
Abstract
The occurrence of drug–drug interactions (DDIs) can significantly affect the safety of a patient, and thus assessing DDI risk is important. Recently, physiologically based pharmacokinetic (PBPK) modeling has been increasingly used to predict DDI potential. Here, we present a PBPK modeling concept and strategy. We also surveyed PBPK-related articles about the prediction of DDI potential in humans published up to October 10, 2017. We identified 107 articles, including 105 drugs that fit our criteria, with a gradual increase in the number of articles per year. Studies on antineoplastic and immunomodulatory drugs (26.7%) contributed the most to published PBPK models, followed by cardiovascular (20.0%) and anti-infective (17.1%) drugs. Models for specific products such as herbal products, therapeutic protein drugs, and antibody–drug conjugates were also described. Most PBPK models were used to simulate cytochrome P450 (CYP)-mediated DDIs (74 drugs, of which 85.1% were CYP3A4-mediated), whereas some focused on transporter-mediated DDIs (15 drugs) or a combination of CYP and transporter-mediated DDIs (16 drugs). Full PBPK, first-order absorption modules and Simcyp® software were predominantly used for modeling. Recently, DDI predictions associated with genetic polymorphisms, special populations, or both have increased. The 107 published articles reasonably predicted the DDI potentials, but further studies of physiological properties and harmonization of in vitro experimental designs are required to extend the application scope, and improvement of DDI predictions using PBPK modeling will be possible in the future.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Outstanding achievements in the pharmaceutical industry have led to the approval of numerous drugs for use in clinical settings, and consequently, patients are frequently exposed to polypharmacy to treat concurrent diseases or to treat a single disease effectively (Hajjar et al. 2007). Co-administration of multiple drugs increases the prevalence of drug–drug interactions (DDIs), and clinically significant DDIs are mainly mediated by pharmacokinetic (PK) mechanisms. PK DDIs are caused by changes in the absorption, distribution, metabolism, and excretion (ADME) properties of a drug due to co-administered drugs, which often involve inhibition or induction of drug metabolizing enzymes, transporters, or both (Varma et al. 2015b). The occurrence of DDIs can reduce the efficacy or safety of a drug. Furthermore, rare but fatal adverse reactions could result from DDIs and be a major cause of the withdrawal of a drug from the market (Zhang et al. 2009). Therefore, understanding and assessing PK DDIs are essential for rational therapeutics and have traditionally been investigated by conducting clinical trials. However, the high expense and potential risks related to conducting clinical trials have necessitated the introduction of alternative approaches to studying DDIs (Von Moltke et al. 1998). Therefore, efforts to develop models that utilize drug parameters for the in vitro prediction DDIs in humans are increasing, and specific detailed information about these approaches are available in previously published studies (Einolf 2007; Boulenc and Barberan 2011; Bohnert et al. 2016).
Briefly, the approaches are generally classified into three categories, simple static, mechanistic static and mechanistic dynamic models. For simple static models, quantification of the DDI potential is mainly based on a single constant inhibitor concentration and inhibition constant derived from in vitro data. The model assumes that the concentration of the inhibitor does not change over time. In addition, the substrate drug is assumed to be metabolized only in the liver, and the fraction metabolized (fm) for the substrate drug is 100%. Hence, the simple static model represents the worst-case scenario, and the DDI magnitude could be overestimated (Einolf 2007). The mechanistic static model includes additional information, and the substrate drug is assumed to be metabolized not only in the liver but also the intestines. The fm of substrate drugs are considered, and the net effect of competitive or mechanism-based inhibition and induction can be incorporated into the model (Fahmi et al. 2008). Nevertheless, the model is not capable of describing the complete dynamic characteristics of drug metabolism in humans because a single constant inhibitor concentration is used and the DDI magnitude difference between staggered and simultaneous dosing cannot be described (Fowler et al. 2017). Above all, the greatest weakness of both simple and mechanistic static models is the associated challenges in applying the most relevant inhibitor concentration, and therefore the DDI magnitude could differ based on the inhibitor concentration (e.g., the maximum concentration [Cmax], average concentration [Caverage], and hepatic inlet concentration) (Boulenc and Barberan 2011; Cho et al. 2014).
Unlike other approaches, mechanistic dynamic models such as the PBPK model aim to explain all PK characteristics of a drug and describe time-variable concentrations of the substrate and inhibitor drug in different organs (Jones et al. 2015). Therefore, temporal profiles of inhibition procedure are defined, and the model has been shown to be more predictive than static models are generally (Einolf 2007). For example, the prediction of the DDI potential of AZD2066 as a perpetrator using the simple static model indicated that the occurrence of clinically significant DDIs is possible (area under the curve [AUC] ratio > 1.1) (Nordmark et al. 2014). However, the likelihood of DDIs occurring in vivo was low when the same in vitro data were analyzed using the PBPK model. The in vivo study also indicated no or low risk for clinically significant DDIs, and this improved accuracy may be partially attributable to the ability of the PBPK model to use time-variable drug concentrations instead of a single inhibitor concentration (Nordmark et al. 2014).
In addition, the PBPK model considers inter-individual variabilities such as the age, sex, ethnicity, and genetic polymorphisms and can assess individual PK variability. Hence, the magnitude and range of DDIs in the virtual population that reflects individual variability can be investigated using the PBPK model (Einolf 2007). Furthermore, the effect of factors such as the dosing regimen and population on changes on DDI potentials can be explored. Overall, the PBPK model is a more powerful strategy for predicting the DDI potential than existing methods, and regulatory agencies have approved DDI studies using PBPK models to replace clinical trials. Consequently, the application of PBPK modeling for DDI prediction has increased widely in recent years, and numerous articles have been published (Huang et al. 2013).
However, the PBPK model has limitations because abundant input data related to the PK characteristics of the drug are required for successful DDI prediction. Thus, it is a time-consuming process compared to static approaches and generally more preferably used in the late drug development stage. In addition, input parameters derived from in vitro assay or in silico prediction methods are highly variable, and uncertainty exists. Therefore, continuous refinement of the model is required as the drug-related knowledge accumulates. In addition, precise input parameter values related to the human physiology are currently lacking, and further studies are necessary for more accurate PBPK modeling (Boulenc and Barberan 2011; Varma et al. 2015b).
Nevertheless, the utility of PBPK modeling has recently been expanded to drug development, and clinical practice and the investigation of DDI potentials accounts for the highest application (Zhang et al. 2009). However, the systemic evaluation of PBPK modeling articles focused on DDI potentials has not been reported yet. In this review, we briefly described the concept of PBPK modeling and its use in predicting DDI potentials, and we examined 107 published articles on PBPK modeling for predicting DDI potentials up to October 10, 2017.
PBPK modeling: concept and methodology
Concept of PBPK modeling
PBPK modeling is a mathematical modeling technique that uses a series of mass balance differential equations to predict the ADME characteristics of drugs in humans and other animal species. The solutions to these differential equations are typically concentrations of a drug in each organ or tissue as a function of time. Indeed, the concept of PBPK modeling is not new. The use of multi-compartmental models that incorporate physicochemical and physiological components in the simulation of PK data was first adapted by Teorell as early as 1937 (Teorell 1937). Despite the long history of PBPK modeling, the expansion of its use has been limited due to its mathematical complexity. However, for several decades, efforts have been made to refine PBPK models so they can be used in drug development and environmental toxicology (Rowland et al. 2011 and references therein). Currently, with the advancements in computing power, improvements in silico/in vitro tools, and knowledge of physiology coupled with the availability of user-friendly software, PBPK modeling is rapidly becoming a powerful tool for predicting human PK (Khalil and Laer 2011; Rowland et al. 2011; Rowland-Yeo et al. 2013; Jones et al. 2015; Zhuang and Lu 2016). Thus, PBPK modeling is becoming increasingly popular and can be used to (1) predict preclinical/clinical PK profiles; (2) determine oral absorption characteristics including food or formulation effects, or both; (3) select the first-in-human dose; (4) predict clinical DDI potentials; (5) predict special population PK characteristics such as pediatric, geriatric, pregnancy, obstetric, and profiles of patients with concurrent disease states; and (6) predict large molecule PK during the drug discovery and development process (Khalil and Laer 2011; Rowland et al. 2011; Baneyx et al. 2012; Wagner et al. 2015). Of these applications, the highest portion was related to the prediction of DDIs (Huang et al. 2013), and a recent literature review reported that DDI-related articles accounted for the highest percentage (28%) of a total of 366 PBPK-related studies (Jones et al. 2013; Vieira et al. 2014; Sager et al. 2015). Presently, PBPK modeling and simulation is recommended by regulatory agencies, e.g., the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Ministry of Health Labor and Welfare of Japan to inform DDI study design and estimate the magnitude (Huang et al. 2013; Jones et al. 2015; Sager et al. 2015).
The full (whole-body) PBPK model consists of a number of compartments that represent different body organs or tissues, connected by the systemic circulation, e.g., the arterial and venous blood. Each organ is generally identified as either perfusion or permeability rate limited (Jones et al. 2006). The perfusion rate limited model is assigned under the assumptions that the tissue membrane is present without a barrier and that the blood flow rate is the rate-limiting factor. In the permeability rate limited model, drug-specific permeability rather than the blood flow rate is the rate-limiting factor. For example, if drugs cross the tissue membrane by an active transport process, the permeability rate limited model incorporating efflux- or influx-related parameters is used to describe the active transport process (Jones et al. 2013; Sager et al. 2015). In contrast, the minimal PBPK model construct reduces the number of compartments to no more than five, and other organs with comparable blood flow rates are grouped as one compartment to simplify the model (Jones et al. 2013; Sager et al. 2015).
Construction of PBPK models
Input parameters included in the PBPK model can be divided into three categories: system, drug, and the study design (Jamei et al. 2009a). System-dependent parameters are related to the physiological properties of the body and are defined by organ volume, mass, blood flow rate, enzyme or transporter abundance, plasma protein abundance, hematocrit, or genetic polymorphisms (Rowland et al. 2011). Information on these physiological properties of humans or other species are now available in the literature, and PBPK modeling also enables the incorporation of the altered physiological properties in different disease states or population groups (Jones et al. 2013).
Drug-dependent parameters consist of physicochemical parameters as well as ADME related parameters of the drug determined from a variety of in vitro, in silico, or in vivo data, or a combination of these (Tsamandouras et al. 2015). To explain the absorption process, mechanistic absorption models are required and rely on various drug-specific parameters including molecular weight, lipophilicity, solubility, and pKa values. Initially, a first-order absorption model was developed based on one-compartment kinetics. In addition, a compartmental absorption and transit (CAT) model, which divided the gastrointestinal tract into nine compartments (the stomach, seven small intestinal compartments, and the colon), has been introduced. Recently, advanced compartmental absorption and transit (ACAT) and advanced dissolution, absorption, and metabolism (ADAM) models have also been developed to supplement the CAT model (Jamei et al. 2009b). Distribution of the drug in each organ is generally described by either a perfusion or permeability rate limited model, as mentioned above. Clearance, which is a key parameter of the PBPK model, has a considerable effect on the PK behavior of the drug. Several approaches have been introduced for the characterization of in vivo clearance, and the in vitro–in vivo extrapolation (IVIVE) method, which was developed to predict the PK profiles of humans before the first dosing, is coupled with PBPK modeling to describe whole-organ clearance. In addition, the retrograde approach, which is a back-calculation method from oral clearance to in vitro intrinsic clearance, or direct incorporation of in vivo clearance could be used. Parameter estimation, which is the estimation of an in vitro intrinsic clearance parameter from observed PK profiles, can also be used when essential in vitro data and scaling factors are not available (Tsamandouras et al. 2015). For hepatic clearance, the application of IVIVE has been well studied, and in vitro data derived from experiments with recombinant enzymes, microsomes, or hepatocytes have been extrapolated to whole-liver clearance using scaling factors (Chen et al. 2012). For non-hepatic clearance, such as renal or biliary excretion, other approaches can be used to predict in vivo organ clearance. When a single approach is insufficient to characterize the in vivo clearance, any combination of the abovementioned information can be used to compensate for the missing clearance details.
Finally, information on the study design such as dose, route, and frequency of administration, the effect of concomitant drugs and food, and formulation properties is required to define a PBPK modeling and simulation.
PBPK modeling software
In the last step, the PBPK model equations and integration algorithms can be written and solved using specific programming languages, simulation software, or spreadsheet programs to simulate the PK profile of a drug in the plasma and tissues (Khalil and Laer 2011). The open and designed software are two main types of software currently used for PBPK modeling and simulation (Khalil and Laer 2011; Bouzom et al. 2012). The open software packages for PBPK modeling such as MATLAB®, NONMEM®, Berkeley Madonna®, SAAM II®, and acslX® require the modeler to write and code their model equations and functions. Thus, they are less suitable for novice modelers (Khalil and Laer 2011; Bouzom et al. 2012). The designed software comprises Simcyp®, GastroPlus®, PK-Sim®, Cloe PK ®, and MoBi®, which have made PBPK modeling more accessible to those without extensive modeling and programming experience. These user-friendly software packages include physiological databases of predefined species and populations that are combined with compound-specific information and are used to parameterize a whole-body PBPK model (Bouzom et al. 2012; Kuepfer et al. 2016). The availability of user-friendly software has broadened the use of PBPK models in the drug discovery and development process (Khalil and Laer 2011; Bouzom et al. 2012; Jones et al. 2015; Kuepfer et al. 2016).
PBPK model verification
A newly developed PBPK model is used to simulate PK profiles of predefined populations and actual clinical trials using the aforementioned user-friendly software packages. The performance of the PBPK model is subsequently verified by comparing the simulated PK parameters (AUC and Cmax) with the observed clinical data and using the visual inspection approach for the concentration–time profiles (Kuepfer et al. 2016). The predictive performance of PBPK models is evaluated using the mean observed/predicted ratio of the AUC and Cmax, and is considered acceptable when the ratios fall within the predefined success range (e.g., 1.25-, 1.5-, or 2-fold) (Guest et al. 2011; Abduljalil et al. 2014; Wagner et al. 2015; Ke et al. 2016). In addition, the visual inspection checks were deemed acceptable if the clinically determined plasma concentrations are within the 5th and 95th percentiles of the predicted profile (Zhou et al. 2016). Although there appears to be a lack of consistency in the acceptance criteria for model verification, it was recently reported that the criteria should be predefined by considering various factor and should be appropriate (Jones et al. 2015). This PBPK model should also adequately predict independent clinical data (e.g., different dose levels, population, and route of administration) that are not used in the model construction, if possible. The PBPK model is refined during this step by parameter optimization and sensitivity analysis is mandatory for model optimization. Sensitivity analysis is informative for identifying key parameters that are likely to affect the model performance. The identified sensitive parameters should be reflected in the model (Zhou et al. 2016; Zhuang and Lu 2016). The verification/modification step is essential for the subsequent use of the model, and afterward, the verified PBPK model can be used to simulate other uninvestigated clinical scenarios or DDI predictions.
PBPK modeling for predicting DDI potentials
The DDI prediction study using PBPK modeling involves the development of PBPK models for both the victim and perpetrator drugs. The developed models are subsequently verified based on clinical data obtained using the dosage regimen planned for the study where possible, followed by model refinement. Then, the DDI prediction is carried out by simulating the substrate-inhibitor interaction according to the study design.
PBPK modeling articles on predicting DDI potentials
Journal search
Journal articles were selected using the PubMed search engine. The search terms were “Physiologically based pharmacokinetic modeling” and “drug–drug interaction,” and 214 articles were identified for the period up to October 10, 2017. Among the articles, a review article and a study on the application of PBPK modeling in animals were excluded, whereas articles on the PK of DDI using PBPK modeling in humans were analyzed. Finally, we identified 107 articles, including 105 involving different drugs that fit our scope and were further categorized by publication year (Fig. 1). The first article on PBPK modeling for DDI prediction was reported in 2008 (Chenel et al. 2008; Hyland et al. 2008), and the number of related articles has consistently increased since then to more than 20 articles per year (Fig. 1). Although most of the articles studied CYP-mediated DDIs, transporter-mediated DDIs were first reported in 2012. Similarly, articles on PBPK modeling for predicting complex DDI potentials that considered the effects of both CYP- and transporter-mediated DDIs have also been reported since 2012 (Fig. 1). The increase in the number of articles on DDI-related PBPK modeling may be attributable to the development of user-friendly software, and the regulatory authorities’ approval of the application of PBPK modeling in the drug development process. The increased knowledge of the body’s physiology and the advancement of in vitro experimental techniques for investigating drug properties and providing improved drug PK profiling are anticipated to increase the number of articles on predicting DDIs using PBPK modeling. Detailed information on DDI mechanisms, PBPK model description, and interactive predictions of DDIs of the 105 classified drugs are summarized in Tables 1, 2, 3, 4, 5, 6, along with references.

PBPK modeling articles per year for predicting DDIs
Classification of selected drugs from articles on DDI-related PBPK modeling
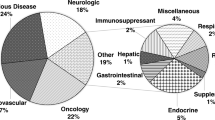
A total of 105 drugs were categorized according to the first level of the Anatomical Therapeutic Chemical (ATC) classification system, which groups drugs according to their main anatomical group, as developed by the World Health Organization (http://www.whocc.no/atcddd/). The developed PBPK models for the drugs were found in the following therapeutic categories at the indicated proportions: antineoplastic and immunomodulatory had the highest contribution (28 drugs, 26.7%), followed by the cardiovascular system (21 drugs, 20.0%), systemic anti-infective (18 drugs, 17.1%), alimentary tract and metabolism (11 drugs, 10.2%), nervous system (11 drugs, 10.5%), blood and blood-forming organs (six drugs, 5.7%), respiratory system, anti-parasitic (three drugs, 2.9%, each), musculoskeletal system (two drugs, 1.9%), systemic hormonal, and various other categories (one drug, 1.0%, each) (Fig. 2).

Classification of 105 drugs selected in the DDI-related articles using PBPK modeling according to the first level of the Anatomical Therapeutic Chemical (ATC) classification system, which groups drugs according to their main anatomical group, as developed by the World Health Organization (http://www.whocc.no/atcddd/)
Among the antineoplastic and immunomodulatory agents, 24 drugs were antineoplastic with the highest contribution. Antineoplastic drugs are highly toxic and have a narrow therapeutic index. In addition, chemotherapy regimens include at least one or more antineoplastic drugs (Chabner and Roberts 2005). Thus, the DDI risk is increased in patients with cancer and, therefore, the evaluation of DDI potential is important under such conditions. However, volunteer recruitment is challenging for clinical trials and, consequently, the use of PBPK modeling for determining antineoplastic DDIs has increased (Table 1). However, this strategy is limited because most simulations were performed in healthy populations and the altered physiological properties in patients with cancer were not incorporated in the models. Nevertheless, it is encouraging that recent articles have included the physiological conditions of patients with cancer and this additional detailed information is required for more reliable prediction (Ono et al. 2017; Einolf et al. 2017a). Cardiovascular drugs had the second highest proportion of agents investigated using PBPK models (Table 2). It has been reported that patients with cardiovascular diseases often present with a higher incidence of DDI that those without these conditions do (Mendel et al. 2011). This observation occurs possibly because these conditions are mainly associated with old age, multiple drug regimens, and the nature of the cardiovascular drug (Mendel et al. 2011). Among the cardiovascular drugs, statins were the most investigated, and PBPK models of atorvastatin (Zhang et al. 2015; Duan et al. 2017), fluvastatin (Yoshikado et al. 2016), pitavastatin (Yoshikado et al. 2016; Duan et al. 2017), pravastatin (Varma et al. 2012), rosuvastatin (Jamei et al. 2014; Wang et al. 2017), and simvastatin (Fenneteau et al. 2010) have been developed (Table 2). The statins were shown to be substrates of CYP3A4 (simvastatin), organic anion-transporting polypeptide 1/3 (OATP1B1/3; fluvastatin, pravastatin, and rosuvastatin), or both CYP isoforms and OATP1B1/3 (atorvastatin and pitavastatin). Furthermore, the inhibitory effects of the statins were simulated using PBPK modeling (Table 2).
Articles on PBPK modeling of DDI for specific products
Of the 105 drugs included in our dataset, two herbal products, silibinin, a semi-purified milk thistle seed extract, and Wuzhi capsule, consisting of Schisandra sphenanthera ethanol extract, were included. Recently, the PBPK models of the individual constituents (silybin A and B) of silibinin were developed (Brantley et al. 2014; Gufford et al. 2015; Table 4). In humans, the inhibitory effects of silibinin on intestinal glucuronidation of raloxifene and the substrates of CYP2C9 (warfarin) and CYP3A4 (midazolam) were accurately predicted using the PBPK modeling and simulation approach (Brantley et al. 2014; Gufford et al. 2015). The PBPK models of two main active components (schisantherin A and schisandrin A) for Wuzhi capsule were also established (Zhang et al. 2017; Table 4). The developed PBPK models adequately predicted the PK of tacrolimus-associated DDIs mediated by CYP3A4 inhibition (Zhang et al. 2017). These studies showed the feasibility of using the PBPK modeling and simulation approach to predict of herb–drug interaction potentials. However, the limited human PK data available for herbal constituents, combined with a lack of herbal product standardization, continues to challenge the development of PBPK models for the prediction of herb–drug interaction potentials (Brantley et al. 2014; Gufford et al. 2015).
PBPK models for cytokine-modulating protein drugs such as blinatumomab and sirukumab have been reported to quantitatively predict the potential clinical DDI between the therapeutic protein and a small-molecule drug in disease states associated with significantly elevated levels of the cytokine, interleukin (IL)-6 (Xu et al. 2015; Jiang et al. 2016; Table 1). The increased level of cytokines such as IL-6, IL-1β, tumor necrosis factor-α, and interferon-α in some inflammatory disease states can downregulate the drug-metabolizing CYP enzymes, decreasing the clearance of co-administered small-molecule drugs that are substrates of the affected CYP enzymes (Xu et al. 2015; Jiang et al. 2016). Cytokine-modulating protein drugs could reverse (or normalize) the suppression of CYP expression and a subsequent increase in the clearance of concomitant small-molecule drugs, referred to as disease-related protein DDIs (Aitken et al. 2006; Xu et al. 2015; Jiang et al. 2016). The alteration of IL-6 levels and its suppression of CYPs before and after therapeutic protein drug treatment in patients were investigated, and the developed PBPK models successfully predicted the perpetuating role of IL-6 (Xu et al. 2015; Jiang et al. 2016).
In addition, a PBPK model of brentuximab vedotin, an antibody–drug conjugate (ADC), has been reported, and the DDI potentials were investigated (Chen et al. 2015c; Table 1). ADCs are monoclonal antibodies attached to cytotoxic small molecules by a chemical link and designed as a targeted therapy to treat cancer. The conversion of the antibody-conjugated cytotoxic small molecules to unconjugated forms was described in the model, and the predicted DDI potential between the unconjugated cytotoxic small molecule and other drugs was comparable to that observed in previous data (Chen et al. 2015c).
PBPK modeling for predicting CYP- or transporter-mediated DDIs or both
The mechanisms of the DDIs associated with the classified drugs were further characterized. Most PBPK models of the drugs were used to simulate CYP-mediated DDIs (74 drugs, 70.5%), whereas some focused on transporter-mediated DDIs (15 drugs, 14.3%), or a combination of CYP and transporter-mediated DDIs (16 drugs, 15.2%).
Among the 74 drugs used to simulate CYP-mediated DDIs, 85.1% (63 drugs) were CYP3A4-mediated (Tables 1, 2, 3, 4, 5). CYP3A4 is the major enzyme involved in drug metabolism, which explains the increased susceptibility to CYP3A4-mediated DDIs (Lynch and Price 2007). The PBPK models for 18 drugs associated with multiple CYP isoforms, which revealed substrates (3/74 drugs), inhibitors (6/74 drugs), and both inducers and inhibitors (5/74 drugs), or both substrate and inducers (2/74 drugs) have been published (Tables 1, 2, 3, 4, 5). As mentioned previously, the PBPK models for two therapeutic protein drugs (2/74 drugs), blinatumomab and sirukumab, reasonably explored the reverse suppressions of multiple IL-6-induced CYP isoform levels and predicted their modulatory effects on the PK of small-molecule drugs metabolized by the multiple CYP isoforms (Table 1). Recently, the PBPK model of all-trans-retinoic acid, which is both a substrate and inducer of CYP26A1, the main all-trans-retinoic acid hydroxylase in human liver, has been developed and published (Jing et al. 2017; Table 1). The developed model was used to quantitatively predict the interaction between all-trans-retinoic acid and liarozole, an inhibitor of CYP26A1 (Jing et al. 2017; Table 1).
In contrast, only one drug, silibinin, was used for a non-CYP-mediated DDI prediction, and the inhibitory effects of silibinin on uridine diphosphate glucuronyltransferase 1A1 (UGT1A1) were evaluated (Gufford et al. 2015; Table 4).
Seven drugs were used to predict OATP1B1/3-mediated DDIs, which accounted for most of the 15 drugs evaluated for transporter-mediated DDI predictions (Tables 1, 2, 3, 4, 5). The OATP family is a well-characterized family of uptake transporters, and, particularly, the subfamily of OATP1B transporters has been reported to be involved in clinically significant DDIs (Koenen et al. 2011). The prediction of OATP1B1/3-mediated DDIs was 42.9 and 57.1% for the substrate and inhibitor, respectively.
Sixteen drugs were used to assess combined CYP- and transporter-mediated DDIs (Tables 1, 2, 3, 4, 5). The developed PBPK models could describe the DDI potentials when the drugs acted as inhibitors or substrates of both CYP isoforms and uptake (OATPs, OATs, OCTs, or MATEs) or efflux (P-glycoprotein [P-gp] or breast cancer resistance protein [BCRP]) transporters simultaneously in the kidney and liver. In contrast to the PBPK model, the simple static model considers the inhibitory or inductive effects on enzymes or transporters separately (Einolf 2007). It has been reported that most drugs act simultaneously as substrates or inhibitors of a specific CYP enzyme and transporter, and the PBPK modeling approach is useful for revealing combinatorial effects of enzymes and transporters (Shugarts and Benet 2008).
PBPK modeling was used to predict DDIs between inhibitory parent–metabolite pairs for 15 of the 105 drugs (14.3%, Tables 1, 2, 3, 4, 5). This integrated PBPK approach for both parent and metabolite(s) based on the availability of in vitro and in vivo metabolite data may increase the accuracy of predictions of all DDIs compared with the predictions based on the parent compounds alone (Chen et al. 2015b).
The limitation of the articles published to date is that the investigations were restricted to a small number of well-known drug metabolizing enzymes or transporters. This may have resulted from the lack of information about the physiological properties of the body such as the absolute abundance of non-CYP enzymes or transporters. This may have consequently led to the unsuccessful use of the IVIVE approaches (Varma et al. 2012). Thus, further investigations are needed to broaden the application scope of PBPK modeling to studies of potential DDIs.
PBPK modeling strategies to predict DDI potentials
The full PBPK model was more commonly constructed (54 drugs, 51.4%) than the minimal PBPK model was (42 drugs, 40.0%), and the PBPK models of nine drugs (8.6%) were constructed using both full and minimal models (Tables 1, 2, 3, 4, 5). To describe the absorption process, the first-order absorption model was predominantly selected (63 drugs, 64.3%), followed by the ADAM model (17 drugs, 17.3%), CAT (three drugs, 3.1%), and ACAT model (5 drugs; 5.1%). The remaining 10.2% involve cases of multiple absorption models in different articles. The elimination process was mainly described using of multiple strategies (41 of 105 drugs, 39.0%), followed by IVIVE (26 of 105 drugs, 24.8%), in vivo clearance parameters (19 of 105 drugs, 18.1%), and retrograde approaches (17 of 105 drugs, 16.2%). The remaining 1.9% were parameter estimation (2/105 drugs). However, it was reported that the in vitro inhibition potencies differed among various literature reports and, thus, the harmonization of in vitro experimental designs is needed for the construction of more precise PBPK model (Gertz et al. 2013). For the clinical trial design, the oral dosing route was more predominant (94 drugs, 89.5%) than the intravenous administration was (seven drugs, 6.7%). Additionally, four drugs (3.8%) were investigated using both oral and intravenous administration routes. As mentioned earlier, several criteria are used for the verification of successful PBPK models. The most frequently used the criteria for evaluating the predictive performance of PBPK, and the most commonly used was values within a two-fold range of the ratio of the predicted to the observed mean AUC or Cmax (49 of 105 drugs, 46.7%). In addition, other criteria determined were values within a 1.25- or 1.5-fold range or 5th and 95th percentiles. However, numerous PBPK models of some drugs (38 of 105 drugs, 36.2%) did not use the specific criteria for the predictive performance. Of the commercially available software packages for PBPK modeling, the most commonly used software to predict DDI potentials was the Simcyp® (76 of 105 drugs, 72.4%).
PBPK modeling articles on effects of body physiology on DDIs
Most of the PBPK modeling articles predicted DDI potentials only in healthy adult populations, which is a limitation of this approach because the magnitude of DDI potentials can be altered by the different physiological properties of various population groups or patients. However, some recent articles have included the effects of physiological properties on the prediction of DDI potentials and examples of these articles are discussed below.
Genetic polymorphisms
Among the 105 drugs on PBPK modeling of DDIs, five (clopidogrel, efavirenz, oxycodone, veliparib, and voriconazole) investigated the effect of genetic polymorphisms associated with DDIs (Table 6). It has been reported that CYP2B6, CYP2C9, CYP2C19, and CYP2D6 polymorphisms account for the most frequent variations in phase I metabolic enzymes, and the identified genetic polymorphisms affect enzyme activities (Zhou et al. 2009). Thus, additional considerations of the effect of gene polymorphism would be required in the evaluation of potential DDIs mediated by these CYPs. Information on genetic polymorphisms of clinically important drug transporters is scarce, in contrast to that on genetic polymorphisms of drug-metabolic CYPs, although several genetic polymorphisms have been identified in efflux (P-gp) and uptake (OATPs) transporters (Sissung et al. 2010). The effects of CYP2B6, CYP2C19, or CYP2D6 polymorphism, or a combination of any of these on the PK behaviors of their substrates with or without inhibitors, were evaluated using PBPK modeling, which captured both the inhibitory potency of the perpetrator and the effect of genetic polymorphisms on the PK properties of the substrate drug (Table 6).
Special populations
Physiological properties such as organ weight, blood flow, plasma binding, and drug metabolic enzyme or transporter activity are dependent on the species or population considered. For example, in the case of special populations (e.g., children, pregnant women, or disease-specific population), these physiological properties differ between the special and the healthy adult population (Hartmanshenn et al. 2016). Previous studies of the altered physiological properties of special populations are insufficient and, therefore, the use of PBPK modeling to predict DDI potentials in special population is limited. However, 12 articles supporting such modeling were found in our dataset (Tables 1, 2, 3, 4, 5); these included patients with impaired renal or hepatic function (Emoto et al. 2013; Lu et al. 2014; Nakamaru et al. 2015; Ono et al. 2017; Wagner et al. 2017), patients who were immunocompromised or had rheumatoid arthritis (Xu et al. 2015; Jiang et al. 2016), patients with cancer (Einolf et al. 2017a; Jing et al. 2017; Ono et al. 2017), pediatrics (Johnson and Rostami-Hodjegan 2011; Jing et al. 2017; Olafuyi et al. 2017a), and a pregnant population (Olafuyi et al. 2017b). The simulated results were comparable to clinical observations in special populations, and further investigations of disease- or age-related physiological properties in humans would be helpful.
Conclusion
The importance of predicting DDI potential has been discussed in many review articles, and PBPK modeling has been increasingly used for DDI predictions. This review provides a brief overview of PBPK model development and its application for DDI predictions. In addition, 107 PBPK modeling articles on the prediction of DDI potentials were identified, and the advantages of PBPK modeling, including capturing time-variable changes and inter-individual variability, have increased the number of articles published yearly. Although the articles reasonably predicted the DDI potentials in humans, investigation of DDI potentials using PBPK modeling was restricted to a limited number of drug metabolizing enzyme-mediated and transporter-mediated DDIs. In addition, the simulations were performed mostly in healthy adult populations. To widen the application scope of PBPK modeling in DDI predictions, more information on the physiological properties of the body and the incorporation of pathophysiological conditions in disease states are required. In addition, a harmonized in vitro experimental design is required for proper PBPK model building, and precise acceptance criteria should be set up for the validation process. Nevertheless, the outstanding achievements and progress in life sciences and computer technologies will soon solve these problems, and DDI predictions will be improved by the incorporation of in vitro data into PBPK models.
References
Abduljalil K, Cain T, Humphries H, Rostami-Hodjegan A (2014) Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos 42:1478–1484
Aitken AE, Richardson TA, Morgan ET (2006) Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol 46:123–149
Almond LM, Mukadam S, Gardner I, Okialda K, Wong S, Hatley O, Tay S, Rowland-Yeo K, Jamei M, Rostami-Hodjegan A, Kenny JR (2016) Prediction of drug-drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab Dispos 44:821–832
Bae SH, Park WS, Han S, Park GJ, Lee J, Hong T, Jeon S, Yim DS (2017) Physiologically based pharmacokinetic predictions of intestinal BCRP-mediated effect of telmisartan on the pharmacokinetics of rosuvastatin in humans. Biopharm Drug Dispos 38:363
Ball K, Jamier T, Parmentier Y, Denizot C, Mallier A, Chenel M (2017) Prediction of renal transporter-mediated drug-drug interactions for a drug which is an OAT substrate and inhibitor using PBPK modelling. Eur J Pharm Sci 106:122–132
Baneyx G, Fukushima Y, Parrott N (2012) Use of physiologically based pharmacokinetic modeling for assessment of drug-drug interactions. Future Med Chem 4:681–693
Baneyx G, Parrott N, Meille C, Iliadis A, Lave T (2014) Physiologically based pharmacokinetic modeling of CYP3A4 induction by rifampicin in human: influence of time between substrate and inducer administration. Eur J Pharm Sci 56:1–15
Bohnert T, Patel A, Templeton I, Chen Y, Lu C, Lai G, Leung L, Tse S, Einolf HJ, Wang YH, Sinz M, Stearns R, Walsky R, Geng W, Sudsakorn S, Moore D, He L, Wahlstrom J, Keirns J, Narayanan R, Lang D, Yang X, International Consortium for Innovation and Quality in Pharmaceutical Development Victim Drug-Drug Interactions Working Group (2016) Evaluation of a new molecular entity as a victim of metabolic drug-drug interactions-an industry perspective. Drug Metab Dispo 44:1399–1423
Boulenc X, Barberan O (2011) Metabolic-based drug-drug interactions prediction, recent approaches for risk assessment along drug development. Drug Metab Drug Interact 26:147–168
Bouzom F, Ball K, Perdaems N, Walther B (2012) Physiologically based pharmacokinetic (PBPK) modelling tools: how to fit with our needs? Biopharm Drug Dispos 33:55–71
Brantley SJ, Gufford BT, Dua R, Fediuk DJ, Graf TN, Scarlett YV, Frederick KS, Fisher MB, Oberlies NH, Paine MF (2014) Physiologically based pharmacokinetic modeling framework for quantitative prediction of an herb-drug interaction. CPT Pharm Syst Pharmacol 3:e107
Budha NR, Ji T, Musib L, Eppler S, Dresser M, Chen Y, Jin JY (2016) Evaluation of cytochrome P450 3A4-mediated drug-drug interaction potential for cobimetinib using physiologically based pharmacokinetic modeling and simulation. Clin Pharmacokinet 55:1435–1445
Bui KH, Zhou D, Agbo F, Guo J (2015) Effect of multiple intravenous doses of lanicemine (AZD6765) on the pharmacokinetics of midazolam in healthy subjects. J Clin Pharmacol 55:1024–1030
Burt HJ, Neuhoff S, Almond L, Gaohua L, Harwood MD, Jamei M, Rostami-Hodjegan A, Tucker GT, Rowland-Yeo K (2016) Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur J Pharm Sci 88:70–82
Chabner BA, Roberts TG Jr (2005) Timeline: chemotherapy and the war on cancer. Nat Rev Cancer 5:65–72
Chen Y, Jin JY, Mukadam S, Malhi V, Kenny JR (2012) Application of IVIVE and PBPK modeling in prospective prediction of clinical pharmacokinetics: strategy and approach during the drug discovery phase with four case studies. Biopharm Drug Dispos 33:85–98
Chen J, Liu D, Zheng X, Zhao Q, Jiang J, Hu P (2015a) Relative contributions of the major human CYP450 to the metabolism of icotinib and its implication in prediction of drug-drug interaction between icotinib and CYP3A4 inhibitors/inducers using physiologically based pharmacokinetic modeling. Expert Opin Drug Metab Toxicol 11:857–868
Chen Y, Mao J, Hop CE (2015b) Physiologically based pharmacokinetic modeling to predict drug-drug interactions involving inhibitory metabolite: a case study of amiodarone. Drug Metab Dispos 43:182–189
Chen Y, Samineni D, Mukadam S, Wong H, Shen BQ, Lu D, Girish S, Hop C, Jin JY, Li C (2015c) Physiologically based pharmacokinetic modeling as a tool to predict drug interactions for antibody-drug conjugates. Clin Pharmacokinet 54:81–93
Chen Y, Ma F, Lu T, Budha N, Jin JY, Kenny JR, Wong H, Hop CE, Mao J (2016) Development of a physiologically based pharmacokinetic model for itraconazole pharmacokinetics and drug-drug interaction prediction. Clin Pharmacokinet 55:735–749
Chenel M, Bouzom F, Aarons L, Ogungbenro K (2008) Drug-drug interaction predictions with PBPK models and optimal multiresponse sampling time designs: application to midazolam and a phase I compound. Part 1: comparison of uniresponse and multiresponse designs using PopDes. J Pharmacokinet Pharmacodyn 35:635–659
Cherkaoui-Rbati MH, Paine SW, Littlewood P, Rauch C (2017) A quantitative systems pharmacology approach, incorporating a novel liver model, for predicting pharmacokinetic drug–drug interactions. PLoS ONE 12:e0183794
Cho DY, Bae SH, Lee JK, Kim YW, Kim BT, Bae SK (2014) Selective inhibition of cytochrome P450 2D6 by Sarpogrelate and its active metabolite, M-1, in human liver microsomes. Drug Metab Dispos 42:33–39
Damle B, Varma MV, Wood N (2011) Pharmacokinetics of voriconazole administered concomitantly with fluconazole and population-based simulation for sequential use. Antimicrob Agents Chemother 55:5172–5177
de Kanter R, Sidharta PN, Delahaye S, Gnerre C, Segrestaa J, Buchmann S, Kohl C, Treiber A (2016) Physiologically-based pharmacokinetic modeling of macitentan: prediction of drug-drug interactions. Clin Pharmacokinet 55:369–380
de Zwart L, Snoeys J, De Jong J, Sukbuntherng J, Mannaert E, Monshouwer M (2016) Ibrutinib dosing strategies based on interaction potential of CYP3A4 perpetrators using physiologically based pharmacokinetic modeling. Clin Pharmacol Ther 100:548–557
Dhuria S, Einolf H, Mangold J, Sen S, Gu H, Wang L, Cameron S (2013) Time-dependent inhibition and induction of human cytochrome P4503A4/5 by an oral IAP antagonist, LCL161, in vitro and in vivo in healthy subjects. J Clin Pharmacol 53:642–653
Djebli N, Fabre D, Boulenc X, Fabre G, Sultan E, Hurbin F (2015) Physiologically based pharmacokinetic modeling for sequential metabolism: effect of CYP2C19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab Dispos 43:510–522
Duan P, Zhao P, Zhang L (2017) Physiologically based pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict drug-drug interactions (DDIs). Eur J Drug Metab Pharmacokinet 42:689–705
Einolf HJ (2007) Comparison of different approaches to predict metabolic drug-drug interactions. Xenobiotica 37:1257–1294
Einolf HJ, Zhou J, Won C, Wang L, Rebello S (2017a) A physiologically-based pharmacokinetic modeling approach to predict drug-drug interactions of Sonidegib (LDE225) with perpetrators of CYP3A in cancer patients. Drug Metab Dispos 45:361–374
Einolf HJ, Lin W, Won CS, Wang L, Gu H, Chun DY, He H, Mangold JB (2017b) Physiologically-based pharmacokinetic model predictions of panobinostat (LBH589) as a victim and perpetrator of drug-drug interactions. Drug Metab Dispos
Emoto C, Fukuda T, Cox S, Christians U, Vinks AA (2013) Development of a physiologically-based pharmacokinetic model for sirolimus: predicting bioavailability based on intestinal CYP3A content. CPT Pharm Syst Pharmacol 2:e59
Fahmi OA, Boldt S, Kish M, Obach RS, Tremaine LM (2008) Prediction of drug-drug interactions from in vitro induction data: application of the relative induction score approach using cryopreserved human hepatocytes. Drug Metab Dispos 36:1971–1974
Fenneteau F, Poulin P, Nekka F (2010) Physiologically based predictions of the impact of inhibition of intestinal and hepatic metabolism on human pharmacokinetics of CYP3A substrates. J Pharm Sci 99:486–514
Fowler S, Morcos PN, Cleary Y, Martin-Facklam M, Parrott N, Gertz M, Yu L (2017) Progress in prediction and interpretation of clinically relevant metabolic drug-drug interactions: a minireview illustrating recent developments and current opportunities. Curr Pharmacol Rep 3:36–49
Freise KJ, Shebley M, Salem AH (2017) Quantitative prediction of the effect of CYP3A inhibitors and inducers on venetoclax pharmacokinetics using a physiologically based pharmacokinetic model. J Clin Pharmacol 57:796–804
Gertz M, Cartwright CM, Hobbs MJ, Kenworthy KE, Rowland M, Houston JB, Galetin A (2013) Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug-drug interaction potential. Pharm Res 30:761–780
Gidal BE, Maganti R, Laurenza A, Yang H, Verbel DA, Schuck E, Ferry J (2017) Effect of enzyme inhibition on perampanel pharmacokinetics: why study design matters. Epilepsy Res 134:41–48
Greupink R, Schreurs M, Benne MS, Huisman MT, Russel FG (2013) Semi-mechanistic physiologically-based pharmacokinetic modeling of clinical glibenclamide pharmacokinetics and drug–drug-interactions. Eur J Pharm Sci 49:819–828
Grillo JA, Zhao P, Bullock J, Booth BP, Lu M, Robie-Suh K, Berglund EG, Pang KS, Rahman A, Zhang L, Lesko LJ, Huang SM (2012) Utility of a physiologically-based pharmacokinetic (PBPK) modeling approach to quantitatively predict a complex drug-drug-disease interaction scenario for rivaroxaban during the drug review process: implications for clinical practice. Biopharm Drug Dispos 33:99–110
Guest EJ, Aarons L, Houston JB, Rostami-Hodjegan A, Galetin A (2011) Critique of the two-fold measure of prediction success for ratios: application for the assessment of drug-drug interactions. Drug Metab Dispos 39:170–173
Gufford BT, Barr JT, Gonzalez-Perez V, Layton ME, White JR Jr, Oberlies NH, Paine MF (2015) Quantitative prediction and clinical evaluation of an unexplored herb-drug interaction mechanism in healthy volunteers. CPT Pharm Syst Pharmacol 4:701–710
Guo J, Zhou D, Li Y, Khanh BH (2015) Physiologically based pharmacokinetic modeling to predict complex drug-drug interactions: a case study of AZD2327 and its metabolite, competitive and time-dependent CYP3A inhibitors. Biopharm Drug Dispos 36:507–519
Gupta N, Hanley MJ, Venkatakrishnan K, Bessudo A, Rasco DW, Sharma S, O’Neil BH, Wang B, Liu G, Ke A, Patel C, Rowland Yeo K, Xia C, Zhang X, Esseltine DL, Nemunaitis J (2017) Effects of strong CYP3A inhibition and induction on the pharmacokinetics of Ixazomib, an oral proteasome inhibitor: Results of drug-drug interaction studies in patients with advanced solid tumors or lymphoma and a physiologically based pharmacokinetic analysis. J Clin Pharmacol
Hajjar ER, Cafiero AC, Hanlon JT (2007) Polypharmacy in elderly patients. Am J Geriatr Pharmacother 5:345e51
Han B, Mao J, Chien JY, Hall SD (2013) Optimization of drug-drug interaction study design: comparison of minimal physiologically based pharmacokinetic models on prediction of CYP3A inhibition by ketoconazole. Drug Metab Dispos 41:1329–1338
Hartmanshenn C, Scherholz M, Androulakis IP (2016) Physiologically-based pharmacokinetic models: approaches for enabling personalized medicine. J Pharmacokinet Pharmacodyn 43:481–504
Hsu V, de LTVM, Zhao P, Zhang L, Zheng JH, Nordmark A, Berglund EG, Giacomini KM, Huang SM (2014) Towards quantitation of the effects of renal impairment and probenecid inhibition on kidney uptake and efflux transporters, using physiologically based pharmacokinetic modelling and simulations. Clin Pharmacokinet 53:283–293
Huang SM, Abernethy DR, Wang Y, Zhao P, Zineh I (2013) The utility of modeling and simulation in drug development and regulatory review. J Pharm Sci 102:2912–2923
Huang W, Nakano M, Sager JE, Ragueneau-Majlessi I, Isoherranen N (2017) Physiologically based pharmacokinetic (PBPK) model of the CYP2D6 Probe atomoxetine: extrapolation to special populations and drug-drug interactions. Drug Metab Dispos 45:1156–1165
Hyland R, Dickins M, Collins C, Jones H, Jones B (2008) Maraviroc: in vitro assessment of drug-drug interaction potential. Br J Clin Pharmacol 66:498–507
Hynes SM, Wickremsinhe E, Zhang W, Decker R, Ott J, Chandler J, Mitchell M (2015) Evaluation of the likelihood of a selective CHK1 inhibitor (LY2603618) to inhibit CYP2D6 with desipramine as a probe substrate in cancer patients. Biopharm Drug Dispos 36:49–63
Jamei M, Dickinson GL, Rostami-Hodjegan A (2009a) A framework for assessing inter-individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: a tale of ‘bottom-up’ vs ‘top-down’ recognition of covariates. Drug Metab Pharmacokinet 24:53–75
Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, Tucker G (2009b) Population-based mechanistic prediction of oral drug absorption. AAPS J 11:225–237
Jamei M, Bajot F, Neuhoff S, Barter Z, Yang J, Rostami-Hodjegan A, Rowland-Yeo K (2014) A mechanistic framework for in vitro-in vivo extrapolation of liver membrane transporters: prediction of drug-drug interaction between rosuvastatin and cyclosporine. Clin Pharmacokinet 53:73–87
Jiang X, Zhuang Y, Xu Z, Wang W, Zhou H (2016) Development of a physiologically based pharmacokinetic model to predict disease-mediated therapeutic protein-drug interactions: modulation of multiple cytochrome p450 enzymes by interleukin-6. AAPS J 18:767–776
Jing J, Nelson C, Paik J, Shirasaka Y, Amory JK, Isoherranen N (2017) Physiologically based pharmacokinetic model of all-trans-retinoic acid with application to cancer populations and drug interactions. J Pharmacol Exp Ther 361:246–258
Johnson TN, Rostami-Hodjegan A (2011) Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatr Anaesth 21:291–301
Jones HM, Parrott N, Jorga K, Lave T (2006) A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet 45:511–542
Jones HM, Mayawala K, Poulin P (2013) Dose selection based on physiologically based pharmacokinetic (PBPK) approaches. AAPS J 15:377–387
Jones HM, Chen Y, Gibson C, Heimbach T, Parrott N, Peters SA, Snoeys J, Upreti VV, Zheng M, Hall SD (2015) Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther 97:247–262
Ke A, Barter Z, Rowland-Yeo K, Almond L (2016) Towards a best practice approach in PBPK Modeling: case example of developing a unified efavirenz model accounting for induction of CYPs 3A4 and 2B6. CPT Pharmacometrics Syst Pharmacol 5:367–376
Khalil F, Laer S (2011) Physiologically based pharmacokinetic modeling: methodology, applications, and limitations with a focus on its role in pediatric drug development. J Biomed Biotechnol 907461
Kim SJ, Toshimoto K, Yao Y, Yoshikado T, Sugiyama Y (2017) Quantitative analysis of complex drug-drug interactions between repaglinide and cyclosporin A/gemfibrozil using physiologically based pharmacokinetic models with in vitro transporter/enzyme inhibition data. J Pharm Sci
Koenen A, Kroemer HK, Grube M, Meyer zu Schwabedissen HE (2011) Current understanding of hepatic and intestinal OATP-mediated drug-drug interactions. Expert Rev Clin Pharmacol 4:729–742
Kudo T, Hisaka A, Sugiyama Y, Ito K (2013) Analysis of the repaglinide concentration increase produced by gemfibrozil and itraconazole based on the inhibition of the hepatic uptake transporter and metabolic enzymes. Drug Metab Dispos 41:362–371
Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, Block M, Eissing T, Teutonico D (2016) Applied concepts in PBPK modeling: how to build a PBPK/PD Model. CPT Pharmacometrics Syst Pharmacol 5:516–531
Li J, Kim S, Sha X, Wiegand R, Wu J, LoRusso P (2014) Complex disease-, gene-, and drug-drug interactions: impacts of renal function, CYP2D6 phenotype, and OCT2 activity on veliparib pharmacokinetics. Clin Cancer Res 20:3931–3944
Lin W, Ji T, Einolf H, Ayalasomayajula S, Lin TH, Hanna I, Heimbach T, Breen C, Jarugula V, He H (2017) Evaluation of drug-drug interaction potential between sacubitril/valsartan (LCZ696) and statins using a physiologically based pharmacokinetic model. J Pharm Sci 106:1439–1451
Lu C, Suri A, Shyu WC, Prakash S (2014) Assessment of cytochrome P450-mediated drug-drug interaction potential of orteronel and exposure changes in patients with renal impairment using physiologically based pharmacokinetic modeling and simulation. Biopharm Drug Dispos 35:543–552
Lynch T, Price A (2007) The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician 76:391–396
Mamidi R, Dallas S, Sensenhauser C, Lim HK, Scheers E, Verboven P, Cuyckens F, Leclercq L, Evans DC, Kelley MF, Johnson MD, Snoeys J (2017) In vitro and physiologically-based pharmacokinetic based assessment of drug-drug interaction potential of canagliflozin. Br J Clin Pharmacol 83:1082–1096
Mano Y, Sugiyama Y, Ito K (2015) Use of a physiologically based pharmacokinetic model for quantitative prediction of drug-drug interactions via CYP3A4 and estimation of the intestinal availability of CYP3A4 substrates. J Pharm Sci 104:3183–3193
Mao J, Fan P, Wong S, Wang J, Ismaili MHA, Dean B, Hop CECA, Wright M, Chen Y (2017) Strategy for CYP3A induction risk assessment from preclinical signal to human: a Case example of a late-stage discovery compound. Pharm Res
Marcantonio EE, Ballard J, Gibson CR, Kassahun K, Palamanda J, Tang C, Evers R, Liu C, Zajic S, Mahon C, Mostoller K, Hreniuk D, Mehta A, Morris D, Wagner JA, Stoch SA (2014) Prednisone has no effect on the pharmacokinetics of CYP3A4 metabolized drugs—midazolam and odanacatib. J Clin Pharmacol 54:1280–1289
Marsousi N, Daali Y, Rudaz S, Almond L, Humphries H, Desmeules J, Samer CF (2014) Prediction of metabolic interactions with oxycodone via CYP2D6 and CYP3A inhibition using a physiologically based pharmacokinetic model. CPT Pharm Syst Pharmacol 3:e152
Marsousi N, Desmeules JA, Rudaz S, Daali Y (2017) Prediction of drug-drug interactions using physiologically-based pharmacokinetic models of CYP450 modulators included in Simcyp software. Biopharm Drug Dispos
Marzolini C, Rajoli R, Battegay M, Elzi L, Back D, Siccardi M (2017) Physiologically based pharmacokinetic modeling to predict drug-drug interactions with efavirenz involving simultaneous inducing and inhibitory effects on cytochromes. Clin Pharmacokinet 56:409–420
Min JS, Kim D, Park JB, Heo H, Bae SH, Seo JH, Oh E, Bae SK (2016) Application of physiologically based pharmacokinetic modeling in predicting drug-drug interactions for sarpogrelate hydrochloride in humans. Drug Des Dev Ther 10:2959–2972
Moj D, Hanke N, Britz H, Frechen S, Kanacher T, Wendl T, Haefeli WE, Lehr T (2017) Clarithromycin, midazolam, and digoxin: application of PBPK modeling to gain new insights into drug-drug interactions and co-medication regimens. AAPS J 19:298–312
Moltó J, Rajoli R, Back D, Valle M, Miranda C, Owen A, Clotet B, Siccardi M (2017) Use of a physiologically based pharmacokinetic model to simulate drug-drug interactions between antineoplastic and antiretroviral drugs. J Antimicrob Chemother 72:805–811
Nakamaru Y, Emoto C, Shimizu M, Yamazaki H (2015) Human pharmacokinetic profiling of the dipeptidyl peptidase-IV inhibitor teneligliptin using physiologically based pharmacokinetic modeling. Biopharm Drug Dispos 36:148–162
Navid A, Ng DM, Wong SE, Lightstone FC (2016) Application of a physiologically based pharmacokinetic model to study theophylline metabolism and its interactions with ciprofloxacin and caffeine. CPT Pharm Syst Pharmacol 5:74–81
Neuhoff S, Yeo KR, Barter Z, Jamei M, Turner DB, Rostami-Hodjegan A (2013) Application of permeability-limited physiologically-based pharmacokinetic models: part II-prediction of P-glycoprotein mediated drug-drug interactions with digoxin. J Pharm Sci 102:3161–3173
Nordmark A, Andersson A, Baranczewski P, Wanag E, Stahle L (2014) Assessment of interaction potential of AZD2066 using in vitro metabolism tools, physiologically based pharmacokinetic modelling and in vivo cocktail data. Eur J Clin Pharmacol 70:167–178
Olafuyi O, Coleman M, Badhan RKS (2017a) Development of a paediatric physiologically based pharmacokinetic model to assess the impact of drug-drug interactions in tuberculosis co-infected malaria subjects: a case study with artemether-lumefantrine and the CYP3A4-inducer rifampicin. Eur J Pharm Sci 106:20–33
Olafuyi O, Coleman M, Badhan RKS (2017b) The application of physiologically-based pharmacokinetic modelling to assess the impact of antiretroviral-mediated drug-drug interactions on piperaquine antimalarial therapy during pregnancy. Biopharm Drug Dispos
Ono C, Hsyu PH, Abbas R, Loi CM, Yamazaki S (2017) Application of physiologically based pharmacokinetic modeling to the understanding of bosutinib pharmacokinetics: prediction of drug-drug and drug-disease interactions. Drug Metab Dispos 45:390–398
Pahwa S, Alam K, Crowe A, Farasyn T, Neuhoff S, Hatley O, Ding K, Yue W (2017) Pretreatment with rifampicin and tyrosine kinase inhibitor dasatinib potentiates the inhibitory effects toward OATP1B1- and OATP1B3-mediated transport. J Pharm Sci 106:2123–2135
Park MH, Shin SH, Byeon JJ, Lee GH, Yu BY, Shin YG (2017) Prediction of pharmacokinetics and drug-drug interaction potential using physiologically based pharmacokinetic (PBPK) modeling approach: a case study of caffeine and ciprofloxacin. Korean J Physiol Pharmacol 2:107–115
Perdaems N, Blasco H, Vinson C, Chenel M, Whalley S, Cazade F, Bouzom F (2010) Predictions of metabolic drug-drug interactions using physiologically based modelling two cytochrome P450 3A4 Substrates coadministered with ketoconazole or verapamil. Clin Pharmacokinet 49:239–258
Pfeifer ND, Goss SL, Swift B, Ghibellini G, Ivanovic M, Heizer WD, Gangarosa LM, Brouwer KL (2013) Effect of ritonavir on (99m)Technetium-mebrofenin disposition in humans: a semi-PBPK modeling and in vitro approach to predict transporter-mediated DDIs. CPT Pharm Syst Pharmacol 2:e20
Posada MM, Bacon JA, Schneck KB, Tirona RG, Kim RB, Higgins JW, Pak YA, Hall SD, Hillgren KM (2015) Prediction of renal transporter mediated drug-drug interactions for pemetrexed using physiologically based pharmacokinetic modeling. Drug Metab Dispos 43:325–334
Posada MM, Cannady EA, Payne CD, Zhang X, Bacon JA, Pak YA, Higgins JW, Shahri N, Hall SD, Hillgren KM (2017) Prediction of transporter-mediated drug-drug interactions for baricitinib. Clin Transl Sci
Qi F, Zhu L, Li N, Ge T, Xu G, Liao S (2017) Influence of different proton pump inhibitors on the pharmacokinetics of voriconazole. Int J Antimicrob Agents 49:403–409
Rangaraj N, Matthew H, Gondi K, Sekhar S (2016) Application of a “fit for purpose” PBPK model to investigate the CYP3A4 induction potential of enzalutamide. Drug Metab Lett 10:172–179
Rekić D, Röshammar D, Mukonzo J, Ashton M (2011) In silico prediction of efavirenz and rifampicin drug-drug interaction considering weight and CYP2B6 phenotype. Br J Clin Pharmacol 71:536–543
Rougee LRA, Mohutsky MA, Bedwell DW, Ruterbories KJ, Hall SD (2017) The impact of the hepatocyte-to-plasma pH gradient on the prediction of hepatic clearance and drug-drug interactions for CYP2C9 and CYP3A4 substrates. Drug Metab Dispos 45:1008–1018
Rowland M, Peck C, Tucker G (2011) Physiologically-based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol 51:45–73
Rowland A, Mangoni AA, Hopkins A, Sorich MJ, Rowland A (2016) Optimized cocktail phenotyping study protocol using physiological based pharmacokinetic modeling and in silico assessment of metabolic drug-drug interactions involving modafinil. Front Pharmacol 27:517
Rowland-Yeo K, Jamei M, Yang J, Tucker GT, Rostami-Hodjegan A (2010) Physiologically based mechanistic modelling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut—the effect of diltiazem on the time-course of exposure to triazolam. Eur J Pharm Sci 39:298–309
Rowland-Yeo K, Jamei M, Rostami-Hodjegan A (2013) Predicting drug-drug interactions: application of physiologically based pharmacokinetic models under a systems biology approach. Expert Rev Clin Pharmacol 6:143–157
Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N (2015) Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos 43:1823–1837
Shebley M, Fu W, Badri P, Bow D, Fischer V (2017a) Physiologically based pharmacokinetic modeling suggests limited drug-drug interaction between clopidogrel and dasabuvir. Clin Pharmacol Ther 102:679–687
Shebley M, Liu J, Kavetskaia O, Sydor J, De Morais SM, Fischer V, Nijsen M, Bo DAJ (2017b) Mechanisms and predictions of drug-drug interactions of the hepatitis C virus three direct-acting antiviral regimen: paritaprevir/ritonavir, ombitasvir, and dasabuvir. Drug Metab Dispos 45:755–764
Shi JG, Fraczkiewicz G, Williams WV, Yeleswaram S (2015) Predicting drug-drug interactions involving multiple mechanisms using physiologically based pharmacokinetic modeling: a case study with ruxolitinib. Clin Pharmacol Ther 97:177–185
Shitara Y, Sugiyama Y (2017) Preincubation-dependent and long-lasting inhibition of organic anion transporting polypeptide (OATP) and its impact on drug-drug interactions. Pharmacol Ther 177:67–80
Shugarts S, Benet LZ (2008) The role of transporters in the pharmacokinetics of orally administered drugs. Pharm Res 26:2039–2054
Siccardi M, Marzolini C, Seden K, Almond L, Kirov A, Khoo S, Owen A, Back D (2013a) Prediction of drug-drug interactions between various antidepressants and efavirenz or boosted protease inhibitors using a physiologically based pharmacokinetic modelling approach. Clinical Pharmacokinetics 52:583–592
Siccardi M, Olagunju A, Seden K, Ebrahimjee F, Rannard S, Back D, Owen A (2013b) Use of a physiologically-based pharmacokinetic model to simulate artemether dose adjustment for overcoming the drug-drug interaction with efavirenz. In Silico Pharmacol 1:4
Sissung TM, Baum CE, Kirkland CT, Gao R, Gardner ER, Figg WD (2010) Pharmacogenetics of membrane transporters: an update on current approaches. Mol Biotechnol 44:152–167
Snoeys J, Beumont M, Monshouwer M, Ouwerkerk-Mahadevan S (2016) Mechanistic understanding of the nonlinear pharmacokinetics and intersubject variability of simeprevir: a PBPK-guided drug development approach. Clin Pharmacol Ther 99:224–234
Templeton I, Ravenstijn P, Sensenhauser C, Snoeys J (2016) A physiologically based pharmacokinetic modeling approach to predict drug-drug interactions between domperidone and inhibitors of CYP3A4. Biopharm Drug Dispos 37:15–27
Teorell T (1937) Studies on the diffusion effect upon ionic distribution: II. Experiments on ionic accumulation. J Gen Physiol 21:107–122
Tornio A, Filppula AM, Kailari O, Neuvonen M, Nyronen TH, Tapaninen T, Neuvonen PJ, Niemi M, Backman JT (2014) Glucuronidation converts clopidogrel to a strong time-dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug-drug interactions. Clin Pharmacol Ther 96:498–507
Tsamandouras N, Rostami-Hodjegan A, Aarons L (2015) Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br J Clin Pharmacol 79:48–55
Varma MV, Lai Y, Feng B, Litchfield J, Goosen TC, Bergman A (2012) Physiologically based modeling of pravastatin transporter-mediated hepatobiliary disposition and drug-drug interactions. Pharm Res 29:2860–2873
Varma MV, Lai Y, Kimoto E, Goosen TC, El-Kattan AF, Kumar V (2013) Mechanistic modeling to predict the transporter- and enzyme-mediated drug-drug interactions of repaglinide. Pharm Res 30:1188–1199
Varma MV, Scialis RJ, Lin J, Bi YA, Rotter CJ, Goosen TC, Yang X (2014) Mechanism-based pharmacokinetic modeling to evaluate transporter-enzyme interplay in drug interactions and pharmacogenetics of glyburide. AAPS J 16:736–748
Varma MV, Lin J, Bi YA, Kimoto E, Rodrigues AD (2015a) Quantitative rationalization of gemfibrozil drug interactions: consideration of transporters-enzyme interplay and the role of circulating metabolite gemfibrozil 1-o-beta-glucuronide. Drug Metab Dispos 43:1108–1118
Varma MV, Pang KS, Isoherranen N, Zhao P (2015b) Dealing with the complex drug-drug interactions: towards mechanistic models. Biopharm Drug Dispos 36:71–92
Varma MV, Kimoto E, Scialis R, Bi Y, Lin J, Eng H, Kalgutkar AS, El-Kattan AF, Rodrigues AD, Tremaine LM (2017) Transporter-mediated hepatic uptake plays an important role in the pharmacokinetics and drug-drug interactions of montelukast. Clin Pharmacol Ther 101:406–415
Vieira ML, Zhao P, Berglund EG, Reynolds KS, Zhang L, Lesko LJ, Huang SM (2012) Predicting drug interaction potential with a physiologically based pharmacokinetic model: a case study of telithromycin, a time-dependent CYP3A inhibitor. Clin Pharmacol Ther 91:700–708
Vieira MD, Kim MJ, Apparaju S, Sinha V, Zineh I, Huang SM, Zhao P (2014) PBPK model describes the effects of comedication and genetic polymorphism on systemic exposure of drugs that undergo multiple clearance pathways. Clin Pharmacol Ther 95:550–557
Von Moltke LL, Greenblatt DJ, Schmider J, Wright CE, Harmatz JS, Shader RI (1998) In vitro approaches to predicting drug interactions in vivo. Biochem Pharmacol 55:113–122
Wagner C, Zhao P, Pan Y, Hsu V, Grillo J, Huang SM, Sinha V (2015) Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA Public Workshop on PBPK. CPT Pharm Syst Pharmacol 4:226–230
Wagner C, Zhao P, Arya V, Mullick C, Struble K, Au S (2017) Physiologically based pharmacokinetic modeling for predicting the effect of intrinsic and extrinsic factors on darunavir or lopinavir exposure coadministered with ritonavir. J Clin Pharmacol
Wang J, Xia S, Xue W, Wang D, Sai Y, Liu L, Liu X (2013) A semi-physiologically-based pharmacokinetic model characterizing mechanism-based auto-inhibition to predict stereoselective pharmacokinetics of verapamil and its metabolite norverapamil in human. Eur J Pharm Sci 50:290–302
Wang Q, Zheng M, Leil T (2017) Investigating transporter-mediated drug-drug interactions using a physiologically based pharmacokinetic model of rosuvastatin. CPT Pharmacometrics Syst Pharmacol 6:228–238
Wisniowska B, Polak S (2016) Virtual Clinical Trial Toward polytherapy safety assessment: combination of physiologically based pharmacokinetic/pharmacodynamic-based modeling and simulation approach with drug-drug interactions involving terfenadine as an example. J Pharm Sci 105:3415–3424
World Health Organization (WHO) Collaborating centre for drug statistics methodology. http://www.whocc.no. Accessed March 1, 2017
Xia B, Barve A, Heimbach T, Zhang T, Gu H, Wang L, Einolf H, Alexander N, Hanna I, Ke J, Mangold JB, He H, Sunkara G (2014) Physiologically based pharmacokinetic modeling for assessing the clinical drug-drug interaction of alisporivir. Eur J Pharm Sci 63:103–112
Xu Y, Hijazi Y, Wolf A, Wu B, Sun YN, Zhu M (2015) Physiologically based pharmacokinetic model to assess the influence of blinatumomab-mediated cytokine elevations on cytochrome P450 enzyme activity. CPT Pharm Syst Pharmacol 4:507–515
Yamashita F, Sasa Y, Yoshida S, Hisaka A, Asai Y, Kitano H, Hashida M, Suzuki H (2013) Modeling of rifampicin-induced CYP3A4 activation dynamics for the prediction of clinical drug-drug interactions from in vitro data. PLoS ONE 8:e70330
Yamazaki S, Johnson TR, Smith BJ (2015) Prediction of drug-drug interactions with crizotinib as the CYP3A substrate using a physiologically based pharmacokinetic model. Drug Metab Dispos 43:1417–1429
Yoshikado T, Yoshida K, Kotani N, Nakada T, Asaumi R, Toshimoto K, Maeda K, Kusuhara H, Sugiyama Y (2016) Quantitative analyses of hepatic OATP-mediated interactions between statins and inhibitors using PBPK modeling with a parameter optimization method. Clin Pharmacol Ther 100:513–523
Yoshikado T, Kazuya M, Kusuhara H, Furihata KI, Sugiyama Y (2017) Quantitative analyses of the influence of parameters governing rate-determining process of hepatic elimination of drugs on the magnitudes of drug-drug interactions via hepatic OATPs and CYP3A using physiologically-based pharmacokinetic models. J Pharm Sci 106:2739–2750
Yu Y, Loi CM, Hoffman J, Wang D (2016) Physiologically based pharmacokinetic modeling of palbociclib. J Clin Pharmacol 57:173–184
Zhang T (2015) Physiologically based pharmacokinetic modeling of disposition and drug-drug interactions for atorvastatin and its metabolites. Eur J Pharm Sci 77:216–229
Zhang L, Zhang YD, Zhao P, Huang SM (2009) Predicting drug-drug interactions: an FDA perspective. AAPS J 11:300–306
Zhang H, Bu F, Li L, Jiao Z, Ma G, Cai W, Zhuang X, Lin HS, Shin JG, Xiang X. (2017) Prediction of drug-drug interaction between tacrolimus and principal ingredients of Wuzhi Capsule in Chinese healthy volunteers using physiologically-based pharmacokinetic modelling. Basic Clin Pharmacol Toxicol
Zhao Y, Hu ZY (2014) Physiologically based pharmacokinetic modelling and in vivo [I]/K(i) accurately predict P-glycoprotein-mediated drug-drug interactions with dabigatran etexilate. Br J Pharmacol 171:1043–1053
Zhou SF, Liu JP, Chowbay B (2009) Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab Rev 41:89–295
Zhou D, Bui K, Sostek M, Al-Huniti N (2016) Simulation and prediction of the drug-drug interaction potential of naloxegol by physiologically based pharmacokinetic modeling. CPT Pharm Syst Pharmacol 5:250–257
Zhuang X, Lu C (2016) PBPK modeling and simulation in drug research and development. Acta Pharm Sin B 6:430–440
Acknowledgements
This research was supported by the Bio & Medical Technology Development Program (No. 2013M3A9B5075838) and the Basic Research Laboratory (BRL) Program (2015R1A4A1042350) through the National Research Foundation of Korea grant funded by the Ministry of Education, Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Min, J.S., Bae, S.K. Prediction of drug–drug interaction potential using physiologically based pharmacokinetic modeling. Arch. Pharm. Res. 40, 1356–1379 (2017). https://doi.org/10.1007/s12272-017-0976-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-017-0976-0