
Abstract
JAKs are a family of intracellular tyrosine kinases consisting of four members, JAK1, JAK2, JAK3, and TYK2. They are key components of the JAK-STAT pathway that transmit signals of many cytokines involved in the pathogenesis of numerous immune-mediated diseases and have been major molecular targets in developing new drugs for the treatment of such diseases. Some small-molecule inhibitors of JAKs have been approved by the FDA for rheumatoid arthritis, psoriatic arthritis, and inflammatory bowel disease. Now, newer JAK inhibitors with isoform-selectivity among the four different JAKs are being developed, with the aim of improving clinical outcomes compared with earlier developed drugs with pan-JAK inhibition. Most of these selective inhibitors target the kinase domains of JAKs, functioning through the traditional inhibition mode of kinases; but recently those that target their pseudokinase domains, allosterically inhibiting the enzymes, have been under development. In this review, key characteristics, efficacy, and safety of FDA-approved and representative drugs in late stages of development are briefly described in order to provide clinical implications with respect to JAK inhibitor selectivity and future development perspectives. The recent development of pseudokinase-targeted inhibitors of JAKs is also included.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cytokines play crucial roles in the regulation of immune responses and are also key drivers in the pathogenesis of immune-mediated diseases. Targeting them has been a major therapeutic approach for the treatment of such diseases (Liu et al. 2013; Jamilloux et al. 2019). Cytokines are categorized according to the type of their binding receptors. Many of them involved in the pathogenesis of immune-mediated diseases bind to type I/II cytokine receptors. These types of receptors use the same mechanism of signal transduction, called the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway (O’Shea et al. 2013b). Binding of cytokines to their receptors leads to activation of intracellular receptor-associated JAKs followed by phosphorylation of multiple substrates including STATs. Phosphorylated STATs dimerize and translocate into the nucleus where they undertake DNA binding and activate transcription of new cytokines and other target genes (Kontzias et al. 2012; Liongue et al. 2016). Because of the major roles of the JAK-STAT pathway in mediating the effect of cytokines in immunoregulation and immunopathology, supported by knockout animal model studies and successful genome-wide association studies for a variety of immune-mediated diseases, intracellular key signaling molecules have been targeted for developing new therapeutics (Plenge 2010; O’Shea et al. 2012; O’Shea et al. 2013a). Until now, three JAK inhibitors, tofacitinib, baricitinib, and upadacitinib, have been approved by the FDA for immune-mediated diseases including rheumatoid arthritis (RA), psoriatic arthritis (PsA), and inflammatory bowel disease (IBD), and they are being tested in clinical trials for other diseases (Fragoulis et al. 2019; Taylor 2019; FDA 2020).
Prior to the emergence of JAK inhibitors, the development of monoclonal antibodies and recombinant proteins that target either some cytokines or their receptors had revolutionized the treatment of such diseases, but not all patients responded to those biological drugs (Gadina 2013). While targeting either a particular extracellular cytokine or its receptor may not give sufficient clinical responses, targeting intracellular JAKs can simultaneously control the signals and actions of multiple JAK-dependent cytokines (Banerjee et al. 2017). Drugs developed earlier are pan-JAK inhibitors, also called non-selective inhibitors, that do not block one particular member of the four different isomers JAK1, JAK2, JAK3, and TYK2 within the JAK family. Rather they block them in an unselective way, which results in the interruption of downstream signals and actions by a wide spectrum of cytokines and causes adverse effects that are mostly predictable based on the simultaneous blocking of multiple cytokines (Hirahara et al. 2016; Gadina et al. 2018). Newer inhibitors of JAKs showing specificity for one particular JAK member have been developed as selective JAK inhibitors with the aim of mitigating the safety concerns of non-selective inhibitors by interrupting signaling of a narrower range of cytokines (Virtanen et al. 2019). Approved JAK inhibitors and most under development, including non-selective and selective inhibitors, target the kinase domains of JAKs, competing with ATP at the active sites of those domains (Banerjee et al. 2017; Choy 2019). In addition to the kinase domain, which is located in the Janus homology 1 (JH1) region of the structure, the JAK family also has a distinctive catalytically defective pseudokinase domain of the JH2 region (Babon et al. 2014). The development of highly selective JAK inhibitors is not easy because of the high sequence homology of their kinase domains. Currently, isoform-selective inhibitors that allosterically bind the pseudokinase domains located outside the kinase domains of JAKs are being developed for multiple immune-mediated diseases. Although one has progressed to late clinical development, it remains to be seen whether this clinical candidate will be the first successful case in the development of pseudokinase-targeted therapeutics (Kung and Jura 2019; Lu et al. 2020). This review summarizes the basic characteristics, efficacy, and safety of kinase-targeted JAK inhibitors that bind to the kinase domain, including non-selective and selective inhibitors that are FDA-approved or under late clinical development for the treatment of immune-mediated diseases and then introduces the recent development of pseudokinase-targeted JAK inhibitors that bind to the pseudokinase domain (Fig. 1). A comparison of clinical outcomes and risks between non-selective and selective JAK inhibitors is also included because the clinical implications of isoform-selective JAK inhibition versus non-selective pan-JAK inhibition have not yet been elucidated.

Structures of JAK inhibitors
Development of drugs targeting the JH1 kinase domain of JAKs
Type I/II cytokine receptors are employed by immunoregulatory cytokines including interleukins (ILs), interferons (IFNs), colony stimulating factors (CSFs), and erythropoietin (EPO); and their intracellular domains selectively associate with the four different JAKs in certain combinations of pairs depending on the cytokine (O’Shea et al. 2013a). In terms of the dependence of different cytokines on the four JAKs, JAK3 has the most limited function, exclusively associating with the common γc family cytokines that use JAK1. JAK1 is also used by other cytokines including the gp130 family, interferons, and the IL-10 family. JAK2 is important for signaling by the βc family, CSF and EPO. TYK2 contributes to the IL-12 family and IFNα/β. JAK1 and JAK2 have broad functions (O’Shea et al. 2013c). Thus, it is predictable that drugs with pan-JAK inhibition can cause adverse effects. Among the type I/II cytokines, IL-6, IL-12, IL-23, INFs and GM-CSF have pathogenic roles in RA, psoriasis, IBD, and other immune-mediated diseases and JAK inhibitors blocking such cytokines have proved their clinical outcomes in treating such diseases (O’Shea et al. 2013b). The first approved drug, tofacitinib, inhibits JAK1, JAK2, and JAK3. Baricitinib is the second approved drug, affecting JAK1 and JAK2. These two drugs are classified as non-selective inhibitors. With the use of this class of drugs, there have been safety concerns such as infection, malignancy, cardiovascular events, thrombosis, and laboratory abnormalities including anemia, neutropenia, and elevated lipids (Schwartz et al. 2017; Gadina et al. 2019). Recently approved upadacitinib was developed to have high JAK1 selectivity so that other JAK-related adverse effects could be minimized (Bechman et al. 2019). With continuous efforts to reduce non-selective pan-JAK inhibition, various isoform-selective inhibitors with high specificity for JAK1 or JAK3 are being developed (Virtanen et al. 2019). The approved JAK inhibitor drugs and majority currently under late clinical development block the JH1 kinase domains of JAKs that are responsible for phosphorylation, followed by interruption of subsequent signal transductions (O’Shea et al. 2015) (Fig. 2). Among these kinase-targeted inhibitors of JAKs, approved drugs and representative drugs under late clinical development stages for immune-mediated diseases listed in Table 1 are described as follows.

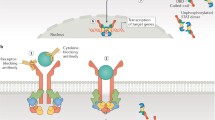
JAK-STAT pathway and mechanism of action of kinase-targeted and pseudokinase-targeted inhibitors of JAKs. Binding of type I/II cytokine to the receptor induces activation and phosphorylation of JAK, which leads to phosphorylation and dimerization of STAT. STAT dimer translocates into the nucleus to regulate the transcription of target genes. Kinase-targeted inhibitors directly inhibit kinase activity by binding to the kinase domain (JH1) of JAKs, thereby preventing phosphorylation of STAT and subsequent STAT-dependent transcription. Pseudokinase-targeted inhibitors allosterically inhibit the kinase domain (JH1) through binding to the pseudokinase domain (JH2) of JAKs, resulting in interruption of downstream signaling transduction and STAT-dependent transcription. The kinase-targeted and pseudokinase-targeted inhibitors of JAKs included herein inhibit one or more isoforms among JAK1, JAK2, JAK3, and TYK2. JH1 Janus homology 1, JH2 Janus homology 2, FERM four-point-one, ezrin, radixin, and moesin, SH2 Src homology 2
Tofacitinib
Tofacitinib has a pyrrolo[2,3-d]pyrimidine scaffold. It is an oral JAK inhibitor with preferential inhibition for JAK1 and JAK3 over JAK2 and TYK2 and acts through binding to the ATP binding site in the JH1 kinase domain (Hodge et al. 2016; Roskoski Jr 2016). The first approval of this drug was for the treatment of RA, where it is indicated for patients with inadequate responses to disease-modifying antirheumatic drugs (DMARDs) based on improvement with tofacitinib in clinical response and physical functioning status compared with placebo as measured by the American College of Rheumatology 20% (ACR20) and Health Assessment Questionnaire-Disability Index (HAQ-DI), respectively, in a series of phase 3 clinical trials. These multiple trials revealed the efficacy and safety of tofacitinib as monotherapy and in combination with other DMARDs in the treatment of RA of patients inadequately responsive to DMARDs. The drug at the dosing regimen approved by the FDA showed greater ACR20 response rates ranging from 42–60% compared with those of placebo of 24–31% (Fleischmann et al. 2012; van Vollenhoven et al. 2012; Burmester et al. 2013; Kremer et al. 2013; van der Heijde et al. 2013). Its effectiveness in methotrexate (MTX)-naïve patients was superior to that of MTX in the ACR70 response and radiographic progression (Lee et al. 2014). The most common adverse events (AEs) of tofacitinib were infections including nasopharyngitis, upper respiratory tract infection, and urinary tract infection. Increased risk of herpes zoster was reported with this drug and its occurrence was more frequent than with bDMARDs (Fragoulis et al. 2018; Kivitz et al. 2018; Harigai 2019). After its initial approval in 2012 for RA, tofacitinib was also approved in 2017 for the treatment of PsA. It is indicated for patients of PsA with poor responses or intolerance to prior DMARDs. Tofacitinib has been recommended for use in combination with MTX or other conventional DMARDs (cDMARDs) for the treatment of PsA because it was tested as a combination therapy in two pivotal trials (Ly et al. 2019). Both pivotal studies met their two primary endpoints including improvements in ACR20 and HAQ-DI score compared with placebo. The drug at the recommended dose achieved ACR20 responses in 50% of patients from both studies, compared with 24–33% with placebo. Its safety profile in PsA patients was analogous to that in RA patients (Gladman et al. 2017; Mease et al. 2017). In 2018, another additional approval of tofacitinib was obtained in ulcerative colitis (UC), where it is indicated for patients active even after previous conventional therapy or treatment with TNF-α antagonists by meeting respective primary endpoints in three pivotal phase 3 trials, two for induction therapy and one for maintenance therapy. Greater remission rates both in the induction trials at 8 weeks and in the maintenance trial at 52 weeks were shown for tofacitinib compared with placebo. Remission was defined as a total Mayo score of 2 points or lower with no subscore exceeding 1 and a rectal bleeding subscore of 0. The drug at the recommended dose achieved 18.5% and 16.6% of remission rates from the two induction trials compared with 8.2% and 3.6% with placebo. Also the remission rate in the maintenance trial was 40.6% with 11.1% for placebo. Across all three trials, the rates of overall infection and herpes zoster infection were higher with tofacitinib than with placebo (Sandborn et al. 2017; Varyani et al. 2019). Besides, clinical developments of tofacitinib in other immune-mediated diseases including ankylosing spondylitis (AS) and juvenile idiopathic arthritis (JIA) are underway.
Baricitinib
Baricitinib is a pyrrolo[2,3-d]pyrimidine derivative like tofacitinib and a potent inhibitor of JAK1 and JAK2 with 100-fold affinity for JAK1 and JAK2 over JAK3 and some activity for TYK2. It was designed to attenuate possible immunosuppressive effects from the inhibition of JAK3 using previously established structure-activity relationship work. Baricitinib inhibits the enzyme by binding to the ATP binding site of the JH1 kinase domain (Fridman et al. 2010; Choy et al. 2019; Xin et al. 2020). The standard efficacy outcomes of baricitinib including ACR20 and HAQ-DI score from multiple phase 3 clinical trials in RA led to FDA approval in 2018 for patients with inadequate responses to one or more TNF-α antagonist therapies. Use as monotherapy or in combination with MTX or other non-biologic DMARDs was approved. The ACR20 response rates of baricitinib at the approved dose by the FDA were 49% and 66% in patients with inadequate responses or intolerance to either TNF-α antagonists or cDMARDs, respectively, compared with 27% and 39% in the respective placebo groups. With background MTX, baricitinib demonstrated its superiority over adalimumab and placebo in patients with inadequate responses to MTX. This drug alone or with MTX was also shown to be superior to MTX monotherapy as initial therapy. Infections such as herpes zoster were mostly more frequent with baricitinib than with control groups. There were decreases in neutrophil counts and increases in LDL levels. Incidence rates of major adverse cardiovascular events (MACE) with baricitinib were low and similar to those with placebo and adalimumab (Genovese et al. 2016; Dougados et al. 2017; Fleischmann et al. 2017; Taylor et al. 2017). Pooled data including the pivotal phase 3 results showed that venous thromboembolism (VTE) including deep vein thrombosis (DVT) and pulmonary embolism (PE) occurred with the higher dose of baricitinib with no cases in placebo (Taylor et al. 2019). There are several ongoing clinical studies in multiple immune diseases including systemic lupus erythematosus (SLE) and alopecia areata (AA).
Upadacitinib
Upadacitinib, unlike tofacitinib and baricitinib, is a JAK1 selective inhibitor with a tricyclic imidazopyrrolopyrazine scaffold. This drug was designed to have increased selectivity for JAK1 and is the first FDA-approved selective JAK inhibitor for the treatment of RA. Its selectivity towards JAK1 is 74-fold and 58-fold higher than that towards JAK2 and JAK3, respectively, and comes from an ability to bind JAK1 at two separate sites that are outside and on the ATP binding site of the JH1 kinase domain (Graff et al. 2014; Bechman et al. 2019). Amino acid sequence identity across the JAK family members is very high within the ATP binding sites, which are the targets for non-selective JAK inhibitors, and the design strategy focus was on the structurally different regions in the active site. Specifically, the canonical glycine-rich loops in JAK1 and JAK2 assume different conformations because of their amino acid sequence differences and the trifluoroethyl group in the structure of upadacitinib was introduced for optimal occupation of the van der Waals interaction space in the loop, providing an induced fit into JAK1 (Parmentier et al. 2018; Xu et al. 2020). Based on data from five pivotal phase three trials, upadacitinib received marketing approval by the FDA in 2019 and is indicated for patients of RA with inadequate responses or intolerance to one or more DMARDs, either as monotherapy or in combination with MTX. This drug met all primary endpoints such as ACR20 and ACR50 in the pivotal trials. Upadacitinib was compared with MTX as monotherapy in patients of RA who were naïve or inadequate responders to MTX (Smolen et al. 2019; van Vollenhoven et al. 2020). It was also compared with placebo as add-on therapy after an insufficient response with a cDMARD (Burmester et al. 2018). When MTX obtained an inadequate response, comparisons of upadacitinib with either adalimumab or placebo on a stable background of MTX were carried out (Fleischmann et al. 2019). Its combination therapy with a cDMARD was compared with placebo in patients with failure of a biologic DMARD (bDMARD) (Genovese et al. 2018). Compared with placebo, the drug proved to be superior across multiple RA patient populations. Collectively, upadacitinib at the approved dose by the FDA achieved ACR20 improvement responses with 64–68% of patients as monotherapy or in combination with a cDMARD. When combined with MTX, it showed superiority over adalimumab in improving ACR20 response when combined with MTX. The most common AEs were infections such as upper respiratory tract infection, nasopharyngitis, and urinary tract infection, and incidence rates were higher than for placebo. The overall safety profile of upadacitinib was similar to that of adalimumab and other JAK inhibitors, but upadacitinib showed higher incidence rates of herpes zoster compared with adalimumab and MTX. The rates of VTE, MACE, and malignancy were comparable with those for adalimumab and MTX (Cohen et al. 2019). Clinical developments for other therapeutic indications including UC, Crohn’s disease (CD) and atomic dermatitis (AD) are ongoing.
Filgotinib
Filgotinib is another JAK1 selective inhibitor that owns a triazolopyridine scaffold, showing a near 30-fold selectivity for JAK1 over JAK2 dependent signaling in whole blood assays. It was discovered after the identification and subsequent optimization process of triazolopyridine-based compounds as selective inhibitors against the kinase domain of JAK1 through kinase-focused screening (Van Rompaey et al. 2013; Menet et al. 2014). There are a series of completed or ongoing phase 3 trials for RA. In one of the phase 3 trials, filgotinib was found to be more effective than placebo as measured by the ACR20 response rate in patients of RA with inadequate responses or intolerance to one or more bDMARDs. Frequencies of anemia were reported to be similar between filgotinib and placebo, and there were no clinically relevant changes in lymphocytes and neutrophils (Genovese et al. 2019). Increases in hemoglobins were also observed with filgotinib in phase 2 studies, which can be attributed to the lack of JAK2 inhibition that is linked to typical hematological adverse effects (Kavanaugh et al. 2017; Wethovens et al. 2017). Positive primary outcome data from two phase 3 trials of filgotinib for other patient populations such as MTX-inadequate responders and MTX-naïve patients have been reported recently. In both studies, filgotinib met the primary endpoints as measured by the ACR20 response rates in 77–81% of patients at the higher dose. It was superior to placebo on background MTX in the MTX-inadequate responders, and its combination therapy with MTX was found to be more effective in MTX-naïve patients than MTX monotherapy (Combe et al. 2019; Westhovens et al. 2019). The pooled safety data across the above phase 3 trials showed that the frequency of AEs with filgotinib was similar to that in control groups. The most common AEs were infections including upper respiratory tract infection and nasopharyngitis. Incidence rates of herpes zoster, MACE, and VTE including DVT and PE were low and similar among groups (Winthrop et al. 2019). Filgotinib is also being investigated for IBD, which includes UC and CD.
Decernotinib
Decernotinib is a JAK3 selective inhibitor with a pyrrolo[2,3-b]pyridine scaffold. It was discovered based on the identification of such scaffold using inhibitory activity screening of a compound library against the kinase domain of JAK3 and showed 5-fold selectivity towards JAK3 compared with other JAKs based on in vitro enzyme assays (Farmer et al. 2015; Mahajan et al. 2015). In principle, selective JAK3 inhibitors block signaling of the common γc cytokines, which is dependent on both JAK1 and JAK3, and have the potential benefit of avoiding unwanted adverse effects by sparing JAK1 and JAK2 functions (Pei et al. 2018). In two phase 2 dose-escalation studies, decernotinib demonstrated efficacy both as monotherapy and in combination with MTX in patients of RA with inadequate responses to MTX by meeting the primary endpoints including ACR20 response rates. Decernotinib showed greater ACR20 response rates than placebo in both monotherapy and combination therapy studies, with roughly 65% at the higher doses and 46.5–68.1% at all doses. AEs reported from both studies were similar to pan-JAK inhibitors (Fleischmann et al. 2015; Genovese et al. 2016). Its ACR20 responses appeared to be comparable to those of pan-JAK inhibitors such as tofacitnib and baricitinib, which were 50–70% in their phase 3 studies with similar patient populations. However, development of neutropenia indicated the possibility of off-target effects by this drug (Gadina et al. 2016). No further clinical data has been available and further development in RA seems to have been discontinued (Westhovens 2019).
Ritlecitinib
Ritlecitinib, also known as PF-06651600, is a JAK3 selective inhibitor having a pyrrolo[2,3-b]pyrimidine scaffold like tofacitinib. It inhibits the kinase activity of JAK3 at an IC50 of 33.1 nM with no effects on other JAKs, which have IC50s higher than 10,000 nM. Retlecitinib was discovered by the optimization of tofacitinib in order to achieve JAK3 isoform selectivity through its covalent interaction with residue Cys909, which is unique in the ATP binding site of JAK3 (Telliez et al. 2016; Xu et al. 2020). In the proof of concept phase 2a trial for RA, the drug revealed significant remission as measured by the simple disease activity index (SDAI), where patients responded inadequately to MTX and up to 50% could have received prior TNF-α inhibitor treatment. Most AEs were mild with no treatment-related serious or severe AEs (Robinson et al. 2020). With regard to alopecia areata (AA), it showed positive results in improving hair growth in an early clinical study, on which phase 2b/3 trials are currently ongoing (Peeva 2019). Upon results from clinical trials of this drug, the clinical potential of highly selective JAK3 inhibition for immune-mediated diseases will emerge.
Development of drugs targeting the JH2 pseudokinase domain of JAKs
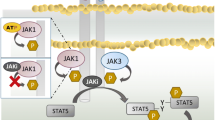
Allosteric kinase inhibitors are molecules binding to an allosteric site outside the ATP-binding sites of kinases, which are highly conserved. Owing to the high homology of the ATP-binding sites, they will have some advantages over ATP-competitive kinase inhibitors in achieving greater selectivity and resultant lower off-target adverse effects. Given the high sequence homology within the respective JH1 kinase domains of JAKs, the JH2 pseudokinase domain can be an ideal allosteric site for designing JAK inhibitors with high selectivity. The advantage of targeting the pseudokinase domains is an increased possibility of identifying inhibitors that are highly selective with decreased side effects (Moslin et al. 2017; Lu et al. 2020). The JH2 pseudokinase domains in JAKs mediate their critical regulatory functions through autoinhibitory interaction with the kinase domains, harboring mutations underlying immune diseases and cancers, which renders them as potential novel drug targets. For such pseudokinase-mutated diseases, targeting the pseudokinase domains could provide mutant-specific inhibitors of JAKs (Byrne et al. 2017; Gadina et al. 2019; Kung and Jura 2019). As pseudokinase-targeted inhibitors, small molecule ligands were identified that bind to and stabilize the JH2 pseudokinase domain of TYK, stabilizing an autoinhibitory interaction with the kinase domain and blocking receptor-mediated kinase activation and downstream signaling (Tokarski et al. 2015) (Fig. 2). One of such pseudokinase-targeted inhibitors of JAKs has been progressed to late clinical developments (Lu et al. 2020) (Table 1).
Deucravacitinib
Deucravacitinib, also known as BMS-986,165, is an allosteric and selective inhibitor of TYK2. Unlike other JAK inhibitors, it allosterically and selectively binds to the JH2 pseudokinase domain of TYK2, giving rise to JAK isoform and kinome selectivity (Catlett et al. 2017). Deucravacitinib was discovered by a systematic structure-based optimization focusing on the unique alanine pocket formed by a rare Ala 671 of the JH2 pseudokinase domain of TYK2 (Chang et al. 2019). By locking the TYK2 pseudokinase domain in a conformation that stabilizes an autoinhibitory interaction with the kinase domain, the drug prevents receptor-mediated activation of TYK2, resulting in the blockade of downstream signaling and STAT-dependent transcription (Fig. 2). Deucravacitinib is a highly selective TYK2 pseudokinase inhibitor with an IC50 of 0.2 nM, representing a 1000-fold selectivity for the TYK2 peudokinase domain in terms of IC50 values among a panel of 249 protein and lipid kinases and pseudokinases as it was bound to only two other kinases or pseudokinases with IC50 values under 200 nM (Burke et al. 2019). This first pseudokinase-targeted clinical candidate demonstrated efficacy compared with placebo in a phase 2 study in patients with psoriasis. The ratio of patients achieving Psoriasis Area and Severity Index (PASI) 75 was 68.9% and 75.0% with the more suitable dosing regimens and 7% with placebo (Papp et al. 2018). Results of an ongoing phase 3 clinical trial comparing this drug with placebo and apremilast as active comparator are awaited (Havnaer et al. 2019). Other clinical trials for SLE, UC and CD are in progress.
Comparison of efficacy and safety between non-selective JAK inhibitors and selective JAK inhibitors
In biochemical and enzyme assays, these JAK inhibitor drugs showed differences in JAK isoform-selectivity, even though target selectivity is not absolute and may be affected by the substrate and drug concentration studied. Tofacitinib is a non-selective inhibitor affecting JAK3, JAK1, and JAK2 while another non-selective inhibitor baricitinib affects JAK1 and JAK2. Upadacitinib and filgotinib are JAK1 selective inhibitors. These drugs all inhibit JAK1. JAK inhibitors, both non-selective and selective, have demonstrated their efficacy in terms of main clinical outcome measures in multiple immune-mediated diseases. RA is such a disease that has been intensively investigated with JAK inhibitors in the most diverse patient populations. Upon a review of phase 3 clinical studies for RA of the four different JAK inhibitors, tofacitinib, baricitinib, upadacitinib, and filgotinib, it is noted that they have been investigated in similar patient populations, which embrace mostly those who are naïve to treatment and those who have shown inadequate responses with prior treatments including MTX, cDMARDs, or bDMARDs (Table 2). Direct comparisons cannot be made and head-to-head clinical trials of JAK inhibitors are needed to obtain further and more accurate information; however, given this lack of head-to-head direct comparisons among JAK inhibitors, indirect comparisons of clinical outcomes with different drugs among similar patient populations will provide clinical implications with respect to JAK isoform selectivity as well as knowledge necessary to treat RA patients with JAK inhibitors. Similar treatment-refractory patient populations have been examined in different phase 3 clinical trials with the four drugs. First, in TNF-α inhibitor or bDMARDs-inadequate responders, tofacitinib, baricitinib, upadacitinib, and filgotinib at the FDA-approved and best doses showed ACR20 response rates of 42%, 49%, 65%, and 66% in ORAL Step, RA BEACON, SELECT-BEYOND, and FINCH-2 trials, respectively (Burmester et al. 2013; Genovese et al. 2016; Genovese et al. 2018; Genovese et al. 2019). The primary endpoints were met in all four studies with the two selective inhibitors showing numerically higher remissions than the non-selective ones. Second, in patients with inadequate responses to cDMARDs, the ACR20 response rates of tofacitinib, baricitinib, and upadacitinib were 52%, 66%, and 64% from ORAL Sync, RA BUILD, and SELECT-NEXT trials, respectively. Although there is no telling difference between the non-selective and selective inhibitors, relatively better remissions are shown with the more JAK1-focused baricitinib and mostly JAK1-targeted upadacitinib (Kremer et al. 2013; Burmester et al. 2017; Dougados et al. 2017). Third, in ORAL Standard, RA BEAM, SELECT-COMPARE, and FINCH-1 trials whose patients have been refractory to MTX, the four drugs on background MTX all met primary endpoints with ACR20 response rates of 52%, 70%, 71%, and 77%, respectively, with tofacitinib being numerically lower than the other three drugs (van Vollenhoven et al. 2012; Taylor et al. 2017; Comber et al. 2019; Fleischmann et al. 2019). The RA BEAM, SELECT-COMPARE, and FINCH-1 studies also compared baricitinib, upadacitinib, and filgotinib with adalimumab and placebo where three JAK inhibitor drugs were better than the two comparators, demonstrating that baricitinib, upadacitinib, and filgotinib outperformed adalimumab for clinical remissions and the poorest outcome was with MTX monotherapy. Finally, tofacitinib, baricitinib, upadacitinib, and filgotinib were compared with MTX in ORAL Start, RA BEGIN, SELECT-EARLY, and FINCH-3 trials where DMARD-naïve patients showed ACR20 response rates of 26%, 77%, 52%, and 78%, respectively with no background drug, demonstrating that these drugs are better than MTX for early RA with filgotinib being numerically higher than the others (Lee et al. 2014; Fleischmann et al. 2017; Westhovens et al. 2019; van Vollenhoven et al. 2020). Overall, these drugs mostly showed superiority over placebo in inadequate responders to bDMARDs, cDMARDs, and MTX. Tofacitinib showed lower clinical responses than baricitinib, upadacitinib, or figotinib while filgotinib showed higher responses than the other drugs in similar patient populations. The efficacy of baricitinib and upadacitinib was similar in inadequate responders to cDMARDs and MTX with no consistency in other populations. Nonetheless, this suggests that inhibition of JAK1 is needed for treating RA. Their superiority as initial therapy over MTX was also observed in treatment-naïve patients. Compared with adalimumab, their clinical responses emerged as being non-inferior or better in combination with MTX in MTX-inadequate responders.
With the use of JAK inhibitors in immune-mediated diseases including RA, significant AEs include serious infections, herpes zoster, malignancy, NMSC, MACE, and VTE. Among these safety concerns, tofacitinib, baricitinib, upadacitinib, and filgotinib were found to increase only the risk of herpes zoster infections from integrated analysis of controlled studies including randomized controlled trials and long term extension studies (Olivera et al. 2020). In the long-term safety analysis of integrated data for the earlier approved tofacitinib and baricitinib for RA, the incidence rates of the two drugs at all their doses did not show differences in the expression per 100 patient years, with, respectively, 2.7 and 2.8 in serious infections, 3.9 and 3.3 in herpes zoster, 0.9 and 0.8 in malignancy excluding NMSC, and 0.6 and 0.4 in NMSC. A data comparison with placebo was provided in the baricitinib analysis and the herpes zoster incidence rate was higher. Their safety profiles were maintained similar to those observed in their phase 3 trials (Cohen et al. 2017; Genovese et al. 2019). There was no significant difference with the use of JAK inhibitors including tofacitinib, baricitinib, and upadacitinib regarding occurrence of MACE, but increased risk of VTE for both tofacitinib and baricitinib at their higher doses was signaled from the results of multiple randomized controlled trials for RA (Xie et al. 2019). Only the lower dose of baricitinib was approved by the FDA. Based on interim data from a post-marketing safety trial that displayed an increased risk of pulmonary thromboembolism and death with tofacitinib at 10 mg BID compared with TNF-α inhibitors, FDA updated the labeling to contain the new boxed warning for RA and PsA in July 2019 (FDA 2019; Olivera et al. 2020). The integrated safety analysis of the SELECT phase 3 trials showed that the safety profile of the recently approved upadacitinib at the recommended dose was comparable to the placebo and adalimumab groups and similar to tofacitinib and baricitinib with four cases of VTE reported and further evaluation will be necessary. The lower dose was recommended with no consistent incremental efficacy benefit of the higher dose and the VTE risk was not dose-dependent (Cohen et al. 2019; Smolen et al. 2019). Filgotinib so far has been found to show an encouraging safety profile in all significant AEs from the FINCH phase 3 trials with lower risk in herpes zoster compared with other JAK inhibitors (Winthrop K et al. 2019). Long-term follow-up is needed. Overall, there have been no significant differences in the safety profile among the different JAK inhibitor drugs. One important issue to be investigated is whether or not the increased risk of VTE is limited to the higher doses of the drugs. These findings suggest their narrow therapeutic window.
Nonetheless, clinical differences in both efficacy and safety profile among these JAK inhibitors can be fully identified by direct comparisons in head-to-head clinical trials and long-term extension studies.
Conclusions and future perspectives
JAK inhibitor therapies have been considered to be acceptable for immune-mediated diseases by being comparable with other therapies even with the class safety issues. Clinical efficacy and safety data of the JAK inhibitors so far have been much more focused on RA. Upon the release of data from their ongoing clinical developments in a variety of immune-mediated diseases, the diseases that may or may not benefit from the inhibition of various JAKs with different isoform-selectivity will emerge more clearly as well as the clinical potential of different particular-isoform selective inhibition. With regard to the recently raised risk of VTE, its underlying mechanisms are currently unknown, and whether it is disease-specific or can be controlled by JAK inhibitor selectivity remains unknown. More data from long-term follow-up and extension studies as well as additional information from ongoing clinical trials will be needed for confirmation. As targeting the regulatory pseudokinase domains of JAKs could be an alternative approach for enhancing the selectivity of JAK inhibitors, clinical development success of the first clinical candidate of TYK pseudokinase-targeted inhibitor will be important. It is anticipated that pseudokinases may become mainstream drug targets for immune-mediated diseases.
References
Babon JJ, Lucet IS, Murphy JM, Nicola NA, Varghese LN (2014) The molecular regulation of Janus kinase (JAK) activation. Biochem J 462:1–13. https://doi.org/10.1042/BJ20140712
Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM (2017) JAK-STAT signaling as a target for inflammatory and autoimmune disease: Current and future prospects. Drugs 77:521–546. https://doi.org/10.1007/s40265-017-0701-9
Bechman K, Yates M, Galloway JB (2019) The new entries in the therapeutic armamentarium: The small molecule JAK inhibitors. Pharmacol Res 147:104392. https://doi.org/10.1016/j.phrs.2019.104392
Burke JR, Cheng L, Gillooly KM, Strnad J, Zupa-Fernandez A, Catlett IM, Zhang Y, Heimrich EM, McIntyre KW, Cunningham MD, Carman JA, Zhou X, Banas D, Chaudhry C, Li S, D’Arienzo C, Chimalakonda A, Yang X, Xie JH, Pang J, Zhao Q, Rose SM, Huang J, Moslin RM, Wrobleski ST, Weinstein DS, Salter-Cid LM (2019) Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med 11:eaaw1736. https://doi.org/10.1126/scitranslmed.aaw1736
Burmester GR, Blanco R, Charles-Schoeman C, Wollenhaupt J, Zerbini C, Benda B, Gruben D, Wallenstein G, Krishnaswami S, Zwillich SH, Koncz T, Soma K, Bradley J, Mebus C (2013) Tofacitinib (CP-690,550) in combination with methotrexate in patients with active rheumatoid arthritis with an inadequate response to tumour necrosis factor inhibitors: a randomised phase 3 trial. Lancet 381:451–460. https://doi.org/10.1016/S0140-6736(12)61424-X
Burmester GR, Kremer JM, den Bosch FV, Kivitz A, Bessette L, Li Y, Zhou Y, Othman AA, Pangan AL, Camp HS (2018) Safety and efficacy of upadacitinib in patients with rheumatoid arthritis and inadequate response to conventional synthetic disease-modifying anti-rheumatic drugs (SELECT-NEXT): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 391:2503–2512. https://doi.org/10.1016/S0140-6736(18)31115-2
Byrne DP, Foulkes DM, Eyers PA (2017) Pseudokiases: update on their functions and evaluation as new drug targets. Future Med Chem 9:245–265. https://doi.org/10.4155/fmc-2016-0207
Catlett I, Aras U, Liu Y, Bei D, Girgis I, Murthy B, Honczarenko M, Rose S (2017) A first-in-human, study of BMS-986165, a selective, potent, allosteric small molecule inhibitor of tyrosine kinase 2. Ann Rheum Dis 76:859.1–859.859. https://doi.org/10.1136/annrheumdis-2017-eular.3809
Chang Y, Xu S, Ding K (2019) Tyrosine kinase 2 (TYK2) allosteric inhibitors to treat autoimmune diseases. J Med Chem 62:8951–8952. https://doi.org/10.1021/acs.jmedchem.9b01612
Choy EH (2019) Clinical significance of Janus kinase inhibitor selectivity. Rheumatology 58:953–962. https://doi.org/10.1093/rheumatology/key339
Choy EHS, Miceli-Richard C, González-Gay MA, Sinigaglia L, Schlichting DE, Meszaros G, de la Torre I, Schulze-Koops H (2019) The effect of JAK/JAK2 inhibition in rheumatoid arthritis: efficacy and safety of baricitinib. Clin Exp Rheumatol 37:694–704
Cohen SB, Tanaka Y, Mariette X, Curtis JR, Lee EB, Nash P, Winthrop KL, Charles-Schoeman C, Thirunavukkarasu K, DeMasi R, Geier J, Kwok K, Wang L, Riese R, Wollenhaupt J (2017) Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5+ years: integrated analysis of data from the global clinical trials. Ann Rheum Dis 76:1253–1262. https://doi.org/10.1136/annrheumdis-2016-210457
Cohen S, van Vollenhoven R, Winthrop K, Zerbini CA, Tanaka Y, Bessette L, Zhang Y, Khan N, Hendrickson B, Enejosa JJ, Burmester G (2019) Safety profile of upadacitinib in rheumatoid arthritis: Integrated analysis from the SELECT phase 3 clinical program. American College of Rheumatology Web. Available at https://acrabstracts.org/abstract/safety-profile-of-upadacitinib-in-rheumatoid-arthritis-integrated-analysis-from-the-select-phase-3-clinical-program/. Accessed 13 Aug 2020
Combe B, Kivitz A, Tanaka Y, van der Heijde D, Matzkies F, Bartok B, Ye L, Guo Y, Tasset C, Sundy J, Mozaffarian N, Landewé R, Bae SC, Keystone E, Nash P. (2019) Efficacy and safety of filgotinib for patients with rheumatoid arthritis with inadequate response to methotrexate: FINCH1 primary outcome results. American College of Rheumatology Web. Available at https://acrabstracts.org/abstract/efficacy-and-safety-of-filgotinib-for-patients-with-rheumatoid-arthritis-with-inadequate-response-to-methotrexate-finch1-primary-outcome-results/. Accessed August 18 2020
Dougados M, van der Heijde D, Chen YC, Greenwald M, Drescher E, Liu J, Beattie S, Witt S, de la Torre I, Gaich C, Rooney T, Schlichting D, de Bono S, Emery P (2017) Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: results from the RA-BUILD study. Ann Rheum Dis 76:88–95. https://doi.org/10.1136/annrheumdis-2016-210094
Farmer LJ, Ledeboer MW, Hoock T, Arnost MJ, Bethiel RS, Bennani YL, Black JJ, Brummel CL, Chakilam A, Dorsch WA, Fan B, Cochran JE, Halas S, Harrington EM, Hogan JK, Howe D, Huang H, Jacobs DH, Laitinen LM, Liao S, Mahajan S, Marone V, Martinez-Botella G, McCarthy P, Messersmith D, Namchuk M, Oh L, Penny MS, Pierce AC, Raybuck SA, Rugg A, Salituro FG, Saxena K, Shannon D, Shlyakter D, Swenson L, Tian SK, Town C, Wang J, Wang T, Wannamaker MW, Winquist RJ, Zuccola HJ (2015) Discovery of VX-509 (Decernotinib): A potent and selective Janus kinase 3 inhibitor for the treatment of autoimmune diseases. J Med Chem 58:7195–7216. https://doi.org/10.1921/acs.jmedchem.5b00301
FDA (US Food & Drug Administration) (2019) FDA approves Boxed Warning about increased risk of blood clots and death with higher dose of arthritis and ulcerative colitis medicine tofacitinib (Xeljanz, Xeljanz XR). US FDA official website. Available at https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-boxed-warning-about-increased-risk-blood-clots-and-death-higher-dose-arthritis-and. Accessed 24 Aug 2020
FDA (US Food & Drug Administration) (2020) Novel Drug Approvals for 2019. US FDA official website. Available at https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2019US. Accessed 24 Aug 2020
Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N (2015) A randomized, double-Blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol 67:334–343. https://doi.org/10.1002/art.38949
Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, Gruben D, Wallenstein GV, Zwillich SH, Kanik KS (2012) Placebo-Controlled Trial of Tofacitinib Monotherapy in Rheumatoid Arthritis. N Engl J Med 367:495–507. https://doi.org/10.1056/NEJMoa1109071
Fleischmann R, Pangan AL, Song IH, Mysler E, Bessette L, Peterfy C, Durez P, Ostor AJ, Li Y, Zhou Y, Othman AA, Genovese MC (2019) Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: Results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol 71:1788–1800. https://doi.org/10.1002/art.41032
Fleischmann R, Schiff M, van der Heijde D, Ramos-Remus C, Spindler A, Stanislav M, Zerbini CAF, Gurbuz S, Dickson C, de Bono S, Schlichting D, Beattie S, Kuo WL, Rooney T, Macias W, Takeuchi T (2017) Baricitinib, methotrexate, or combination in patients with rheumatoid arthritis and no or limited prior disease-modifying antirheumatic drug treatment. Arthritis Rheumatol 69:506–517. https://doi.org/10.1002/art.39953
Fragoulis GE, McInnes IB, Siebert S (2019) JAK-inhibitors, New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology 58:i43–i54. https://doi.org/10.1093/rheumatology/key276
Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, Haley PJ, Shepard S, Rodgers JD, Yeleswaram S, Hollis G, Newton RC, Metcalf B, Friedman SM, Vaddi K (2010) Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol 184:5298–5307. https://doi.org/10.4049/jimmunol.0902819
Gadina M (2013) Janus kinases: An ideal target for the treatment of autoimmune diseases. J Investig Dermatol Symp Proc 16:S70–S72. https://doi.org/10.1038/jidysmp.2013.29
Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, Changelian P, Laurence A, O’Shea JJ (2018) Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J Leukoc Biol 104:499–514. https://doi.org/10.1002/JLB.5RI0218-084R
Gadina M, Le MT, Schwartz DM, Silvennoinen O, Nakayamada S, Yamaoka K, O’Shea JJ (2019) Janus kinases to jakinibs: from basic insights to clinical practice. Rheumatology 58:i4–i16. https://doi.org/10.1093/rheumatology/key432
Gadina M, Schwartz DM, O’Shea JJ (2016) Decernotinib: A next-generation Jakinb. Arthritis Rheumatol 68:31–34. https://doi.org/10.1002/art.39463
Genovese MC, Fleischmann R, Combe B, Hall S, Rubbert-Roth A, Zhang Y, Zhou Y, Mohamed MEF, Meerwein S, Pangan AL (2018) Safety and efficacy of upadacitinib in patients with active rheumatoid arthritis refractory to biologic disease-modifying anti-rheumatic drugs (SELECT-BEYOND): a double-blind, randomised controlled phase 3 trial. Lancet 391:2513–2524. https://doi.org/10.1016/S0140-6736(18)31116-4
Genovese MC, Kalunian K, Gottenberg J, Mozaffarian N, Bartok B, Matzkies F, Gao J, Guo Y, Tasset C, Sunday JS, de Vlam K, Walker D, Takeuchi T (2019) Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: The FINCH 2 randomized clinical trial. JAMA 322:315–325. https://doi.org/10.1001/jama.2019.9055
Genovese MC, Kremer J, Zamani O, Ludivico C, Krogulec M, Xie L, Beattie SD, Koch AE, Cardillo TE, Rooney TP, Macias WL, de Bono S, Schlichting DE, Smolen JS (2016) Baricitinib in patients with refractory rheumatoid arthritis. N Engl J Med 374:1243–1252. https://doi.org/10.1056/NEJMoa1507247
Genovese M, Smolen J, Takeuchi T, Burmester G, Brinker D, Rooney T, Zhong J, Mo D, Saifan C, Cardoso A, Issa M, Wu WS, Winthrop K. (2019) Safety Profile of Baricitinib for the Treatment of Rheumatoid Arthritis up to 7 Years: An updated integrated safety analysis. American College of Rheumatology Web. Available at https://acrabstracts.org/abstract/safety-profile-of-baricitinib-for-the-treatment-of-rheumatoid-arthritis-up-to-7-years-an-updated-integrated-safety-analysis/. Accessed August 14 2020
Genovese MC, van Vollenhoven RF, Pacheco-Tena C, Zhang Y, Kinnman N (2016) VX-509 (decernotinib), an oral selective JAK-3 inhibitor, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheumatol 68:46–55. https://doi.org/10.1002/art.39473
Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, Kudlacz E, Wang C, Menon S, Hendrikx T, Kanik KS (2017) Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med 377:1525–1536. https://doi.org/10.1056/NEJMoa1615977
Graff C, Schwartz A, Voss J, Wishart N, Olson L, George J, Hyland D, Camp H (2014) Characterization of ABT-494, a second generation JAK1 selective inhibitor. American College of Rheumatology Web. Available at https://acrabstracts.org/abstract/characterization-of-abt-494-a-second-generation-jak1-selective-inhibitor/. Accessed 25 Aug 2020
Harigai M (2019) Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology 58:i34–i42. https://doi.org/10.1093/rheumatology/key287
Havnaer A, Weinberg JM, Han G (2019) Systemic therapies in psoriasis: An update on newly approved and pipeline biologics and oral treatments. Cutis 104:17–20
Hirahara K, Schwartz D, Gadina M, Kanno Y, O’Shea JJ (2016) Targeting cytokine signaling in autoimmunity: back to the future and beyond. Curr Opin Immunol 43:89–97. https://doi.org/10.1016/j.coi.2016.10.001
Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, Menon S, Lamba M, Zwillich S (2016) The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin Exp Rheumatol 34:318–328
Jamilloux Y, Jammal TE, Vuitton L, Gerfaud-Valentin M, Kerever S, Sève P (2019) JAK inhibitors for the treatment of autoimmune and inflammatory diseases. Autoimmun Rev 18:102390. https://doi.org/10.1016/j.autrev.2019.102390
Kavanaugh A, Kremer J, Ponce L, Cseuz R, Reshetko OV, Stanislavchuk M, Greenwald M, Van der Aa A, Vanhoutte F, Tasset C, Harrison P (2017) Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose-finding study (DARWIN 2). Ann Rheum Dis 76:1009–1019. https://doi.org/10.1136/annrheumdis-2016-210105
Kivitz AJ, Cohen S, Keystone E, van Vollenhoven RF, Haraoui B, Kaine J, Fan H, Connell CA, Bananis E, Takiya L, Fleischmann R (2018) A pooled analysis of the safety of tofacitinib as monotherapy or in combination with background conventional synthetic disease-modifying antirheumatic drugs in a Phase 3 rheumatoid arthritis population. Semin Arthritis Rheum 48:406–415
Kontzias A, Kotlyar A, Laurence A, Changelian P, O’Shea JJ (2012) Jakinibs: A new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol 12:464–470. https://doi.org/10.1016/j.coph.2012.06.008
Kremer J, Li ZG, Hall S, Fleischmann R, Genovese M, Martin-Mola E, Isaacs JD, Gruben D, Wallenstein G, Krishnaswami S, Zwillich SH, Koncz T, Riese R, Bradley J (2013) Tofacitinib in combination with nonbiologic disease-modifying antirheumatic drugs in patients with active rheumatoid arthritis: a randomized trial. Ann Intern Med 159:253–261. https://doi.org/10.7326/0003-4819-159-4-201308200-00006
Kung JE, Jura N (2019) Prospects for pharmacological targeting of pseudokinases. Nat Rev Drugs Discov 18:501–526. https://doi.org/10.1038/s41573-019-0018-3
Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, Koncz T, Krishnaswami S, Wallenstein GV, Zang C, Zwillich SH, van Vollenhoven RF (2014) Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 370:2377–2386. https://doi.org/10.1056/NEJMoa1310476
Liongue C, Sertori R, Ward AC (2016) Evolution of cytokine receptor signaling. J Immunol 197:11–18. https://doi.org/10.4049/jimmunol.1600372
Liu X, Fang L, Guo TB, Mei H, Zhang JZ (2013) Drug targets in the cytokine universe for autoimmune disease. Trends Immunol 34:120–128. https://doi.org/10.1016/j.it.2012.10.003
Lu X, Smaill JB, Ding K (2020) New promise and opportunities for allosteric kinase inhibitors. Angew Chem Int Ed 59:2–15. https://doi.org/10.1002/anie.201914525
Ly K, Beck KM, Smith MP, Orbai AM, Liao W (2019) Tofacitinib in the management of active psoriatic arthritis: patient selection and perspectives. Psoriasis (Auckl) 9:97–107. https://doi.org/10.2147/PTT.S161453
Mahajan S, Hogan JK, Shlyakhter D, Oh L, Salituro FG, Farmer L, Hoock TC (2015) VX-509 (decernotinib) is a potent and selective Janus kinase 3 inhibitor that attenuates inflammation in animal models of autoimmune diseases. J Pharmacol Exp Ther 353:405–414. https://doi.org/10.1124/jpet.114.221176
Mease P, Hall S, FitzGerald O, van der Heijde D, Merola JF, Avila–Zapata F, Cieślak D, Graham D, Wang C, Menon S, Hendrikx T, Kanik KS (2017) Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med 377:1537–1550. https://doi.org/10.1056/NEJMoa1615975
Menet CJ, Fletcher SR, Van Lommen G, Geney R, Blanc J, Smits K, Jouannigot N, Deprez P, van der Aar EM, Clement-Lacroix P, Lepescheux L, Galien R, Vayssiere B, Nelles L, Christophe T, Brys R, Uhring M, Ciesielski F, van Rompaey L (2014) Triazolopyridines as selective JAK1 inhibitors: From hit identification to GLPG0634. J Med Chem 57:9323–9342. https://doi.org/10.1021/jm501262q
Moslin R, Gardner D, Santella J, Zhang Y, Duncia JV, Liu C, Lin J, Tokarski JS, Strnad J, Pedicord D, Chen J, Blat Y, Zupa-Fernandez A, Cheng L, Sun H, Chaudhry C, Huang C, D’Arienzo C, Sack JS, Muckelbauer JK, Chang C, Tredup J, Xie D, Aranibar N, Burke JR, Carter PH, Weinstein DS (2017) Identification of imidazo[1,2-b]pyridazine TYK2 pseudokinase ligands as potent and selective allosteric inhibitors of TYK2 signalling. Med Chem Commun 8:700–712. https://doi.org/10.1039/c6md00560h
Olivera PA, Lasa JS, Bonovas S, Danese S, Peyrin-Biroulet L (2020) Safety of Janus kinase inhibitors in patients with inflammatory bowel diseases or other immune-mediated diseases: A systematic review and meta-analysis. Gastroenteroloy 158:1554–1573. https://doi.org/10.1053/j.gastro.2020.01.001
O’Shea JJ, Holland SM, Staudt LM (2013a) JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med 368:161–170. https://doi.org/10.1056/NEJMra1202117
O’Shea JJ, Kontzias A, Yamaoka K, Tanaka Y, Laurence A (2013b) Janus kinase inhibitors in autoimmune diseases. Ann Rheum Dis 72:ii111–ii115. https://doi.org/10.1136/annrheumdis-2012-202576
O’Shea JJ, Laurence A, McInnes IB (2013c) Back to the future: Oral targeted therapy for RA and other autoimmune diseases. Nat Rev Rheumatol 9:173–182. https://doi.org/10.1038/nrrheum.2013.7
O’Shea JJ, Plenge R (2012) JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36:542–550. https://doi.org/10.1016/j.immuni.2012.03.014
O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A (2015) The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu Rev Med 66:311–328. https://doi.org/10.1046/annurev-med-051113-024537
Papp KA, Gordon KB, Thaçi D, Morita A, Gooderham M, Foley P, Girgis IG, Kundu S, Banerjee S (2018) Efficacy and safety of a potent and highly selective oral tyrosine kinase 2 inhibitor, BMS986165, in patients with moderate-to-severe plaque psoriasis: A phase II, randomized, placebo-controlled trial. American College of Rheumatology Web. Available at https://acrabstracts.org/abstract/efficacy-and-safety-of-a-potent-and-highly-selective-oral-tyrosine-kinase-2-inhibitor-bms-986165-in-patients-with-moderate-to-severe-plaque-psoriasis-a-phase-ii-randomized-placebo-controlled-tria/. Accessed August 2 2020
Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, Camp HS, Padley RJ, George JS, Hyand D, Resebraugh M, Wishart N, Olson L, Long AJ (2018) In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatol 2:23. https://doi.org/10.1186/s41927-018-0031-x
Peeva E (2019) Oral Janus kinase inhibitors PF-06700841 and PF-06651600 provide clinically evident therapeutic effect at 4 and 6 weeks in patients with alopecia areata and greater efficacy over 24 weeks in patients with a shorter duration of their current alopecia episode: Results of a randomized phase 2a trial. American Academy of Dermatology Web. Available at https://www.aadhighlights2019.com/articles/jak-inhibitors-pf-06700841-and-pf-06651600/read. Accessed August 25 2020
Pei H, He L, Shao M, Yang Z, Ran Y, Li D, Zhou Y, Tang M, Wang T, Gong Y, Chen X, Yang S, Xiang M, Chen L (2018) Discovery of a highly selective JAK3 inhibitor for the treatment of rheumatoid arthritis. Sci Rep 8:5273. https://doi.org/10.1038/s41598-018-23569-y
Plenge R (2010) GWASs and the age of human as the model organism for autoimmune genetic research. Genome Biol 11:212. https://doi.org/10.1186/gb-2010-11-5-212
Robinson MF, Damjanov N, Stamenkovic B, Radunovic G, Kivitz A, Cox L, Manukyan Z, Banifield C, Saunders M, Chandra D, Vincent MS, Mancuso J, Peeva E, Beebe JS (2020) Efficacy and safety of PF-06651600 (ritlecitinib), a novel JAK3/TEC inhibitor in patients with moderate to severe rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Rheumatol. https://doi.org/10.1002/art.41316
Roskoski R Jr (2016) Janus kinas (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res 111:784–803. https://doi.org/10.1016/j.phrs.2016.07.038
Sandborn WJ, Su C, Sands BE, D’Haens GR, Vermeire S, Schreiber S, Danese S, Feagan BG, Reinisch W, Niezychowski W, Friedman G, Lawendy N, Yu D, Woodworth D, Mukherjee A, Zhang H, Healey P, Panés J (2017) Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med 376:1723–1736. https://doi.org/10.1056/NEJMoa1606910
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ (2017) JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 16:843–862. https://doi.org/10.1038/nrd.2017.201
Smolen JS, Pangan AL, Emery P, Rigby W, Tanaka Y, Vargas JI, Zhang Y, Damjanov N, Friedman A, Othman AA, Camp HS, Cohen S (2019) Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet 393:2303–2311. https://doi.org/10.1016/S0140-6736(19)30419-2
Taylor PC (2019) Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology 58:i17–i26. https://doi.org/10.1093/rheumatology/key225
Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, del Carmen Morales L, Gonzaga JR, Yakushin S, Ishii T, Emoto K, Beattie S, Arora V, Gaich C, Rooney T, Schlichting D, Macias WL, de Bono S, Tanaka Y (2017) Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med 376:652–662. https://doi.org/10.1056/NEJMoa1608345
Taylor PC, Weinblatt ME, Burmester GR, Rooney TP, Witt S, Walls CD, Issa M, Salinas CA, Saifan C, Zhang X, Cardoso A, González-Gay MA, Takeuchi T (2019) Cardiovascular safety during treatment with baricitinib in rheumatoid arthritis. Arthritis Rheumatol 71:1042–1055. https://doi.org/10.1002/art.40841
Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, Moy E, Balbo P, Li W, Zhao Y, Crouse K, Dickinson C, Symanowicz P, Hegen M, Banker ME, Vincent F, Unwalla R, Liang S, Gilbert AM, Brown MF, Hayward M, Montgomery J, Yang X, Bauman J, Trujillo JI, Casmiro-Garcia A, Vajdos FF, Leung L, Geoghegan KF, Quazi A, Xuan D, Jones L, Hett E, Wright K, Clark JD, Thorarensen A (2016) Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem Biol 11:3442–3451. https://doi.org/10.1021/acschembio.6b00677
Tokarski JS, Zupa-Fernandez A, Tredup JA, Pike K, Chang CY, Xie D, Cheng L, Pedicord D, Muckelbauer J, Johnson SR, Wu S, Edavettal SC, Hong Y, Witmer MR, Elkin LL, Blat Y, Pitts WJ, Weinstein DS, Burke JR (2015) Tyrosine kinase 2-mediated signal transduction in T lymphocytes is blocked by pharmacological stabilization of its pseudokinase domain. J Biol Chem 290:11061–11074. https://doi.org/10.1074/jbc.M114.619502
van der Heijde D, Tanaka Y, Fleischmann R, Keystone E, Kremer J, Zerbini C, Cardiel MH, Cohen S, Nash P, Song YW, Tegzová D, Wyman BT, Gruben D, Benda B, Wallenstein G, Krishnaswami S, Zwillich SH, Bradley JD, Connell CA (2013) Tofacitinib (CP-690,550) in patients with rheumatoid arthritis receiving methotrexate: Twelve-month data from a twenty-four-month phase III randomized radiographic study. Arthritis Rheum 65:559–570. https://doi.org/10.1002/art.37816
van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, Smets B, Lepescheux L, Christophe T, Conrath K, Vandeghinste N, Vayssiere B, De Vos S, Fletcher S, Brys R, van’t Klooster G, Feyen JHM, Menet C (2013) Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol 191:3568–3577. https://doi.org/10.4049/jimmunol.1201348
van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, Forejtova S, Zwillich SH, Gruben D, Koncz T, Wallenstein GV, Krishnaswami S, Bradley JD, Wilkinson B (2012) Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 367:508–519. https://doi.org/10.1056/NEJMoa1112072
van Vollenhoven R, Takeuchi T, Pangan AL, Friedman A, Mohamed MEF, Chen S, Rischmueller M, Blanco R, Xavier RM, Strand V (2020) Efficacy and safety of upadacitinib monotherapy in methotrexate-naïve patients with moderately to severely active rheumatoid arthritis (SELECT‐EARLY): A randomized, double‐blind, active‐comparator, multi‐center, multi‐country trial. Arthritis Rheumatol. https://doi.org/10.1002/art.41384
Varyani F, Argyriou K, Phillips F, Tsakiridou E, Moran GW (2019) Profile of tofacitinib in the treatment of ulcerative colitis: An evidence-based review of recent data. Drug Des Devel Ther 13:4091–4105. https://doi.org/10.2147/DDDT.S182891
Virtanen AT, Haikarainen T, Raivola J, Silvennoinen O (2019) Selective JAKinibs: Prospects in inflammatory and autoimmune diseases. BioDrugs 33:15–32. https://doi.org/10.1007/s40259-019-00333-w
Westhovens R (2019) Clinical efficacy of new JAK inhibitors under development. Just more of the same? Rheumatology 58:i27–i33. https://doi.org/10.1093/rheumatology/key256
Westhovens R, Rigby WFC, van der Heijde D, Ching DWT, Bartok B, Matzkies F, Yin Z, Guo Y, Tasset C, Sundy J, Mozaffarian N, Messina OD, Landewé RBM, Atsumi T, Burmester G (2019) Efficacy and safety of filgotinib for patients with rheumatoid arthritis naïve to methotrexate therapy: FINCH3 primary outcome results. American College of Rheumatology Web. Available at https://acrabstracts.org/abstract/efficacy-and-safety-of-filgotinib-for-patients-with-rheumatoid-arthritis-naive-to-methotrexate-therapy-finch3-primary-outcome-results/. Accessed August 18 2020
Westhovens R, Taylor PC, Alten R, Pavlova D, Enríquez-Sosa F, Mazur M, Greenwald M, Van der Aa A, Vanhoutte F, Tasset C, Harrison P (2017) Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheum Dis 76:998–1008. https://doi.org/10.1136/annrheumdis-2016-210104
Winthrop K, Genovese M, Combe B, Tanaka Y, Kivitz A, Matzkies F, Bartok B, Ye L, Guo Y, Tasset C, Sundy J, Keystone E, Westhovens R, Rigby WFC, Burmester G. (2019) Pooled safety analyses from phase 3 studies of filgotinib in patients with rheumatoid arthritis. American College of Rheumatology Web. Available at https://acrabstracts.org/abstract/pooled-safety-analyses-from-phase-3-studies-of-filgotinib-in-patients-with-rheumatoid-arthritis/. Accessed August 14 2020
Xie W, Huang Y, Xiao S, Sun X, Fan Y, Zhang Z (2019) Impact of Janus kinase inhibitors on risk of cardiovascular events in patients with rheumatoid arthritis: systematic review and meta-analysis of randomised controlled trials. Ann Rheum Dis 78:1043–1054. https://doi.org/10.1136/annrheumdis-2018-214846
Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, Ma H, Wei D, Sun S (2020) The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol 80:106210. https://doi.org/10.1016/j.intimp.2020.106210
Xu P, Shen P, Yu B, Xu X, Ge R, Cheng X, Chen Q, Bian J, Li Z, Wang J (2020) Janus kinases (JAKs): The efficient therapeutic targets for autoimmune diseases and myeloproliferative disorders. Eur J Med Chem 192:112155. https://doi.org/10.1016/j.ejmech.2020.112155
Acknowledgements
This work was supported by the Konyang University Research Fund in 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
There is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kim, HO. Development of JAK inhibitors for the treatment of immune-mediated diseases: kinase-targeted inhibitors and pseudokinase-targeted inhibitors. Arch. Pharm. Res. 43, 1173–1186 (2020). https://doi.org/10.1007/s12272-020-01282-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-020-01282-7