1126/science.aaf1648
Hsu SY, Liang SG, Hsueh AJ (1998) Characterization of two LGR genes homologous to gonadotropin and thyrotropin receptors with extracellular leucine-rich repeats and a G protein-coupled, seven-transmembrane region. Mol Endocrinol 12(12):1830–1845. https://doi.org/10.1210/mend.12.12.0211
Hsu YC, Li L, Fuchs E (2014) Transit-amplifying cells orchestrate stem cell activity and tissue regeneration. Cell 157(4):935–949. https://doi.org/10.1016/j.cell.2014.02.057
Ikuta T, Eguchi H, Tachibana T, Yoneda Y, Kawajiri K (1998) Nuclear localization and export signals of the human aryl hydrocarbon receptor. J Biol Chem 273(5):2895–2904. https://doi.org/10.1074/jbc.273.5.2895
Iyer SS, Gensollen T, Gandhi A, Oh SF, Neves JF, Collin F, Lavin R, Serra C, Glickman J, De Silva PSA, Sartor RB, Besra G, Hauser R, Maxwell A, Llebaria A, Blumberg RS (2018) Dietary and microbial oxazoles induce intestinal inflammation by modulating aryl hydrocarbon receptor responses. Cell 173(5):1123-1134 e11. https://doi.org/10.1016/j.cell.2018.04.037
Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC (2008) The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105(39):15064–15069. https://doi.org/10.1073/pnas.0803124105
Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, Stappenbeck TS (2016) The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165(7):1708–1720. https://doi.org/10.1016/j.cell.2016.05.018
Kamdar K, Johnson AMF, Chac D, Myers K, Kulur V, Truevillian K, Depaolo RW (2018) Innate recognition of the microbiota by TLR1 promotes epithelial homeostasis and prevents chronic inflammation. J Immunol 201(1):230–242. https://doi.org/10.4049/jimmunol.1701216
Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH (2004) Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest 114(8):1098–1106. https://doi.org/10.1172/JCI21086
Kawasaki T, Kawai T (2014) Toll-like receptor signaling pathways. Front Immunol 5:461. https://doi.org/10.3389/fimmu.2014.00461
Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP (2015) Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17(5):662–671. https://doi.org/10.1016/j.chom.2015.03.005
Kim S-H, Jang Y-S (2020) Recent insights into cellular crosstalk in respiratory and gastrointestinal mucosal immune systems. Immune Netw 20(6):e44. https://doi.org/10.4110/in.2020.20.e44
Knudsen KA, Soler AP, Johnson KR, Wheelock MJ (1995) Interaction of alpha-actinin with the cadherin/catenin cell-cell adhesion complex via alpha-catenin. J Cell Biol 130(1):67–77. https://doi.org/10.1083/jcb.130.1.67
Kong S, Zhang YH, Zhang W (2018) Regulation of intestinal epithelial cells properties and functions by amino acids. Biomed Res Int 2018:2819154. https://doi.org/10.1155/2018/2819154
Kordjazy N, Haj-Mirzaian A, Haj-Mirzaian A, Rohani MM, Gelfand EW, Rezaei N, Abdolghaffari AH (2018) Role of toll-like receptors in inflammatory bowel disease. Pharmacol Res 129:204–215. https://doi.org/10.1016/j.phrs.2017.11.017
Lai Y, Gallo RL (2009) AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol 30(3):131–141. https://doi.org/10.1016/j.it.2008.12.003
Lanis JM, Alexeev EE, Curtis VF, Kitzenberg DA, Kao DJ, Battista KD, Gerich ME, Glover LE, Kominsky DJ, Colgan SP (2017) Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol 10(5):1133–1144. https://doi.org/10.1038/mi.2016.133
Laroui H, Ingersoll SA, Liu HC, Baker MT, Ayyadurai S, Charania MA, Laroui F, Yan Y, Sitaraman SV, Merlin D (2012) Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS One 7(3):e32084. https://doi.org/10.1371/journal.pone.0032084
Le Duc Q, Shi Q, Blonk I, Sonnenberg A, Wang N, Leckband D, De Rooij J (2010) Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol 189(7):1107–1115. https://doi.org/10.1083/jcb.201001149
Lee B, Moon KM, Kim CY (2018) Tight junction in the intestinal epithelium: its association with diseases and regulation by phytochemicals. J Immunol Res 2018:2645465. https://doi.org/10.1155/2018/26454652018/2645465
Lee WJ, Hase K (2014) Gut microbiota-generated metabolites in animal health and disease. Nat Chem Biol 10(6):416–424. https://doi.org/10.1038/nchembio.1535
Lee YS, Kim TY, Kim Y, Lee SH, Kim S, Kang SW, Yang JY, Baek IJ, Sung YH, Park YY, Hwang SW, Kim OE, Liu KS, Kamada S, Gao N, Kweon N (2018) Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe 24(6):833-846 e6. https://doi.org/10.1016/j.chom.2018.11.002
Linden SK, Sutton P, Karlsson NG, Korolik V, Mcguckin MA (2008) Mucins in the mucosal barrier to infection. Mucosal Immunol 1(3):183–197. https://doi.org/10.1038/mi.2008.5
Lopez J, Grinspan A (2016) Fecal microbiota transplantation for inflammatory bowel disease. Gastroenterol Hepatol (N Y) 12(6):374–379
Lu Z, Ding L, Lu Q, Chen YH (2013) Claudins in intestines: distribution and functional significance in health and diseases. Tissue Barriers 1(3):e24978. https://doi.org/10.4161/tisb.24978
Lueschow SR, Mcelroy SJ (2020) The paneth cell: the curator and defender of the immature small intestine. Front Immunol 11:587. https://doi.org/10.3389/fimmu.2020.00587
Ma J, Rubin BK, Voynow JA (2018) Mucins, mucus, and goblet cells. Chest 154(1):169–176. https://doi.org/10.1016/j.chest.2017.11.008
Mabbott NA, Donaldson DS, Ohno H, Williams IR, Mahajan A (2013) Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol 6(4):666–677. https://doi.org/10.1038/mi.2013.30
Macdonald TT (1992) Epithelial proliferation in response to gastrointestinal inflammation. Ann NY Acad Sci 664:202–209. https://doi.org/10.1111/j.1749-6632.1992.tb39761.x
Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, Ferrante M, Verhaegen J, Rutgeerts P, Vermeire S (2014) A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63(8):1275–1283. https://doi.org/10.1136/gutjnl-2013-304833
Madara JL (1982) Cup cells: structure and distribution of a unique class of epithelial cells in guinea pig, rabbit, and monkey small intestine. Gastroenterology 83(5):981–994. https://doi.org/10.1016/S0016-5085(82)80064-4
Marinelli L, Martin-Gallausiaux C, Bourhis JM, Beguet-Crespel F, Blottiere HM, Lapaque N (2019) Identification of the novel role of butyrate as AhR ligand in human intestinal epithelial cells. Sci Rep 9(1):643. https://doi.org/10.1038/s41598-018-37019-2
Marks E, Goggins BJ, Cardona J, Cole S, Minahan K, Mateer S, Walker MM, Shalwitz R, Keely S (2015) Oral delivery of prolyl hydroxylase inhibitor: AKB-4924 promotes localized mucosal healing in a mouse model of colitis. Inflamm Bowel Dis 21(2):267–275. https://doi.org/10.1097/MIB.0000000000000277
Martin-Gallausiaux C, Beguet-Crespel F, Marinelli L, Jamet A, Ledue F, Blottiere HM, Lapaque N (2018) Butyrate produced by gut commensal bacteria activates TGF-beta1 expression through the transcription factor SP1 in human intestinal epithelial cells. Sci Rep 8(1):9742. https://doi.org/10.1038/s41598-018-28048-y
Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, Siddiqui S, Basso AS, Otterbein LE, Pardoll DM, Pan F, Priel A, Clish CB, Robson SC, Quintana FJ (2015) Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med 21(6):638–646. https://doi.org/10.1038/nm.3868
Medema JP, Vermeulen L (2011) Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 474(7351):318–326. https://doi.org/10.1038/nature10212
Metidji A, Omenetti S, Crotta S, Li Y, Nye E, Ross E, Li V, Maradana MR, Schiering C, Stockinger B (2018) The environmental sensor AHR protects from inflammatory damage by maintaining intestinal stem cell homeostasis and barrier integrity. Immunity 49(2):353-362 e5. https://doi.org/10.1016/j.immuni.2018.07.010
Michielan A, D’inca R, (2015) Intestinal permeability in inflammatory bowel disease: pathogenesis, clinical evaluation, and therapy of leaky gut. Mediators Inflamm 2015:628157. https://doi.org/10.1155/2015/6281572015/628157
Middelhoff M, Westphalen CB, Hayakawa Y, Yan KS, Gershon MD, Wang TC, Quante M (2017) Dclk1-expressing tuft cells: critical modulators of the intestinal niche? Am J Physiol Gastrointest Liver Physiol 313(4):G285–G299. https://doi.org/10.1152/ajpgi.00073.2017
Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, Macdonald TT, Pallone F, Monteleone G (2011) Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology 141(1):237–248. https://doi.org/10.1053/j.gastro.2011.04.0072011.04.007
Mukherjee S, Karmakar S, Babu SP (2016) TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Braz J Infect Dis 20(2):193–204. https://doi.org/10.1016/j.bjid.2015.10.011
Mukherjee S, Zheng H, Derebe MG, Callenberg KM, Partch CL, Rollins D, Propheter DC, Rizo J, Grabe M, Jiang QX, Hooper LV (2014) Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature 505(7481):103–107. https://doi.org/10.1038/nature12729
Naganuma M, Sugimoto S, Mitsuyama K, Kobayashi T, Yoshimura N, Ohi H, Tanaka S, Andoh A, Ohmiya N, Saigusa K, Yamamoto T, Morohoshi Y, Ichikawa H, Matsuoka K, Hisamatsu T, Watanabe K, Mizuno S, Suda W, Hattori M, Fukuda S, Hirayama A, Abe T, Watanabe M, Hibi T, Suzuki Y, Kanai T, Group IS (2018) Efficacy of Indigo naturalis in a multicenter randomized controlled trial of patients with ulcerative colitis. Gastroenterology 154(4):935–947. https://doi.org/10.1053/j.gastro.2017.11.024
Neurath MF (2017) Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 14(5):269–278. https://doi.org/10.1038/nrgastro.2016.208
Nighot M, Al-Sadi R, Guo S, Rawat M, Nighot P, Watterson MD, Ma TY (2017) Lipopolysaccharide-induced increase in intestinal epithelial tight permeability is mediated by toll-like receptor 4/myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol 187(12):2698–2710. https://doi.org/10.1016/j.ajpath.2017.08.005
Parada Venegas D, De La Fuente MK, Landskron G, Gonzalez MJ, Quera R, Dijkstra G, Harmsen HJM, Faber KN, Hermoso MA (2019) Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol 10:277. https://doi.org/10.3389/fimmu.2019.00277
Peterson CY, Costantini TW, Loomis WH, Putnam JG, Wolf P, Bansal V, Eliceiri BP, Baird A, Coimbra R (2010) Toll-like receptor-4 mediates intestinal barrier breakdown after thermal injury. Surg Infect (Larchmt) 11(2):137–144. https://doi.org/10.1089/sur.2009.053
Porter EM, Bevins CL, Ghosh D, Ganz T (2002) The multifaceted Paneth cell. Cell Mol Life Sci 59(1):156–170. https://doi.org/10.1007/s00018-002-8412-z
Priyadarshini M, Kotlo KU, Dudeja PK, Layden BT (2018) Role of short chain fatty acid receptors in intestinal physiology and pathophysiology. Compr Physiol 8(3):1091–1115. https://doi.org/10.1002/cphy.c170050
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118(2):229–241. https://doi.org/10.1016/j.cell.2004.07.002
Ramanan D, Cadwell K (2016) Intrinsic defense mechanisms of the intestinal epithelium. Cell Host Microbe 19(4):434–441. https://doi.org/10.1016/j.chom.2016.03.0032016.03.003
Rankin EB, Giaccia AJ (2008) The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 15(4):678–685. https://doi.org/10.1038/cdd.2008.21
Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP (2008) Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology 134(1):145–155. https://doi.org/10.1053/j.gastro.2007.09.033
Rothhammer V, Quintana FJ (2019) The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 19(3):184–197. https://doi.org/10.1038/s41577-019-0125-8
Santos AJM, Lo YH, Mah AT, Kuo CJ (2018) The intestinal stem cell niche: homeostasis and adaptations. Trends Cell Biol 28(12):1062–1078. https://doi.org/10.1016/j.tcb.2018.08.001
Sasaki N, Sachs N, Wiebrands K, Ellenbroek SI, Fumagalli A, Lyubimova A, Begthel H, Van Den Born M, Van Es JH, Karthaus WR, Li VS, Lopez-Iglesias C, Peters PJ, Van Rheenen J, Van Oudenaarden A, Clevers H (2016) Reg4 + deep crypt secretory cells function as epithelial niche for Lgr5 + stem cells in colon. Proc Natl Acad Sci USA 113(37):E5399–E5407. https://doi.org/10.1073/pnas.1607327113
Sato T, Van Es JH, Snippert HJ, Stange DE, Vries RG, Van Den Born M, Barker N, Shroyer NF, Van De Wetering M, Clevers H (2011) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469(7330):415–418. https://doi.org/10.1038/nature09637
Sato T, Vries RG, Snippert HJ, Van De Wetering M, Barker N, Stange DE, Van Es JH, Abo A, Kujala P, Peters PJ, Clevers H (2009) Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459(7244):262–265. https://doi.org/10.1038/nature07935
Schiering C, Wincent E, Metidji A, Iseppon A, Li Y, Potocnik AJ, Omenetti S, Henderson CJ, Wolf CR, Nebert DW, Stockinger B (2017) Feedback control of AHR signalling regulates intestinal immunity. Nature 542(7640):242–245. https://doi.org/10.1038/nature21080
Serra D, Mayr U, Boni A, Lukonin I, Rempfler M, Challet Meylan L, Stadler MB, Strnad P, Papasaikas P, Vischi D, Waldt A, Roma G, Liberali P (2019) Self-organization and symmetry breaking in intestinal organoid development. Nature 569(7754):66–72. https://doi.org/10.1038/s41586-019-1146-y
Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, Gonzalez FJ (2008) Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology 134(7):2036–2048. https://doi.org/10.1053/j.gastro.2008.03.0092008.03.009
Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, He B, Cassis L, Bigas A, Cols M, Comerma L, Huang B, Blander JM, Xiong H, Mayer L, Berin C, Augenlicht LH, Velcich A, Cerutti A (2013) Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 342(6157):447–453. https://doi.org/10.1126/science.1237910
Shimotoyodome A, Meguro S, Hase T, Tokimitsu I, Sakata T (2000) Short chain fatty acids but not lactate or succinate stimulate mucus release in the rat colon. Comp Biochem Physiol A Mol Integr Physiol 125(4):525–531. https://doi.org/10.1016/s1095-6433(00)00183-5
Siegert I, Schodel J, Nairz M, Schatz V, Dettmer K, Dick C, Kalucka J, Franke K, Ehrenschwender M, Schley G, Beneke A, Sutter J, Moll M, Hellerbrand C, Wielockx B, Katschinski DM, Lang R, Galy B, Hentze MW, Koivunen P, Oefner PJ, Bogdan C, Weiss G, Willam C, Jantsch J (2015) Ferritin-mediated iron sequestration stabilizes hypoxia-inducible factor-1alpha upon LPS activation in the presence of ample oxygen. Cell Rep 13(10):2048–2055. https://doi.org/10.1016/j.celrep.2015.11.005
Singh R, Chandrashekharappa S, Bodduluri SR, Baby BV, Hegde B, Kotla NG, Hiwale AA, Saiyed T, Patel P, Vijay-Kumar M, Langille MGI, Douglas GM, Cheng X, Rouchka EC, Waigel SJ, Dryden GW, Alatassi H, Zhang HG, Haribabu B, Vemula PK, Jala VR (2019) Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun 10(1):89. https://doi.org/10.1038/s41467-018-07859-7
Sivaprakasam S, Bhutia YD, Yang S, Ganapathy V (2017) Short-chain fatty acid transporters: role in colonic homeostasis. Compr Physiol 8(1):299–314. https://doi.org/10.1002/cphy.c170014
Spit M, Koo BK, Maurice MM (2018) Tales from the crypt: intestinal niche signals in tissue renewal, plasticity and cancer. Open Biol. https://doi.org/10.1098/rsob.180120
Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, Geboes K, Ceuppens JL, Rutgeerts P (2002) Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol 97(8):2000–2004. https://doi.org/10.1111/j.1572-0241.2002.05914.x
Tachibana K, Nakanishi H, Mandai K, Ozaki K, Ikeda W, Yamamoto Y, Nagafuchi A, Tsukita S, Takai Y (2000) Two cell adhesion molecules, nectin and cadherin, interact through their cytoplasmic domain-associated proteins. J Cell Biol 150(5):1161–1176. https://doi.org/10.1083/jcb.150.5.1161
Takahashi T, Shiraishi A (2020) Stem cell signaling pathways in the small intestine. Int J Mol Sci. https://doi.org/10.3390/ijms21062032
Takai Y, Nakanishi H (2003) Nectin and afadin: novel organizers of intercellular junctions. J Cell Sci 116(Pt 1):17–27. https://doi.org/10.1242/jcs.00167
Takeichi M (2014) Dynamic contacts: rearranging adherens junctions to drive epithelial remodelling. Nat Rev Mol Cell Biol 15(6):397–410. https://doi.org/10.1038/nrm3802
Tambuwala MM, Cummins EP, Lenihan CR, Kiss J, Stauch M, Scholz CC, Fraisl P, Lasitschka F, Mollenhauer M, Saunders SP, Maxwell PH, Carmeliet P, Fallon PG, Schneider M, Taylor CT (2010) Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 139(6):2093–2101. https://doi.org/10.1053/j.gastro.2010.06.068
Taylor CT, Dzus AL, Colgan SP (1998) Autocrine regulation of epithelial permeability by hypoxia: role for polarized release of tumor necrosis factor alpha. Gastroenterology 114(4):657–668. https://doi.org/10.1016/s0016-5085(98)70579-7
Tomic G, Morrissey E, Kozar S, Ben-Moshe S, Hoyle A, Azzarelli R, Kemp R, Chilamakuri CSR, Itzkovitz S, Philpott A, Winton DJ (2018) Phospho-regulation of ATOH1 is required for plasticity of secretory progenitors and tissue regeneration. Cell Stem Cell 23(3):436-443 e7. https://doi.org/10.1016/j.stem.2018.07.002
Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV (2008) Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA 105(52):20858–20863. https://doi.org/10.1073/pnas.0808723105
Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, Ley R, Wakeland EK, Hooper LV (2011) The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334(6053):255–258. https://doi.org/10.1126/science.1209791
Van Es JH, Sato T, Van De Wetering M, Lyubimova A, Yee Nee AN, Gregorieff A, Sasaki N, Zeinstra L, Van Den Born M, Korving J, Martens ACM, Barker N, Van Oudenaarden A, Clevers H (2012) Dll1 + secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 14(10):1099–1104. https://doi.org/10.1038/ncb2581
Van Itallie CM, Anderson JM (2006) Claudins and epithelial paracellular transport. Annu Rev Physiol 68:403–429. https://doi.org/10.1146/annurev.physiol.68.040104.131404
Van Itallie CM, Anderson JM (2014) Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol 36:157–165. https://doi.org/10.1016/j.semcdb.2014.08.011
Vancamelbeke M, Vermeire S (2017) The intestinal barrier: a fundamental role in health and disease. Expert Rev Gastroenterol Hepatol 11(9):821–834. https://doi.org/10.1080/17474124.2017.1343143
Vinolo MA, Rodrigues HG, Hatanaka E, Hebeda CB, Farsky SH, Curi R (2009) Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci (Lond) 117(9):331–338. https://doi.org/10.1042/CS20080642
Volk N, Lacy B (2017) Anatomy and physiology of the small bowel. Gastrointest Endosc Clin N Am 27(1):1–13. https://doi.org/10.1016/j.giec.2016.08.001
Xiao T, Wu S, Yan C, Zhao C, Jin H, Yan N, Xu J, Wu Y, Li C, Shao Q, Xia S (2018) Butyrate upregulates the TLR4 expression and the phosphorylation of MAPKs and NK-kappaB in colon cancer cell in vitro. Oncol Lett 16(4):4439–4447. https://doi.org/10.3892/ol.2018.9201
Xue X, Ramakrishnan S, Anderson E, Taylor M, Zimmermann EM, Spence JR, Huang S, Greenson JK, Shah YM (2013) Endothelial PAS domain protein 1 activates the inflammatory response in the intestinal epithelium to promote colitis in mice. Gastroenterology 145(4):831–841. https://doi.org/10.1053/j.gastro.2013.07.010
Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY (2001) Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 294(5549):2155–2158. https://doi.org/10.1126/science.1065718
Yu M, Wang Q, Ma Y, Li L, Yu K, Zhang Z, Chen G, Li X, Xiao W, Xu P, Yang H (2018) Aryl hydrocarbon receptor activation modulates intestinal epithelial barrier function by maintaining tight junction integrity. Int J Biol Sci 14(1):69–77. https://doi.org/10.7150/ijbs.22259
Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’angelo C, Massi-Benedetti C, Fallarino F, Carvalho A, Puccetti P, Romani L (2013) Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39(2):372–385. https://doi.org/10.1016/j.immuni.2013.08.003
Zhang YZ, Li YY (2014) Inflammatory bowel disease: pathogenesis. World J Gastroenterol 20(1):91–99. https://doi.org/10.3748/wjg.v20.i1.91
Zhao Y, Chen F, Wu W, Sun M, Bilotta AJ, Yao S, Xiao Y, Huang X, Eaves-Pyles TD, Golovko G, Fofanov Y, D’souza W, Zhao Q, Liu Z, Cong Y (2018) GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol 11(3):752–762. https://doi.org/10.1038/mi.2017.118
Zhao Y, Ma T, Chen W, Chen Y, Li M, Ren L, Chen J, Cao R, Feng Y, Zhang H, Shi R (2016) MicroRNA-124 promotes intestinal inflammation by targeting aryl hydrocarbon receptor in Crohn’s disease. J Crohns Colitis 10(6):703–712. https://doi.org/10.1093/ecco-jcc/jjw010
Zhu K, Meng Q, Zhang Z, Yi T, He Y, Zheng J, Lei W (2019) Aryl hydrocarbon receptor pathway: role, regulation and intervention in atherosclerosis therapy (Review). Mol Med Rep 20(6):4763–4773. https://doi.org/10.3892/mmr.2019.10748
Zihni C, Mills C, Matter K, Balda MS (2016) Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 17(9):564–580. https://doi.org/10.1038/nrm.2016.80
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT, and Future Planning (Grant Nos. NRF-2019R1I1A1A01057559, NRF-2020R1A2B5B01001690).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare they have no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Seo, K., Seo, J., Yeun, J. et al. The role of mucosal barriers in human gut health. Arch. Pharm. Res. 44, 325–341 (2021). https://doi.org/10.1007/s12272-021-01327-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-021-01327-5