
Abstract
Innate lymphoid cells (ILCs) are a recently defined type of innate-immunity cells that belong to the lymphoid lineage and have lymphoid morphology but do not express an antigen-specific B cell or T-cell receptor. ILCs regulate immune functions prior to the formation of adaptive immunity and exert effector functions through a cytokine release. ILCs have been classified into three groups according to the transcription factors that regulate their development and function and the effector cytokines they produce. Of note, ILCs resemble T helper (Th) cells, such as Th1, Th2, and Th17 cells, and show a similar dependence on transcription factors and distinct cytokine production. Despite their short history in immunology, ILCs have received much attention, and numerous studies have revealed biological functions of ILCs including host defense against pathogens, inflammation, tissue repair, and metabolic homeostasis. Here, we describe recent findings about the roles of ILCs in the pathogenesis of various diseases and potential therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The immune system has long been dichotomized into innate immunity, which consists of myeloid cells and other nonlymphoid cells, and acquired (adaptive) immunity, which is composed of the B cells and T cells that mediate humoral and cellular immunity. Nonetheless, recent studies have discovered a previously unrecognized group of innate immune cells that lack antigen specificity but belong to the lymphoid lineage, so-called innate lymphoid cells (ILCs) (Spits and Cupedo 2012). Unlike T and B cells, ILCs do not express antigen-specific T- or B-cell receptors, and do not possess markers of myeloid or dendritic cells (Walker et al. 2013). ILCs are mainly enriched at mucosal and nonmucosal barriers acting as first-line defenders, but they are also found in lymphoid and nonlymphoid tissues (Diefenbach et al. 2014). ILCs originate from common lymphoid progenitors (CLPs) and depend on a transcriptional repressor, inhibitor of DNA binding 2 (ID2) and cytokines that signal through the common γ-chain of IL-2 (Lim et al. 2017). The lack of common lineage markers and IL-7Rα are also shared features of this family of cells.
ILCs have been classified into three distinct groups according to the transcription factors that regulate their development, function, and cytokine production (Spits et al. 2013; Diefenbach et al. 2014; McKenzie et al. 2014) (Fig. 1). Furthermore, ILCs closely resemble T helper (Th) cells, such as Th1, Th2, and Th17 lineages, and show a similar dependence on transcription factors and produce distinct cytokines (Spits and Di Santo 2011). Group 1 ILCs (ILC1s) express T-box transcription factor (T-bet) and produce IFN-γ, as Th1 cells do (Bernink et al. 2013). ILC2s depend on GATA-binding protein 3 (GATA3) and retinoic acid receptor–related orphan receptor α (ROR-α) and produce Th2 cytokines, such as IL-4, IL-5, IL-9, and IL-13 (Turner et al. 2013; Walker et al. 2013). ILC3s, like Th17 cells, express RORγt and secrete IL-17A and IL-22 (Sanos and Diefenbach 2013).

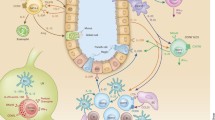
Development and transdifferentiation of the ILC family. All ILC groups are derived from common lymphoid progenitors (CLPs). CLPs develop into NK cell precursors (NKPs) or common helper innate lymphoid precursors (CHILPs). CHILPs next differentiate into ILC progenitors (ILCPs) or LTi precursors (LTiPs). T-bet+ ILC1s consist of NK cells derived from NKPs and ILC1s from ILCPs. ILC1s are further categorized into two subgroups: CD127+ ILC1s and CD103+ intraepithelial ILC1s (ieILC1s). All RORα+GATA3+ ILC2s are derived from ILCPs. ILC2s express various cytokine receptors, such as CD25, CD127, CD132, ST2, IL-17RB, and TSLPR. RORγt+ ILC3s consist of NCR+ ILC3s from ILCPs, CCR6+ LTi cells from LTiPs, and heterogeneous NCR−CCR6− double-negative (DN)-ILC3s. IL-1β and IL-4 drive the transdifferentiation of ILC1s into ILC2s, whereas IL-23, IL-1β, and retinoic acid promote the transdifferentiation of ILC1s into ILC3s. Transdifferentiation can also be reversed from ILC2s and ILC3s to ILC1s in response to IL-12
Of note, studies indicate that ILC2s and ILC3s have the ability to transdifferentiate into T–bet+ IFN-γ–secreting ILC1s in the presence of IL-12, at the cost of GATA3 and RORγt expression, respectively (Bernink et al. 2013; Klose et al. 2013, 2014). Conversely, IL-1β and IL-4 drive the transdifferentiation of ILC1s into ILC2s, whereas IL-23, IL-1β, and retinoic acid promote the transdifferentiation of ILC1s into ILC3s (Bernink et al. 2015; Bal et al. 2016; Ohne et al. 2016; Silver et al. 2016). Nevertheless, transdifferentiation between ILC2s and ILC3s has not been reported.
Since the discovery of ILCs, various functions of these cells in host defense against pathogens and in the regulation of inflammation, carcinogenesis, tissue repair, and metabolic homeostasis have been extensively studied (Spits and Di Santo 2011; Spits and Cupedo 2012; McKenzie et al. 2014). In this review, we summarize the latest research on ILCs and their various pathogenic roles and discuss their potential as therapeutic targets.
General features of ILC1s
ILC1s expressing T-bet and secreting IFN-γ can be subdivided into three subpopulations (Vonarbourg et al. 2010; Bernink et al. 2013; Fuchs et al. 2013; Klose et al. 2014). One expresses CD56 and CD103 and is found in tonsils and in the intestinal intraepithelial space. CD103 binds to integrin β7, a ligand of e-cadherin, and enhances epithelial-cell interactions. CD103+ ILC1s or intraepithelial ILC1s (ieILC1s) produce IFN-γ when stimulated with IL-12, IL-15, and IL-18 (Vosshenrich et al. 2006; Chiossone et al. 2009; Klose et al. 2014) and have granules containing perforin and granzyme B that can lyse target cells. Another cell population expresses CD127 and CD161 but is deficient in CD56, CD94, granzyme B, and perforin (Bernink et al. 2013). These CD127+ ILC1s express T-bet but lack Eomes and reside in the lamina propria (Gordon et al. 2012). CD127+ ILC1s produce IFN-γ in response to IL-12 and IL-18 and are significantly involved in host protection from intracellular pathogens.
From the viewpoint of IFN-γ production, which is the unifying feature of ILC1s, conventional natural killer (cNK) cells can also be classified as ILC1s (McKenzie et al. 2014). Both cNK cells and ieILC1s produce IFN-γ in response to IL-12, IL-15, and IL-18 and exert cytotoxic action through the production of perforin and granzyme B, whereas CD127+ ILC1s are devoid of perforin and granzyme B and are noncytotoxic. cNK cells and other ILC1 populations in mice share cell surface markers such as NKG2D, NK1.1, and NKp46 and transcription factors T-bet and Eomes (Cortez et al. 2014; Daussy et al. 2014; Sojka et al. 2014). In humans, cNK cells and other ILC1s also express transcription factors T-bet and Eomes (Nielsen et al. 2012; Bernink et al. 2015; Bjorklund et al. 2016; Michel et al. 2016; Roan et al. 2016). In this regard, precise discrimination of cNK cells from other ILC1 populations by means of the current criteria is complicated and hard despite ongoing identification of cell surface markers for discrimination (Table 1).
The role of ILC1s in infections
ILC1s participate in the innate immune response to intracellular bacteria, viruses, and parasites. In mice, CD127+ ILC1s prevent infection by the protozoan parasite Toxoplasma gondii by recruiting myeloid cells (Klose et al. 2014; McKenzie et al. 2014). Defense against acute Clostridium difficile infection also appears to be mediated by ILC1s and IFN-γ because mice lacking ILC1s or deficient in IFN-γ are more susceptible to lethal C. difficile infection (Abt et al. 2015). Yang et al. (2015) reported that in humans, the number of ILC1s and their IFN-γ production increase during hepatitis B virus infection, and this upregulation of ILC1 is significantly associated with liver damage in patients with chronic hepatitis B, indicating proinflammatory effects of ILC1s on the pathogenesis of chronic hepatitis B (Table 2).
ILC1s as a disease factor
IFN-γ-secreting ILC1s are likely to be involved in inflammation because an increase in the numbers of ILC1s has been observed in many inflammatory diseases. ILC1 numbers are increased in inflamed intestinal samples from patients with Crohn’s disease (CD) and in bronchial samples from patients with chronic obstructive pulmonary disease (COPD), whereas the ILC3 and ILC2 populations are smaller in CD and COPD patients, respectively. Similarly, Braudeau et al. (2016) reported an increase in ILC1 numbers, at the cost of ILC2s and ILC3s, in acute-phase anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV).
CD56bright ILC1–like cells have been detected in the synovial fluid and synovial tissue of patients with inflammatory arthritis, most of whom had rheumatoid arthritis (RA). These ILC1-like cells produce IFN-γ when they are exposed to IL-2, IL-12, and IL-15 (Dalbeth and Callan 2002). Upon stimulation by IL-12, IL-15, and IL-18, activated ILC1-like cells cause CD14+ monocytes to produce TNF-α through direct cell-to-cell contact. Conversely, CD14+ monocytes synergize with cytokines to promote IFN-γ production by ILC1-like cells (Dalbeth et al. 2004). In addition, increased proportions of ILC1-like cells in the peripheral blood of patients with systemic lupus erythematosus (SLE) have been reported (Schepis et al. 2009). Nonetheless, it has not been determined whether the observed CD56bright cells are cNK cells or noncytotoxic ILC1s. Given the results of a recent study showing the enrichment of ILC1s in the synovial fluid of RA patients as compared to the levels in patients with psoriatic arthritis (Leijten et al. 2015), future studies are needed to determine whether these CD56bright cells are ILC1s.
An increase in ILC1 numbers has also been found in the joints of patients with spondyloarthritis and in peripheral blood of patients with systemic sclerosis (SSc) along with NKp44− ILC3s and NKp44+ ILC3s, respectively (Yeremenko et al. 2015; Roan et al. 2016). In SSc, the frequency of ILC1s increases due to significant changes in CD4+ ILC1s, and these CD4+ ILC1s, unlike CD4− ILC1s, produce TNF-α and GM-CSF, which are potent cytokines that promote SSc.
ILC1s as therapeutic targets
ILC1s have been shown to cause inflammation in several cases; therefore, block-ing IFN-γ is a possible way to weaken an ILC1-associated inflammatory disease. Thus, administration of an anti–IFN-γ antibody (Ab) may relieve the disease, and this approach is considered a promising therapeutic strategy against various inflammatory diseases, such as CD, RA, SLE, and SSc. Fontolizumab (marketed under the trade name HuZAF™), a humanized monoclonal Ab against IFN-γ, is an immunosuppressive therapeutic used for the treatment of autoimmune diseases such as CD and RA. Studies have shown that fontolizumab has excellent tolerability and clinical activity in patients with moderate to severe CD and is currently in a phase II clinical trial (Hommes et al. 2006). Nevertheless, its use against RA in a phase II clinical trial (NCT00281294) was terminated due to failure to meet the endpoint of the first phase. AMG-811 is a human monoclonal Ab against IFN-γ developed by Amgen, and its pharmacokinetic and pharmacodynamic profile in patients with SLE has been determined (Chen et al. 2015). AMG-811 has no significant therapeutic effects in patients with discoid lupus erythematosus despite its tolerability and the changes induced in biomarkers associated with IFN-γ (Werth et al. 2017).
In addition to IFN-γ, another ILC1-targeting stimulatory molecule, IL-12, is an alternative target for the treatment of CD. Ustekinumab (Stelara®) and briakinumab are monoclonal Abs targeting IL-12p40 and are potential therapeutic agents for CD (Sandborn et al. 2012; Niederreiter et al. 2013).
General features of ILC2s
CD4+ Th2 cells have long been considered the major regulators of allergic inflammation because of their production of Th2 cytokines, such as IL-4, IL-5, and IL-13 (Doherty and Broide 2015; Halim 2016). On the other hand, ILC2s recently emerged as crucial players promoting Th2 immunity, which provides protection from helminth infections but causes allergic inflammatory diseases (van Rijt et al. 2016). Mouse and human ILC2s are phenotypically similar because they are CD45+ but lack lineage markers (Walker et al. 2013). Instead, ILC2s express various cytokine receptors (R), such as common γ-chain (CD132 or IL-2Rγ), IL-2Rα (CD25), IL-7Rα (CD127), ST2 (IL-33R or IL-1R-like 1), IL-17RB, and thymic stromal lymphopoietin receptor (TSLPR), and are activated in response to IL-25, IL-33, and TSLP derived from epithelial cells (Licona-Limón et al. 2013). Furthermore, mouse ILC2s express Sca1, ICOS, and KLRG1 (McKenzie et al. 2014), whereas human ILC2s express cysteinyl leukotriene receptor 1 (CysLT1R), CD161 (C-type lectin receptor), and chemoattractant-receptor–homologous molecule expressed on Th2 cells (CRTH2) (Mjösberg et al. 2011; Konya and Mjosberg 2016). Once activated, ILC2s rapidly release large amounts of IL-5 and IL-13, which act on different types of cells and lead to Th2 inflammatory diseases in the airway, skin, and intestine (Diefenbach et al. 2014; McKenzie et al. 2014; Artis and Spits 2015). In addition, ILC2s mediate tissue repair and regulate metabolic homeostasis of glucose and lipids (Hams et al. 2013; Kim et al. 2013; Molofsky et al. 2013; Artis and Spits 2015).
ILC2s in allergic inflammatory diseases
Allergic asthma
Allergic asthma is a common airway disease caused by inappropriate immune responses to allergens. Asthma is characterized by airway hyperresponsiveness (AHR) and inflammation, leading to bronchoconstriction, excessive mucus production, and airway narrowing (Bousquet et al. 2000). Allergic asthma has long been considered a disease of the Th2 immune response; however, emerging studies have revealed that asthma is a more complex disease involving Th17 cells, ILC2s, and ILC3s as well as Th2 cells (Kim et al. 2016).
In addition to Th2 cells, ILC2s are another major source of IL-5 and IL-13, which perform critical functions in the pathogenesis of allergen-induced asthma by stimulating the growth and differentiation of eosinophils and by inducing airway epithelial cells and smooth muscle cells to contract. Halim (2016) demonstrated that in Rag2−/−Il2rg−/− mice, which lack all types of ILCs [or lung natural helper cells as described by (Halim et al. 2014)], intranasal administration of a protease allergen (papain) does not induce eosinophil infiltration and mucus hyperproduction, whereas adoptive transfer of ILC2s induces airway inflammation after papain treatment. The protease activity of papain may cause epithelial cell damage and secretion of TSLP, IL-25, and IL-33, leading to activation of ILC2s. Additionally, a fungal allergen, Alternaria alternata, promotes IL-33 secretion in lungs and activates ILC2 proliferation in mice (Doherty et al. 2012). In a mouse model of ovalbumin (OVA)-induced asthma, OVA sensitization and exposure induces ILC2s to release IL-5 and IL-13 (Klein Wolterink et al. 2012). Nevertheless, OVA-driven ILC2 activation is a controversial topic because an OVA challenge has not always led to ILC2 activation and cytokine production in other studies (Li et al. 2016a).
In humans, ILC2s are present in lungs and bronchoalveolar lavage (BAL) fluid (Drake and Kita 2014). Prior to the identification of ILC2s, Allakhverdi et al. (2009) reported the presence of non-B, non-T lymphocytes that produce IL-5 and IL-13 in the sputum of asthmatic patients after an airway allergen challenge; however, it is not known whether these cells are ILC2s. In addition, both the number of ILC2 and IL-33 levels increase in BAL fluid while upstream cytokines and mediators, such as IL-25, IL-33, TSLP, leukotrienes, and prostaglandins, are upregulated in human asthma. Furthermore, genomewide association studies indicate that genetic polymorphisms near the TSLP, IL-33, IL1RL1 (IL-33R), and RORA loci are strongly linked to asthma, thus pointing to the critical participation of lung ILC2s in human asthma (Kabata et al. 2015). Nonetheless, studies on the contribution of ILC2s to allergic asthma in humans are relatively limited, and this area needs to be further investigated.
In addition to environmental allergens, respiratory viral infections can trigger asthma or aggravate pre-existing asthma in humans (Jackson and Johnston 2010). Research shows that influenza infection activates ILC2s in an IL-33–dependent manner, and rhinovirus infection induces IL-33 and IL-25 and upregulates Th2 cytokines in asthmatic patients (Chang et al. 2011; Jackson et al. 2014). Hong et al. (2014) investigated the effect of rhinovirus infection on ILC2 responses in neonatal mice and found that neonatal infection with rhinovirus increases mucous metaplasia and AHR via IL-25 and IL-2. In contrast, these IL-25-driven Th2 responses to rhinovirus are not observed in adult mice, suggesting that the contribution of rhinovirus infection to asthma development is age-dependent.
In mice, ILC2s present in lungs were shown to contribute to AHR upon influenza virus infection (Chang et al. 2011). ILC2-depleted RAG2 knockout mice manifest reduced AHR after influenza infection. Nonetheless, transfer of wild-type ILC2s, but not IL-13 knockout ILC2s, into IL-13 knockout mice restores AHR, indicating that IL-13 secreted by ILC2s is required for influenza-induced AHR. In addition to AHR induction, ILC2s have been demonstrated to stimulate tissue repair responses after influenza infection in mice (Monticelli et al. 2011). ILC2 microarray analysis has revealed that ILC2s produce large amounts of amphiregulin, a ligand for epithelial growth factor receptor (EGFR), which promotes epithelial cell growth and wound healing. Adoptive transfer of ILC2s or amphiregulin treatment of ILC2-depleted mice restores airway integrity and lung function after influenza infection, indicating that ILC2s participate in both the pathogenesis and repair responses after influenza infection.
Rhinosinusitis
Chronic rhinosinusitis (CRS) is a persistent inflammatory disease involving paranasal sinuses and the lining of nasal passages that lasts for 12 weeks or longer. CRS can be subdivided into CRS with nasal polyps (CRSwNP) and CRS without nasal polyps (CRSsNP) based on the results of endoscopic examination. Symptoms of CRSwNP are similar to those of CRSsNP and include nasal congestion, postnasal drip, facial pain, and headache; however, symptom severity varies with the size of the polyp masses within the nasal cavity (Hamilos 2011; Akdis et al. 2013). Mjösberg et al. (2011) and Mjösberg et al. (2012) demonstrated that ILC2s are enriched in the polyps of patients with CRS and that ILC2s present in polyps secrete Th2 cytokines in response to TSLP, IL-33, and cysteinyl leukotrienes (CysLT), which were also found to be upregulated in patients with CRS. In addition, ILC2s express CRTH2, and activation of CRTH2 through PGD2 ligation stimulates ILC2 chemotaxis in CRS (Xue et al. 2014). CRS can also be categorized into eosinophilic CRS (eCRS) and noneosinophilic CRS (non-eCRS), on the basis of the underlying inflammation (Sakuma et al. 2011). Studies suggest that eCRS is associated with severer symptoms, development of nasal polyps, and poorer treatment outcomes as compared to non-eCRS (Haruna et al. 2009; Soler et al. 2009). The number of ILC2s in polyps was found to increase as numbers of local and circulating eosinophils increased, pointing to the potential role of ILC2s in the activation and survival of eosinophils in CRS (Ho et al. 2015).
Allergic rhinitis (AR), or hay fever, is inflammation of nasal mucous membranes that is characterized by sneezing, itching, nasal congestion, postnasal drip, and rhinorrhea (Greiner et al. 2011). AR is commonly comorbid with CRS and is a predisposing factor for CRS development. Just as CRS, AR is associated with an increase in ILC2 numbers after an allergen challenge in sensitized patients. Doherty et al. (2014) demonstrated that a nasal cat allergen challenge increases the percentage of peripheral-blood CRTH2+ ILC2s 4 h after the allergen challenge in humans. In addition, an increase in the number of peripheral-blood ILC2s is observed in patients with AR during pollen season, indicating the contribution of ILC2s to inflammatory responses in AR (Lao-Araya et al. 2014).
Atopic dermatitis (AD)
ILC2s have been detected in human and mouse skin and are known to be deeply involved in skin homeostasis. In mice, ILC2s residing in the skin are phenotypically distinct from the ILC2s found in other organs because skin ILC2s express CD103 but lack c-Kit; thus, skin ILC2s are designated as dermal ILC2s (dILC2s) (Roediger et al. 2013). In humans, skin ILC2s often express skin-homing chemokine receptors, such as CCR4, CCR10, and CLA, which distinguish these cells from circulating ILC2s. Once activated, dILC2s produce amphiregulin and stimulate the proliferation and migration of epithelial cells, leading to wound healing and skin repair (Salimi et al. 2013). In addition, IL-13 derived from dILC2s interacts with mast cells and represses the production of IL-6 and TNF-α after antigen exposure in mice (Roediger et al. 2013).
DILC2s are also involved in AD, a chronic inflammatory skin disease characterized by a defect in barrier function, infiltration of eosinophils, and elevated serum IgE levels (Roediger et al. 2014). In mice with established AD, dILC2 numbers are increased in the dermis, and these cells produce IL-4, IL-5, and IL-13 in response to TSLP and/or IL-33 (Salimi et al. 2013). Upregulation of TSLP and IL-33 has also been observed in human AD, and dILC2s are enriched in the skin lesions of patients with AD (Salimi et al. 2013). Moreover, prostaglandins can stimulate human dILC2s, whereas activating NK receptor (NKp30 or CD337)-positive human dILC2s can be activated by the tumor-associated surface molecule B7-H7, which is upregulated in the skin of patients with AD (Salimi et al. 2016).
ILC2s in helminth infections
Host protection from helminthic parasites is dependent on the production of Th2 cytokines. In mice infected with Nippostrongylus brasiliensis, this murine parasitic worm triggers the secretion of alarmins, such as IL-25, IL-33, and TSLP, which mediate activation of ILC2s (Moro et al. 2010; Neill et al. 2010; Price et al. 2010). In cooperation with Th2 cells, the activated ILC2s secrete a large amount of Th2 cytokines and induce Th2 immune responses, which are essential for successful clearance of parasitic infections (Williams et al. 2012). In response to parasitic infection, IL-4 activates human B cells and promotes isotype switching to IgE, generating immunity to parasites in vitro (Gascan et al. 1991). IL-5 stimulates the growth and activation of eosinophils, which kill helminth larvae in the presence of Ab or complement and regulate the tissue homeostasis and metabolism that are crucial for the establishment and maintenance of parasites in the host (Hall et al. 1998; Behm and Ovington 2000; Klion and Nutman 2004; Huang and Appleton 2016). IL-13 induces mucus secretion by goblet cells and smooth-muscle contraction, leading to expulsion of the parasites, and increases the migration and turnover of epithelial cells in the mouse mucosa (Sun et al. 2016).
In addition to parasite expulsion, tissue repair and suppression of local inflammation are important for efficient coping with active parasitic infection because such infections cause tissue damage resulting from parasite invasion and infection-induced inflammatory responses (Allen and Sutherland 2014). Accordingly, ILC2-derived IL-9 is necessary for repairing damaged epithelial cells. Furthermore, ILC2s as well as Th2 cells and mast cells produce amphiregulin (AREG), which is a member of the epidermal growth factor (EGF) family of proteins (Zaiss et al. 2006, 2015). Amphiregulin interacts with EGF receptor to promote the proliferation of epithelial cells and mediates antihelminth immunity, inflammation suppression, and wound repair.
ILC2s as therapeutic targets
As robust stimulators of Th2 immunity, ILC2s have been suggested as potential therapeutic targets in allergic diseases, such as asthma, AD, and rhinosinusitis.
Lipoxin A4, an arachidonic-acid–derived negative regulator of ILC2s, is an endogenous lipid mediator with anti-inflammatory and proresolution properties (Konya and Mjosberg 2016) that binds to lipoxin A4 receptor or formyl peptide receptor 2 (AXL/FPR2) expressed on ILC2s and NK cells (Barnig et al. 2013). It inhibits IL-13 production by ILC2s and enhances the ability of NK cells to induce eosinophils in humans, thus pointing to new therapeutic strategies against asthma, such as AXL/FPR2 receptor agonists, which are possible drugs for allergic diseases (Christie et al. 1992; Wu et al. 2013).
Prostaglandin I2 (PGI2) or prostacyclin is also derived from arachidonic acid and also has inhibitory effects on ILC2 function (Karta et al. 2016; Konya and Mjosberg 2016). Zhou et al. (2016) reported that PGI2 analog cicaprost inhibits IL-33–induced ILC2 proliferation in mice and decreases IL-5 and IL-13 secretion in response to IL-2 and IL-33 stimulation in humans. When cicaprost-treated mice are challenged with the allergy-causing fungus Alternaria, cicaprost reduces the number of ILC2s, and this action is accompanied by decreased IL-5 and IL-13 expression in lungs, suggesting that PGI2 regulates airway inflammation by reducing ILC2 responses to aeroallergen (Zhou et al. 2016). Moreover, CRTH2, a receptor for prostaglandin D2 (PGD2) is highly expressed on ILC2s and Th2 cells, and PGD2 ligation promotes CRTH2+ ILC2 migration and IL-13 production in human experimental in vitro models (Martinez-Gonzalez et al. 2015; Wojno et al. 2015). In a murine model of helminth-induced Th2 inflammation, CRTH2-deficient mice show a decrease in the ILC2 response and in inflammation in lungs, suggesting that CRTH2 may be a promising therapeutic target in inflammatory diseases of lungs. Selective CRTH2 antagonists, such as fevipiprant and timapiprant, are currently being tested for the treatment of allergic diseases (Schuligoi et al. 2010; Townley and Agrawal 2012). In addition, leukotriene receptor antagonists, such as montelukast and zafirlukast, have been shown to prevent ILC2 activation in mice (Doherty et al. 2013).
In addition to lipid mediators and their receptors, upstream alarmins, including IL-25, IL-33, and TSLP, are attractive targets for treating allergic diseases (Divekar and Kita 2015). Antibodies blocking IL-25, IL-33, or TSLP, such as tezepelumab, have been reported to be beneficial for treating patients with COPD (Lau et al. 2017). On the other hand, blocking of alarmins may affect multiple cell types or have no effects owing to their redundant activities toward ILC2s (Cayrol and Girard 2014; Vannella et al. 2016). In addition, blocking of cytokines downstream of ILC2s, such as IL-4, IL-5, and IL-13, may be therapeutically effective (Bel and Ten Brinke 2017). Research has revealed that a humanized monoclonal Ab against IL-5 (mepolizumab) and a human Ab against IL-4Rα (dupilumab) have beneficial effects against allergic diseases in humans (Beck et al. 2014; Ortega et al. 2014).
General features of ILC3s
ILC3s comprise at least three subtypes: lymphoid tissue inducer (LTi) cells, natural cytotoxicity receptor (NCR)+ ILC3s, and NCR−CCR6− double-negative ILC3s. All the subtypes have the common feature Lin−RORγt+CD127+CD90+ (Sanos and Diefenbach 2013), and each subtype can be classified according to the expression of CCR6 and NCR (NKp46 in mice and NKp44 in humans). NKp46−CCR6+ LTi cells were initially described as cells that are necessary for the development and formation of lymphoid tissue during the fetal period and after birth in mice (Melo-Gonzalez and Hepworth 2017). Recent studies, however, indicate that LTi cells can secrete cytokines such as IL-17 (Uhlig et al. 2006; Elson et al. 2007). NCR+ ILC3s are the major populations of ILC3s in the murine small intestinal mucosal tissue, and these cells produce IL-22 (Luci et al. 2009; Vonarbourg et al. 2010). NKp46−CCR6−/lo DN-ILC3s are a heterogeneous cell population that secretes IL-17, IL-22, and IFN-γ (Melo-Gonzalez and Hepworth 2017). NCR− ILC3s possess considerable plasticity and can be converted into NCR+ ILC3s or IFN-γ–producing ILC1s (Vonarbourg et al. 2010; Melo-Gonzalez and Hepworth 2017). Just as in Th17 cells, the transcription factor RORγt plays an important role in the differentiation of all ILC3 subsets (Cording et al. 2014).
ILC3s secrete proinflammatory or anti-inflammatory cytokines, such as IL-17 and IL-22, in response to IL-1β, IL-6, and IL-23 (Sedda et al. 2014). IL-17 is a well-studied proinflammatory cytokine that recruits neutrophils and macrophages to induce inflammation. IL-22 is a double-edged cytokine that is known to have either proinflammatory or anti-inflammatory effects in inflammatory diseases (Neurath 2014). ILC3s can also take up and present antigens, indicating that ILC3s can regulate adaptive immunity (Hepworth et al. 2013). Through cytokine production and antigen presentation, ILC3 act as mediators of diverse inflammatory diseases, cancer, and even preeclampsia (Hepworth et al. 2013; Sonnenberg and Artis 2015). Accordingly, ILC3s are being studied as potential therapeutic targets in several diseases (Perry et al. 2012; Li et al. 2016b; Konya et al. 2017).
ILC3s in inflammatory diseases
Acute intestinal inflammation, i.e., inflammatory bowel disease (IBD)
IBD is a chronic relapsing inflammatory disease originating from acute colitis that has not been properly resolved. ILC3s are the most common ILC subtype present in the human intestine in the steady state, and these cells are a major source of GM-CSF upon IL-23 stimulation in mice and humans (Pearson et al. 2016). GM-CSF regulates acute intestinal inflammation by recruiting and maintaining inflammatory monocytes. In addition, ILC3s actively participate in chronic inflammatory diseases, such as CD and ulcerative colitis (UC). The frequency of inflammatory ILC3s is significantly elevated in the inflamed intestinal tissue of patients with IBD and in a mouse model of IBD (Geremia et al. 2011; Hepworth et al. 2013); besides, Longman et al. (2014) reported that ILC3s from the colon tissue of IBD patients produce more IL-22 than control tissue does. Moreover, Lo et al. (2016) demonstrated that ILC3s play a pivotal role in gut fibrosis in salmonella-induced gut fibrosis (which serves as a mouse model of CD) by secreting IL-22 and IL-17A. The pathogenic functions of IL-22, IL-17, and IFN-γ from ILC3s have been well studied in a mouse model of colitis. Eken et al. (2014) demonstrated that IL-22 produced by ILC3s promotes the development of colitis in a Rag1−/− mouse model of anti-CD40 Ab-induced colitis. Treatment with an anti-IL-22 Ab protects anti-CD40–injected mice from colitis, whereas addition of IL-22 via hydrodynamic gene delivery exacerbates colitis symptoms. In addition, IL-7Rα+ ILCs producing IL-17A promote colitis in a Th1-specific T-box transcription factor (Tbx21)−/−Rag2−/− model of UC (Powell et al. 2012). H. hepaticus–infected Rag−/− mice develop chronic colitis through induction of ILC3-derived IL-17A and IFN-γ (Hue et al. 2006; Buonocore et al. 2010), and depletion of all Thy1+ ILCs with an anti-Thy1 Ab alleviates both acute and chronic colitis, while RORγt-deficient mice receiving an anti-CD40 Ab do not develop colitis; these data emphasize the pathological involvement of ILC3s in colitis (Buonocore et al. 2010). The above results show the importance of ILC3-mediated secretion of IL-22, IL-17, and IFN-γ in colitis.
Psoriasis
This is a common, chronic inflammatory autoimmune skin disease characterized by erythematous scaly plaques resulting from excess keratinocytes. Studies indicate that the number of NKp44+ ILC3s is higher in the skin and peripheral blood of patients with psoriasis as compared to patients with AD or healthy volunteers (Teunissen et al. 2014; Villanova et al. 2014). In addition, the frequency of NKp44+ ILC3s in the nonlesional skin of psoriasis patients is higher than that in healthy volunteers, indicating the possible role of NKp44+ ILC3s in the pathogenesis (Villanova et al. 2014). Furthermore, the frequency of NKp44+ ILC3s in the skin correlates with the severity of psoriasis. When patients with psoriasis receive anti-TNF therapy, decreases in inflammatory skin lesions and circulating NKp44+ ILC3 are observed, indicating a correlation between the frequency of NKp44+ ILC3s in blood and the clinical response to anti-TNF therapy. In Aldara-induced skin inflammation in a mouse model of psoriasis, ILC3s were also reported to be a major source of IL-17 and IL-22 in the skin and were shown to induce psoriatic plaque formation (Ward and Umetsu 2014).
RA and ankylosing spondylitis (AS)
RA is characterized by immune-cell infiltration and chronic inflammation caused by inflammatory cytokines such as IL-1, IL-6, IL-17, and TNF-α (McInnes and Schett 2007). Before the nomenclature for ILC was established, Ren et al. (2011) observed that numbers of CD3−CD56+NKp44+CCR6+ cells producing IL-22 are increased in the synovial fluid and peripheral blood of RA patients, and patients with large numbers of CD3−CD56+NKp44+CCR6+ cells show worse 28-joint disease activity scores. In addition, Koo et al. (2013) reported that increased lymphocyte infiltration of RA synovial fluid correlates with upregulation of LTi-like cells and the expression of chemokines that recruit LTi or LTi-like cells expressing CXCR5 and CCR7.
AS is an autoimmune disease that induces chronic inflammation in the joints of the spine. The numbers of lin−RORc−Tbet+NKp44+ ILC3s are significantly increased in the gut, synovial fluid, and bone marrow of patients with AS in comparison with these numbers in healthy volunteers. lin−RORc−Tbet+NKp44+ ILC3s also express the inflammatory cytokines IL-17 and IL-22 as well as α4β7 integrin, pointing to recirculation of ILC3s between the gut and extraintestinal sites (Ciccia et al. 2015). Besides, a significant reduction in intestinal and circulating ILC3 numbers was observed in AS patients after anti-TNF treatment. These results are suggestive of a potential contribution of intestinal ILC3s to the development of AS in relation to their migration between the gut and inflamed sacroiliac joints and induction of inflammation.
Multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE)
MS is an inflammatory disease of the nervous system. The exact causes of MS are not known; however, a combination of genetic and environmental factors seems to contribute to the inflammatory process. Although the function of ILCs in MS is poorly understood, Perry et al. (2012) showed that LTi-like ILCs in cerebrospinal fluid initiate and sustain the formation of focal aggregates. Numbers of ILC3s, including LTi cells, are higher in the blood of patients with MS. In addition, stronger expression of IL-17 is observed in the brain lesions of MS patients according to gene microarray analysis, supporting the potential role of ILC3s in MS (Lock et al. 2002).
ILC3s reside in the meninges of naïve mice in the steady state. ILC3s produce IL-17 and GM-CSF and express CD30L and OX40L, which keep memory T cells alive. In EAE, a mouse model of MS, disease-induced T-cell trafficking in the meninges is impaired in ILC3-deficient Rorc−/− mice with transferred wild-type T cells. In addition, c-kit-mutant (KitW/Wv) mice that are EAE resistant have a lower number of LTi cells, indicating ILC3s’ involvement in EAE (Hatfield and Brown 2015).
Asthma
Emerging studies have shown that asthma is also associated with ILC3s and Th2 immune responses. IL-17-expressing ILC3s are more frequently detected in human BAL fluid samples from patients with severe asthma than in BAL fluid samples from patients with mild asthma or healthy volunteers. Furthermore, in vitro culture of BAL fluid cells with IL-1β, IL-7, and IL-2 results in an increased number of IL-17-producing ILC3s, implying that IL-17-producing ILC3s can be found in lungs and play a role in the pathogenesis of asthma (Barnie et al. 2015). In addition, Nagakumar et al. (2017) found that the number of ILC3s expressing IL-17 is significantly higher in blood, sputum, and BAL fluid of patients with severe treatment-resistant asthma, whereas the number of Th17 cells is unchanged.
In mice, ILCs are rarely detected in lungs, representing only 0.4–1% of all live cells in the steady state (Monticelli et al. 2011). By contrast, in a model of obesity-induced asthma, IL-17A–producing ILC3s are upregulated 3- to 4-fold by IL-1β produced by macrophages (Kim et al. 2014).
ILC3s in cancer
A recent study revealed that ILC3s are directly related to the development of colorectal cancer (CRC) through IL-22. In a mouse model of H. hepaticus azoxymethane (AOM)-induced CRC, colonic ILCs producing IL-17 and IL-22 accumulate in inflamed colon tissue. Kirchberger et al. (2013) reported that in a CRC model, IL-22 blockade with an anti-IL-22 Ab can significantly reduce tumor burden, whereas an anti-IL-17 Ab does not have an antitumorigenic effect but does decrease inflammation, suggesting that IL-22 is a novel therapeutic target in CRC. In addition, an increase in IL-22 production is observed in human CRC tissue (Jiang et al. 2013; Kirchberger et al. 2013). Jiang et al. (2013) demonstrated that IL-22 enhances tumor development and metastasis via activation of STAT3 in human UC and CRC, confirming the participation of IL-22 in CRC.
Carrega et al. (2015) reported that human non–small cell lung cancer (NSCLC) is related to NCR+ ILC3s. The latter cells are present in the lymphoid infiltrates of human NSCLC and produce proinflammatory and chemotactic or angiogenic cytokines. These NCR+ ILC3s can interact with tumor cells and tumor-associated fibroblasts and are more frequently detected in early-stage NSCLC than in advanced-stage NSCLC, implying a role for NCR+ ILC3s in the formation of tertiary lymphoid structures (TLSs). In fact, a positive correlation is observed between TLS density and the number of NCR+ ILC3s in tumor infiltrates. This result, along with the finding that TLSs correlate with better survival, suggests that NCR+ ILC3s may be an effective prognostic factor of NSCLC (Carrega et al. 2015).
ILC3s as therapeutic targets
Dysregulation of ILC3s has been implicated in many inflammatory diseases and cancers; consequently, ILC3s have been suggested as therapeutic targets.
Aryl hydrocarbon receptor, a ligand-dependent transcription factor, is involved in the regulation of differentiation and in the functioning of murine ILCs in the gut (Qiu et al. 2012). Maintaining the intestinal ILC composition is critical for homeostasis and immunity, and dysregulated ILC composition can lead to destruction of the intestinal barrier and excessive immune reactions, resulting in inflammation or cancer. In humans, NKp44+ ILC3s, NKp44− ILC3s, and ILC1s differentially express aryl hydrocarbon receptor (its expression in NKp44+ ILC3s is the highest, expression in NKp44− ILC3s is intermediate, and ILC1s do not express this receptor), and downregulation of aryl hydrocarbon receptor is required for conversion from ILC3s to ILC1s. NKp44+ ILC3s play a protective role against the pathogenesis of CD, and aryl hydrocarbon receptor signaling is essential for the maintenance of NKp44+ ILC3s in the intestine. Li et al. (2016b) observed that ILC1s accumulate at the expense of NKp44+ ILC3s in the inflamed tissue of patients with CD, indicating impairment of the aryl hydrocarbon receptor signaling pathway followed by ILC3-to-ILC1 conversion. Furthermore, Zhao et al. (2016) reported that microRNA-124 induces intestinal inflammation by inhibiting aryl hydrocarbon receptor in CD patients. A knockdown of microRNA-124 and aryl hydrocarbon receptor activation repress inflammation in lipopolysaccharide-stimulated cells in vitro and in a mouse model of TNBS-induced colitis, supporting the notion that aryl hydrocarbon receptor on ILCs is a potential target for the treatment of CD.
Vitamin D deficiency is a risk factor of IBD (Ardesia et al. 2015). Konya et al. (2017) isolated ILC3s from human tonsils and the gut, and demonstrated that ILC3stimulation under the influence of IL-23 and IL-1β induces expression of vitamin D receptor and downregulates the IL-23 receptor pathway in response to the active form of vitamin D, 1α,25-dihydroxy vitamin D3 (1,25D) through a negative feedback loop in vitro. As a result, vitamin D decreases the secretion of such cytokines as IL-17F, IL-22, and GM-CSF from ILC3s, implying that vitamin D is an interesting therapeutic agent that modulates the IL-23 receptor pathway and ILC3s in intestinal inflammation.
Treatment with daclizumab (Zinbryta®), a humanized anti-CD25 monoclonal Ab, decreases the inflammation associated with MS without affecting activated T cells. Daclizumab reduces the number of circulating ILC3s and modifies the phenotype of LTi cells toward NK cells by modulating the differentiation of hematopoietic progenitors in patients with MS (Perry et al. 2012). In May 2016, the FDA approved daclizumab for the treatment of relapsing MS.
IL-23—expressing ILC3s perform essential functions in intestinal immunity, and therapeutic strategies targeting downstream cytokines, such as IL-23, are promising approaches to IBD therapy. Elson et al. (2007) reported that administration of an anti–IL-23 monoclonal Ab prevents and alleviates active colitis in a mouse model of T-cell–mediated colitis. Administration of an anti–IL-23 monoclonal Ab downregulates inflammatory cytokines and chemokines in the inflamed colon.
In addition, targeting the IL-23–IL-17 axis with the anti–IL-17A monoclonal Ab secukinumab (Cosentyx®) and an anti-p40 (subunit of IL-12 and IL-23) monoclonal Ab ustekinumab (Stelara®) has therapeutic effects in both AS and psoriasis (Leonardi et al. 2008; Baeten et al. 2013; Baeten et al. 2015). Although these antibodies, which target the IL-23–IL-17 axis, have initially been intended to target Th17 cells, ILC3s are also an important component of this axis. Despite the contribution of the IL-23–IL-17 axis control to host immunity, neutralizing these cytokines can also have adverse effects, such as tuberculosis, fungal infections, and upper respiratory tract effects (Griffiths et al. 2010; Langley et al. 2014; Baeten et al. 2015; Feagan et al. 2016).
Closing remarks
Numerous studies on ILCs have been conducted in the past 7 years since their discovery, resulting in the identification of three ILC subsets and their development and functions. Despite the beneficial roles of ILCs in immunity and homeostasis, they can also have detrimental effects and are involved in various diseases, from allergic rhinitis to autoimmune inflammatory diseases and cancer; therefore, ILCs are a critical factor in some diseases.
Despite intensive research, much remains to be learned about how each type of ILCs influences the onset, progression, and prognosis of ILC-associated diseases as well as the immune response. In addition, the potential therapeutic targets in ILC-associated diseases need to be determined.
References
Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B, Ling L, Leiner I, Pamer EG (2015) Innate immune defenses mediated by two ILC subsets are critical for protection against acute clostridium difficile infection. Cell Host Microbe 18:27–37
Akdis CA, Bachert C, Cingi C, Dykewicz MS, Hellings PW, Naclerio RM, Schleimer RP, Ledford D (2013) Endotypes and phenotypes of chronic rhinosinusitis: a PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol 131:1479–1490
Allakhverdi Z, Comeau MR, Smith DE, Toy D, Endam LM, Desrosiers M, Liu Y-J, Howie KJ, Denburg JA, Gauvreau GM (2009) CD34+ hemopoietic progenitor cells are potent effectors of allergic inflammation. J Allergy Clin Immunol 123(472–478):e471
Allen JE, Sutherland TE (2014) Host protective roles of type 2 immunity: parasite killing and tissue repair, flip sides of the same coin. Semin Immunol 26:329–340
Ardesia M, Ferlazzo G, Fries W (2015) Vitamin D and inflammatory bowel disease. Biomed Res Int 2015:470805
Artis D, Spits H (2015) The biology of innate lymphoid cells. Nature 517:293–301
Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, McInnes I, van Laar JM, Landewé R, Wordsworth P, Wollenhaupt J, Kellner H, Paramarta J, Wei J, Brachat A, Bek S, Laurent D, Li Y, Wang YA, Bertolino AP, Gsteiger S, Wright AM, Hueber W (2013) Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet 382:1705–1713
Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, Deodhar A, Porter B, Martin R, Andersson M, Mpofu S, Richards HB, Group MS (2015) Secukinumab, an interleukin-17A inhibitor, in Ankylosing Spondylitis. N Engl J Med 373:2534–2548
Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, van Drunen CM, Lutter R, Jonkers RE, Hombrink P, Bruchard M, Villaudy J, Munneke JM, Fokkens W, Erjefalt JS, Spits H, Ros XR (2016) IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol 17:636–645
Barnie PA, Lin X, Liu Y, Xu H, Su Z (2015) IL-17 producing innate lymphoid cells 3 (ILC3) but not Th17 cells might be the potential danger factor for preeclampsia and other pregnancy associated diseases. Int J Clin Exp Pathol 8:11100–11107
Barnig C, Cernadas M, Dutile S, Liu X, Perrella MA, Kazani S, Wechsler ME, Israel E, Levy BD (2013) Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med 5:174ra126
Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, Ming JE, Ren H, Kao R, Simpson E, Ardeleanu M, Weinstein SP, Pirozzi G, Guttman-Yassky E, Suarez-Farinas M, Hager MD, Stahl N, Yancopoulos GD, Radin AR (2014) Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 371:130–139
Behm CA, Ovington KS (2000) The role of eosinophils in parasitic helminth infections: insights from genetically modified mice. Parasitol Today 16:202–209
Bel EH, Ten Brinke A (2017) New anti-eosinophil drugs for asthma and COPD: targeting the Trait. Chest. doi:10.1016/j.chest.2017.05.019
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjosberg JM, Spits H (2013) Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14:221–229
Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, Munneke JM, Hazenberg MD, Villaudy J, Buskens CJ, Bemelman WA, Diefenbach A, Blom B, Spits H (2015) Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity 43:146–160
Bjorklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, Sandberg R, Mjosberg J (2016) The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol 17:451–460
Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM (2000) Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med 161:1720–1745
Braudeau C, Amouriaux K, Neel A, Herbreteau G, Salabert N, Rimbert M, Martin JC, Hemont C, Hamidou M, Josien R (2016) Persistent deficiency of circulating mucosal-associated invariant T (MAIT) cells in ANCA-associated vasculitis. J Autoimmun 70:73–79
Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464:1371–1375
Carrega P, Loiacono F, Di Carlo E, Scaramuccia A, Mora M, Conte R, Benelli R, Spaggiari GM, Cantoni C, Campana S, Bonaccorsi I, Morandi B, Truini M, Mingari MC, Moretta L, Ferlazzo G (2015) NCR(+)ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat Commun 6:8280
Cayrol C, Girard JP (2014) IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol 31:31–37
Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, Dekruyff RH, Umetsu DT (2011) Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol 12:631–638
Chen P, Vu T, Narayanan A, Sohn W, Wang J, Boedigheimer M, Welcher AA, Sullivan B, Martin DA, Ruixo JJ, Ma P (2015) Pharmacokinetic and pharmacodynamic relationship of AMG 811, an anti-IFN-gamma IgG1 monoclonal antibody, in patients with systemic lupus erythematosus. Pharm Res 32:640–653
Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T (2009) Maturation of mouse NK cells is a 4-stage developmental program. Blood 113:5488–5496
Christie PE, Spur BW, Lee T (1992) The effects of Lipoxin A4 on airway responses in asthmatic subjects 1-3. Am Rev Respir Dis 145:1281–1284
Ciccia F, Guggino G, Rizzo A, Saieva L, Peralta S, Giardina A, Cannizzaro A, Sireci G, De Leo G, Alessandro R, Triolo G (2015) Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann Rheum Dis 74:1739–1747
Cording S, Medvedovic J, Cherrier M, Eberl G (2014) Development and regulation of RORgammat(+) innate lymphoid cells. FEBS Lett 588:4176–4181
Cortez VS, Fuchs A, Cella M, Gilfillan S, Colonna M (2014) Cutting edge: salivary gland NK cells develop independently of Nfil3 in steady-state. J Immunol 192:4487–4491
Dalbeth N, Callan MF (2002) A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum 46:1763–1772
Dalbeth N, Gundle R, Davies RJ, Lee YC, McMichael AJ, Callan MF (2004) CD56bright NK cells are enriched at inflammatory sites and can engage with monocytes in a reciprocal program of activation. J Immunol 173:6418–6426
Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA, Marvel J, Yoh K, Takahashi S, Prinz I, de Bernard S, Buffat L, Walzer T (2014) T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med 211:563–577
Diefenbach A, Colonna M, Koyasu S (2014) Development, differentiation, and diversity of innate lymphoid cells. Immunity 41:354–365
Divekar R, Kita H (2015) Recent advances in epithelium-derived cytokines (IL-33, IL-25, and thymic stromal lymphopoietin) and allergic inflammation. Curr Opin Allergy Clin Immunol 15:98–103
Doherty TA, Broide DH (2015) Group 2 innate lymphoid cells: new players in human allergic diseases. J Investig Allergol Clin Immunol 25:1–11
Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, Broide DH (2012) STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol Lung Cell Mol Physiol 303:L577–L588
Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH (2013) Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol 132:205–213
Doherty TA, Scott D, Walford HH, Khorram N, Lund S, Baum R, Chang J, Rosenthal P, Beppu A, Miller M, Broide DH (2014) Allergen challenge in allergic rhinitis rapidly induces increased peripheral blood type 2 innate lymphoid cells that express CD84. J Allergy Clin Immunol 133:1203–1205
Drake LY, Kita H (2014) Group 2 innate lymphoid cells in the lung. Adv Immunol 124:1–16
Eken A, Singh AK, Treuting PM, Oukka M (2014) IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol 7:143–154
Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA (2007) Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132:2359–2370
Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, Blank MA, Johanns J, Gao LL, Miao Y, Adedokun OJ, Sands BE, Hanauer SB, Vermeire S, Targan S, Ghosh S, de Villiers WJ, Colombel JF, Tulassay Z, Seidler U, Salzberg BA, Desreumaux P, Lee SD, Loftus EV Jr, Dieleman LA, Katz S, Rutgeerts P, Group U-I-US (2016) Ustekinumab as induction and maintenance therapy for crohn’s disease. N Engl J Med 375:1946–1960
Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M (2013) Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38:769–781
Gascan H, Gauchat JF, Roncarolo MG, Yssel H, Spits H, de Vries JE (1991) Human B cell clones can be induced to proliferate and to switch to IgE and IgG4 synthesis by interleukin 4 and a signal provided by activated CD4 + T cell clones. J Exp Med 173:747–750
Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, Travis SP, Powrie F (2011) IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med 208:1127–1133
Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, Lindsten T, Reiner SL (2012) The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36:55–67
Greiner AN, Hellings PW, Rotiroti G, Scadding GK (2011) Allergic rhinitis. Lancet 378:2112–2122
Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, Guzzo C, Xia Y, Zhou B, Li S, Dooley LT, Goldstein NH, Menter A, Group AS (2010) Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med 362:118–128
Halim TY (2016) Group 2 innate lymphoid cells in disease. Int Immunol 28:13–22
Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, Takei F (2014) Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40:425–435
Hall LR, Mehlotra RK, Higgins AW, Haxhiu MA, Pearlman E (1998) An essential role for interleukin-5 and eosinophils in helminth-induced airway hyperresponsiveness. Infect Immun 66:4425–4430
Hamilos DL (2011) Chronic rhinosinusitis: epidemiology and medical management. J Allergy Clin Immunol 128:693–707
Hams E, Locksley RM, McKenzie AN, Fallon PG (2013) Cutting edge: IL-25 elicits innate lymphoid type 2 and type II NKT cells that regulate obesity in mice. J Immunol 191:5349–5353
Haruna S, Shimada C, Ozawa M, Fukami S, Moriyama H (2009) A study of poor responders for long-term, low-dose macrolide administration for chronic sinusitis. Rhinology 47:66–71
Hatfield JK, Brown MA (2015) Group 3 innate lymphoid cells accumulate and exhibit disease-induced activation in the meninges in EAE. Cell Immunol 297:69–79
Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM, Wherry EJ, Koni PA, Bushman FD, Elson CO, Eberl G, Artis D, Sonnenberg GF (2013) Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498:113–117
Ho J, Bailey M, Zaunders J, Mrad N, Sacks R, Sewell W, Harvey RJ (2015) Group 2 innate lymphoid cells (ILC2s) are increased in chronic rhinosinusitis with nasal polyps or eosinophilia. Clin Exp Allergy 45:394–403
Hommes DW, Mikhajlova TL, Stoinov S, Štimac D, Vucelic B, Lonovics J, Zákuciová M, D’Haens G, Van Assche G, Ba S (2006) Fontolizumab, a humanised anti-interferon γ antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn’s disease. Gut 55:1131–1137
Hong JY, Bentley JK, Chung Y, Lei J, Steenrod JM, Chen Q, Sajjan US, Hershenson MB (2014) Neonatal rhinovirus induces mucous metaplasia and airways hyperresponsiveness through IL-25 and type 2 innate lymphoid cells. J Allergy Clin Immunol 134:429–439
Huang L, Appleton JA (2016) Eosinophils in Helminth infection: defenders and dupes. Trends Parasitol 32:798–807
Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ (2006) Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med 203:2473–2483
Jackson DJ, Johnston SL (2010) The role of viruses in acute exacerbations of asthma. J Allergy Clin Immunol 125:1178–1187
Jackson DJ, Makrinioti H, Rana BM, Shamji BW, Trujillo-Torralbo MB, Footitt J, Jerico D-R, Telcian AG, Nikonova A, Zhu J, Aniscenko J, Gogsadze L, Bakhsoliani E, Traub S, Dhariwal J, Porter J, Hunt D, Hunt T, Hunt T, Stanciu LA, Khaitov M, Bartlett NW, Edwards MR, Kon OM, Mallia P, Papadopoulos NG, Akdis CA, Westwick J, Edwards MJ, Cousins DJ, Walton RP, Johnston SL (2014) IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med 190:1373–1382
Jiang R, Wang H, Deng L, Hou J, Shi R, Yao M, Gao Y, Yao A, Wang X, Yu L, Sun B (2013) IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 13:59
Kabata H, Moro K, Koyasu S, Asano K (2015) Group 2 innate lymphoid cells and asthma. Allergol Int 64:227–234
Karta M, Doherty T, Broide D (2016) Lipid mediator regulation of group 2 innate lymphoid Cells. Immun Res 12:1
Kim BS, Wojno ED, Artis D (2013) Innate lymphoid cells and allergic inflammation. Curr Opin Immunol 25:738–744
Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT (2014) Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 20:54–61
Kim HY, Umetsu DT, Dekruyff RH (2016) Innate lymphoid cells in asthma: will they take your breath away? Eur J Immunol 46:795–806
Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F (2013) Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med 210:917–931
Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, Hendriks RW (2012) Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol 42:1106–1116
Klion AD, Nutman TB (2004) The role of eosinophils in host defense against helminth parasites. J Allergy Clin Immunol 113:30–37
Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y, Diefenbach A (2013) A T-bet gradient controls the fate and function of CCR6-RORgammat+innate lymphoid cells. Nature 494:261–265
Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, Diefenbach A (2014) Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157:340–356
Konya V, Mjosberg J (2016) Lipid mediators as regulators of human ILC2 function in allergic diseases. Immunol Lett 179:36–42
Konya V, Czarnewski P, Forkel M, Rao A, Kokkinou E, Villablanca EJ, Almer S, Lindforss U, Friberg D, Hoog C, Bergman P, Mjosberg J (2017) Vitamin D downregulates the IL-23 receptor pathway in human mucosal group 3 innate lymphoid cells. J Allergy Clin Immunol. doi:10.1016/j.jaci.2017.01.045
Koo J, Kim S, Jung WJ, Lee YE, Song GG, Kim KS, Kim MY (2013) Increased lymphocyte infiltration in rheumatoid arthritis is correlated with an increase in LTi-like cells in synovial fluid. Immune Netw 13:240–248
Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, Tsai TF, Wasel N, Tyring S, Salko T, Hampele I, Notter M, Karpov A, Helou S, Papavassilis C, Group ES, Group FS (2014) Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med 371:326–338
Lao-Araya M, Steveling E, Scadding GW, Durham SR, Shamji MH (2014) Seasonal increases in peripheral innate lymphoid type 2 cells are inhibited by subcutaneous grass pollen immunotherapy. J Allergy Clin Immunol 134(1193–1195):e1194
Lau EMT, Roche NA, Reddel HK (2017) Therapeutic approaches to asthma-chronic obstructive pulmonary disease overlap. Expert Rev Clin Immunol 13:449–455
Leijten EF, van Kempen TS, Boes M, Michels-van Amelsfort JM, Hijnen D, Hartgring SA, van Roon JA, Wenink MH, Radstake TR (2015) Brief report: enrichment of activated group 3 innate lymphoid cells in psoriatic arthritis synovial fluid. Arthritis Rheumatol 67:2673–2678
Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, Li S, Dooley LT, Gordon KB (2008) Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 371:1665–1674
Li BW, de Bruijn MJ, Tindemans I, Lukkes M, KleinJan A, Hoogsteden HC, Hendriks RW (2016a) T cells are necessary for ILC2 activation in house dust mite-induced allergic airway inflammation in mice. Eur J Immunol 46:1392–1403
Li J, Doty A, Glover SC (2016b) Aryl hydrocarbon receptor signaling involves in the human intestinal ILC3/ILC1 conversion in the inflamed terminal ileum of Crohn’s disease patients. Inflamm Cell Signal 3:e1404
Licona-Limón P, Kim LK, Palm NW, Flavell RA (2013) TH2, allergy and group 2 innate lymphoid cells. Nat Immunol 14:536–542
Lim AI, Verrier T, Vosshenrich CA, Di Santo JP (2017) Developmental options and functional plasticity of innate lymphoid cells. Curr Opin Immunol 44:61–68
Lo BC, Gold MJ, Hughes MR, Antignano F, Valdez Y, Zaph C, Harder KW, McNagny KM (2016) The orphan nuclear receptor ROR alpha and group 3 innate lymphoid cells drive fibrosis in a mouse model of Crohn’s disease. Sci Immunol 1:eaaf8864
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8:500–508
Longman RS, Diehl GE, Victorio DA, Huh JR, Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ, Littman DR (2014) CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med 211:1571–1583
Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D, Dalod M, Littman DR, Vivier E, Tomasello E (2009) Influence of the transcription factor RORgammat on the development of NKp46 + cell populations in gut and skin. Nat Immunol 10:75–82
Martinez-Gonzalez I, Steer CA, Takei F (2015) Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol 36:189–195
McInnes IB, Schett G (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol 7:429–442
McKenzie AN, Spits H, Eberl G (2014) Innate lymphoid cells in inflammation and immunity. Immunity 41:366–374
Melo-Gonzalez F, Hepworth MR (2017) Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology 150:265–275
Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, Zimmer J (2016) Human CD56bright NK cells: an update. J Immunol 196:2923–2931
Mjösberg J, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, Spits H (2011) Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol 12:1055–1062
Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, te Velde AA, Fokkens WJ, van Drunen CM, Spits H (2012) The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37:649–659
Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, Chawla A, Locksley RM (2013) Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 210:535–549
Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12:1045–1054
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S (2010) Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463:540–544
Nagakumar P, Artusio L, Fainardi V, Fleming L, Bush A, Lloyd CM, Saglani S (2017) Role Of airway ILC2 And ILC3 compared To Th2 And Th17 cells in paediatric severe therapy resistant asthma (STRA). Am J Respir Crit Care Med 195:A4915
Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, Jolin HE, McKenzie AN (2010) Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464:1367–1370
Neurath MF (2014) Cytokines in inflammatory bowel disease. Nat Rev Immunol 14:329–342
Niederreiter L, Adolph TE, Kaser A (2013) Anti-IL-12/23 in Crohn’s disease: bench and bedside. Curr Drug Targets 14:1379–1384
Nielsen N, Odum N, Urso B, Lanier LL, Spee P (2012) Cytotoxicity of CD56(bright) NK cells towards autologous activated CD4+T cells is mediated through NKG2D, LFA-1 and TRAIL and dampened via CD94/NKG2A. PLoS ONE 7:e31959
Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, Copenhaver AM, Humbles AA, Liu YJ (2016) IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol 17:646–655
Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, Humbert M, Katz LE, Keene ON, Yancey SW, Chanez P, Investigators M (2014) Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med 371:1198–1207
Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, West N, Tung S, Seddon BP, Uhlig HH, Powrie F (2016) ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife 5:e10066
Perry JS, Han S, Xu Q, Herman ML, Kennedy LB, Csako G, Bielekova B (2012) Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med 4:145ra106
Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, Jackson I, Hashim A, Curtis MA, Jenner RG, Howard JK, Parkhill J, MacDonald TT, Lord GM (2012) The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+innate lymphoid cells. Immunity 37:674–684
Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, Locksley RM (2010) Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A 107:11489–11494
Qiu J, Heller JJ, Guo X, Chen ZM, Fish K, Fu YX, Zhou L (2012) The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 36:92–104
Ren J, Feng Z, Lv Z, Chen X, Li J (2011) Natural killer-22 cells in the synovial fluid of patients with rheumatoid arthritis are an innate source of interleukin 22 and tumor necrosis factor-alpha. J Rheumatol 38:2112–2118
Roan F, Stoklasek TA, Whalen E, Molitor JA, Bluestone JA, Buckner JH, Ziegler SF (2016) CD4+Group 1 innate lymphoid cells (ILC) form a functionally distinct ILC subset that is increased in systemic sclerosis. J Immunol 196:2051–2062
Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, Mitchell AJ, Tay SS, Jain R, Forbes-Blom E, Chen X, Tong PL, Bolton HA, Artis D, Paul WE, de St Fazekas, Groth B, Grimbaldeston MA, Le Gros G, Weninger W (2013) Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nat Immunol 14:564–573
Roediger B, Kyle R, Le Gros G, Weninger W (2014) Dermal group 2 innate lymphoid cells in atopic dermatitis and allergy. Curr Opin Immunol 31:108–114
Sakuma Y, Ishitoya J, Komatsu M, Shiono O, Hirama M, Yamashita Y, Kaneko T, Morita S, Tsukuda M (2011) New clinical diagnostic criteria for eosinophilic chronic rhinosinusitis. Auris Nasus Larynx 38:583–588
Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST, McKenzie AN, Fallon PG, Ogg GS (2013) A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 210:2939–2950
Salimi M, Xue L, Jolin H, Hardman C, Cousins DJ, McKenzie AN, Ogg GS (2016) Group 2 innate lymphoid cells express functional NKp30 receptor inducing Type 2 cytokine production. J Immunol 196:45–54
Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, Sands BE, Hanauer SB, Targan S, Rutgeerts P, Ghosh S, de Villiers WJ, Panaccione R, Greenberg G, Schreiber S, Lichtiger S, Feagan BG, Group CS (2012) Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med 367:1519–1528
Sanos SL, Diefenbach A (2013) Innate lymphoid cells: from border protection to the initiation of inflammatory diseases. Immunol Cell Biol 91:215–224
Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, Eberl G (2010) Lineage relationship analysis of RORgammat+innate lymphoid cells. Science 330:665–669
Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, Cua D, Di Santo JP, Eberl G (2011) RORgammat+innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol 12:320–326
Schepis D, Gunnarsson I, Eloranta ML, Lampa J, Jacobson SH, Karre K, Berg L (2009) Increased proportion of CD56bright natural killer cells in active and inactive systemic lupus erythematosus. Immunology 126:140–146
Schuligoi R, Sturm E, Luschnig P, Konya V, Philipose S, Sedej M, Waldhoer M, Peskar BA, Heinemann A (2010) CRTH2 and D-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology 85:372–382
Sedda S, Marafini I, Figliuzzi MM, Pallone F, Monteleone G (2014) An overview of the role of innate lymphoid cells in gut infections and inflammation. Mediators Inflamm 2014:235460
Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, Pritchard GH, Berlin AA, Hunter CA, Bowler R, Erjefalt JS, Kolbeck R, Humbles AA (2016) Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol 17:626–635
Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, Riley JK, Zhu J, Tian Z, Yokoyama WM (2014) Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife 3:e01659
Soler ZM, Sauer DA, Mace J, Smith TL (2009) Relationship between clinical measures and histopathologic findings in chronic rhinosinusitis. Otolaryngol Head Neck Surg 141:454–461
Sonnenberg GF, Artis D (2015) Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21:698–708
Spits H, Cupedo T (2012) Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol 30:647–675
Spits H, Di Santo JP (2011) The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12:21–27
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E (2013) Innate lymphoid cells—a proposal for uniform nomenclature. Nat Rev Immunol 13:145–149
Sun R, Urban JF Jr, Notari L, Vanuytsel T, Madden KB, Bohl JA, Ramalingam TR, Wynn TA, Zhao A, Shea-Donohue T (2016) Interleukin-13 receptor alpha1-dependent responses in the intestine are critical to parasite clearance. Infect Immun 84:1032–1044
Teunissen MB, Munneke JM, Bernink JH, Spuls PI, Res PC, Te Velde A, Cheuk S, Brouwer MW, Menting SP, Eidsmo L, Spits H, Hazenberg MD, Mjosberg J (2014) Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol 134:2351–2360
Townley RG, Agrawal S (2012) CRTH2 antagonists in the treatment of allergic responses involving TH2 cells, basophils, and eosinophils. Ann Allergy Asthma Immunol 109:365–374
Turner JE, Morrison PJ, Wilhelm C, Wilson M, Ahlfors H, Renauld JC, Panzer U, Helmby H, Stockinger B (2013) IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med 210:2951–2965
Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, Robinson N, Buonocore S, Tlaskalova-Hogenova H, Cua DJ, Powrie F (2006) Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25:309–318
van Rijt L, von Richthofen H, van Ree R (2016) Type 2 innate lymphoid cells: at the cross-roads in allergic asthma. Semin Immunopathol 38:483–496
Vannella KM, Ramalingam TR, Borthwick LA, Barron L, Hart KM, Thompson RW, Kindrachuk KN, Cheever AW, White S, Budelsky AL, Comeau MR, Smith DE, Wynn TA (2016) Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci Transl Med 8:337ra365
Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, Chapman A, Smith CH, Di Meglio P, Nestle FO (2014) Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ILC3 in psoriasis. J Invest Dermatol 134:984–991
Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, Honig M, Pannicke U, Schwarz K, Ware CF, Finke D, Diefenbach A (2010) Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 33:736–751
Vosshenrich CA, Garcia-Ojeda ME, Samson-Villeger SI, Pasqualetto V, Enault L, Richard-Le Goff O, Corcuff E, Guy-Grand D, Rocha B, Cumano A, Rogge L, Ezine S, Di Santo JP (2006) A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol 7:1217–1224
Walker JA, Barlow JL, McKenzie AN (2013) Innate lymphoid cells—how did we miss them? Nat Rev Immunol 13:75–87
Ward NL, Umetsu DT (2014) A new player on the psoriasis block: IL-17A- and IL-22-producing innate lymphoid cells. J Invest Dermatol 134:2305–2307
Werth VP, Fiorentino D, Sullivan BA, Boedigheimer MJ, Chiu K, Wang C, Arnold GE, Damore MA, Bigler J, Welcher AA, Russell CB, Martin DA, Chung JB (2017) Brief report: pharmacodynamics, safety, and clinical efficacy of AMG 811, a human anti-interferon-gamma antibody, in patients with discoid lupus erythematosus. Arthritis Rheumatol 69:1028–1034
Williams CM, Rahman S, Hubeau C, Ma HL (2012) Cytokine pathways in allergic disease. Toxicol Pathol 40:205–215
Wojno ET, Monticelli L, Tran S, Alenghat T, Osborne L, Thome J, Willis C, Budelsky A, Farber D, Artis D (2015) The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol 8:1313
Wu SH, Chen XQ, Liu B, Wu HJ, Dong L (2013) Efficacy and safety of 15(R/S)-methyl-lipoxin A(4) in topical treatment of infantile eczema. Br J Dermatol 168:172–178
Xue L, Salimi M, Panse I, Mjosberg JM, McKenzie AN, Spits H, Klenerman P, Ogg G (2014) Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy Clin Immunol 133:1184–1194
Yang Z, Tang T, Wei X, Yang S, Tian Z (2015) Type 1 innate lymphoid cells contribute to the pathogenesis of chronic hepatitis B. Innate Immun 21:665–673
Yeremenko N, Noordenbos T, Blijdorp I, Hreggvidsdottir H, Germar K, Bernink J, Spits H, Baeten D (2015) AB0049 human type 1 innate lymphoid cells accumulate in the inflamed synovium in spondyloarthritis. Ann Rheum Dis 74:906
Zaiss DM, Yang L, Shah PR, Kobie JJ, Urban JF, Mosmann TR (2006) Amphiregulin, a TH2 cytokine enhancing resistance to nematodes. Science 314:1746
Zaiss DM, Gause WC, Osborne LC, Artis D (2015) Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42:216–226
Zhao Y, Ma T, Chen W, Chen Y, Li M, Ren L, Chen J, Cao R, Feng Y, Zhang H, Shi R (2016) MicroRNA-124 promotes intestinal inflammation by targeting aryl hydrocarbon receptor in Crohn’s disease. J Crohns Colitis 10:703–712
Zhou W, Toki S, Zhang J, Goleniewksa K, Newcomb DC, Cephus JY, Dulek DE, Bloodworth MH, Stier MT, Polosuhkin V, Gangula RD, Mallal SA, Broide DH, Peebles RS Jr (2016) Prostaglandin I2 signaling and inhibition of group 2 innate lymphoid cell responses. Am J Respir Crit Care Med 193:31–42
Acknowledgements
This work was supported by the National Research Foundation (NRF), funded by the Ministry of Science, ICT, and Future Planning [2015R1C1A2A01054457 to H.M.].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Rights and permissions
About this article

Cite this article
Kim, J., Kim, G. & Min, H. Pathological and therapeutic roles of innate lymphoid cells in diverse diseases. Arch. Pharm. Res. 40, 1249–1264 (2017). https://doi.org/10.1007/s12272-017-0974-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-017-0974-2