
Abstract
Insulin resistance is a characteristic feature of type 2 diabetes mellitus (T2DM) and is characterized by defects in insulin signaling. This study investigated the modulatory effects of fucosterol on the insulin signaling pathway in insulin-resistant HepG2 cells by inhibiting protein tyrosine phosphatase 1B (PTP1B). In addition, molecular docking simulation studies were performed to predict binding energies, the specific binding site of fucosterol to PTP1B, and to identify interacting residues using Autodock 4.2 software. Glucose uptake was determined using a fluorescent d-glucose analogue and the glucose tracer 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxyglucose, and the signaling pathway was detected by Western blot analysis. We found that fucosterol enhanced insulin-provoked glucose uptake and conjointly decreased PTP1B expression level in insulin-resistant HepG2 cells. Moreover, fucosterol significantly reduced insulin-stimulated serine (Ser307) phosphorylation of insulin receptor substrate 1 (IRS1) and increased phosphorylation of Akt, phosphatidylinositol-3-kinase, and extracellular signal- regulated kinase 1 at concentrations of 12.5, 25, and 50 µM in insulin-resistant HepG2 cells. Fucosterol inhibited caspase-3 activation and nuclear factor kappa B in insulin-resistant hepatocytes. These results suggest that fucosterol stimulates glucose uptake and improves insulin resistance by downregulating expression of PTP1B and activating the insulin signaling pathway. Thus, fucosterol has potential for development as an anti-diabetic agent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus (T2DM) consists of an array of dysfunctions characterized by hyperglycemia resulting from a combination of resistance to insulin action, inadequate insulin secretion, and excessive or inappropriate glucagon secretion. Insulin action is regulated by binding to insulin receptors (IR) on insulin-sensitive tissues, such as the liver, adipose tissue, and skeletal muscle (Leng et al. 2004). Therefore, insulin resistance is characterized by inadequate regulation of nutrient metabolism and glucose uptake in numerous tissues and organs, including the liver. Stimulation of glucose uptake by organs and tissue as well as specific molecular targets in the insulin signaling pathway is a novel therapeutic approach for T2DM (Moller 2001; Saltiel 2001). Thus, agents that stimulate glucose uptake and improve insulin resistance are generally useful for managing T2DM (Xie et al. 2006). Insulin signal transduction is initiated when insulin binds to the IR, which stimulates IR intrinsic kinase activity and activates a number of kinases, including Akt and extracellular signal- regulated kinase (ERK). This also triggers the phosphatidylinositol-3-kinase (PI3K) pathway, which activates protein kinase B and inhibits glycogen synthase kinase-3 by phosphorylation, resulting in promoted glucose transport (Klover and Mooney 2004). Phosphorylated Akt signaling cascades also have a critical role in cell survival and apoptosis regulation, whereas Akt promotes cell survival by inhibiting programmed cell death through phosphorylation and inactivation of particular members of caspase family, such as caspase-3 (Franke et al. 2003). ERK is activated via the IR-SHC-RAF-MEK1-ERK signal transduction pathway (Saltiel and Pessin 2002).
Protein tyrosine phosphatase 1B (PTP1B) is a negative regulator of the insulin signaling cascade and has attracted intensive investigation in a recent T2DM therapy study (Johnson et al. 2002; Zhang and Zhang 2007). PTP1B is localized in the cytoplasmic face membrane of the endoplasmic reticulum and is widely expressed in insulin-sensitive tissues, such as the liver, muscle, and adipose tissues (Tonks 2003). PTP1B dephosphorylates the IR as well as the tyrosine residues of IRS1, and increase serine 307 phosphorylation leading to insulin resistance (Zabolotny et al. 2004; Xie et al. 2006). Deletion of PTP1B in liver, muscle and fat tissues enhances insulin sensitivity (Clampit et al. 2003; Wu et al. 2001). In vitro and in vivo evidence confirms that PTP1B is an exciting target for T2DM treatment and drug development. Although the significance of PTP1B in the regulation of insulin signaling has been widely reported, the role of PTP1B as an apoptosis modulator has only been reported a few times. A deficiency in PTP1B protects hepatocytes against serum depletion-induced apoptosis (Gonzalez-Rodriguez et al. 2007). Downregulation of PTP1B effectively protects against apoptotic cell death (Song et al. 2008). Furthermore, PTP1B-null mice are more resistant to Fas-induced liver damage than that of wild-type mice (Sangwan et al. 2006). However, it remains unclear whether a PTP1B inhibitor can attenuate HepG2 cell apoptosis. One study showed that overexpression of PTP1B results in an insulin-resistant state. Thus, PTP1B has been considered a promising insulin-sensitive drug target for preventing and treating T2DM (He et al. 2014).
A marine source has served as a rich source of health-promoting components (Barrow and Shahidi 2008). For many years, traditional Western pharmacognosy has focused on identifying and investigating medically important plants and animals in the terrestrial environment, although many marine organisms are used in traditional Chinese medicine. Among marine sources, edible seaweeds are an underexploited plant resource of biologically active secondary metabolites and exhibit a wide range of therapeutic properties (Li and Kim 2011). Seaweeds should receive increasing acceptance worldwide with the current trend for consumers to embrace organically grown and natural foods from clean environment. Several Asian cultures (mainly Korea, China, and Japan) have a strong tradition of using different varieties of seaweed extensively in cooking as well as in herbal medicine preparations. Various edible marine algae sometimes referred to as seaweed, have attracted interest as good source of nutrients. Indeed, one notable feature of edible seaweeds is their richness in phlorotannins, sulfated polysaccharides, carotenoid pigments, phytosterols, and bioactive peptides (Chandini et al. 2008). Fucosterol, isolated from Ecklonia stolonifera, is the predominant sterol in brown seaweeds, comprising 83–97 % of total sterol content (Sanchez-Machado et al. 2004). Fucosterol has numerous biological activities, such as anti-cancer, cholesterol-reducing, antidiabetic, antioxidant, anti-adipogenic, anti-fungal, anti-histaminic, anti-cholinergic, anti-inflammatory, and butyrylcholinesterase inhibitory activities (Jung et al. 2014; Abdul et al. 2016). Although fucosterol exhibits antidiabetic activity, no detailed information is available about the specific PTP1B binding site or the molecular mechanism of fucosterol against T2DM. Therefore, we report herein the effects of fucosterol on insulin-resistant HepG2 cells and possible mechanism in vitro.
Materials and methods
Chemicals
Fucosterol (Fig. 1) was isolated from Ecklonia stolonifera as described by Jung et al. (2013) and its purity (99 %) was determined by HPLC as described elsewhere (Hwang et al. 2012). The fluorescent d-glucose analogue and glucose tracer 2-[N-(7-nitrobenz-2-oxa-1, 3-diazol-4-yl) amino]-2-deoxy-d-glucose (2-NBDG) was purchased from Life Technologies (Carlsbad, CA, USA). Metformin (hydrochloride), phenylmethylsulfonyl fluoride (PMSF), and bovine serum albumin (BSA) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Insulin from bovine pancreas was purchased from Sigma–Aldrich (St. Louis, MO, USA). Minimum essential medium (MEM), penicillin–streptomycin, 0.25 % trypsin-ethylenediaminetetraacetic acid (EDTA), fetal bovine serum (FBS), sodium pyruvate, and nonessential amino acids were purchased from Gibco-BRL Life Technologies (Grand Island, NY, USA). Phospho-Akt (Ser473) (D9E) rabbit monoclonal antibody, and the caspse-3 antibody were obtained from Cell Signaling Technology (Danvers, MA, USA). PTP1B, IRS-1 antibody, p-IRS-1 (Ser307), Akt, PI3-kinase, p-PI3-kinase p85 (Tyr508), ERK, p-ERK, β-actin, and all secondary antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). All other reagents were purchased from Sigma–Aldrich.

Structure of fucosterol
Cell culture
The human hepatocarcinoma HepG2 cell line was purchased from the American Type Culture Collection (HB-8065; Manassas, VA, USA). Cells were maintained in MEM containing 2.0 mM l-glutamine, 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate, and 10 % FBS at 37 °C in a humidified atmosphere with 5 % CO2. The medium was changed every 48 h. Samples were dissolved in DMSO before being added to the cells; the final concentration of DMSO did not exceed 0.1 %.
Cell viability assay
Cell viability was assessed using the MTT assay as described previously (Mossman 1983). In brief, HepG2 cells were seeded in a 96-well plate at a density of 2 × 105 cells/well and incubated at 37 °C for 24 h. The cells were fed fresh serum-free MEM containing various concentrations (25, 50, 100, and 200 μM) of fucosterol, and incubated for 24 and 48 h. Then, the cells were incubated with the 100 μl of MTT solution in 0.5 mg/ml in phosphate-buffered saline (PBS) and incubated for 2 h. To measure the proportion of surviving cells, the medium was replaced with 100 μl DMSO (100 %). Control cells were treated with 0.1 % DMSO; this concentration was not cytotoxic as measured by this assay. The resulting absorbance values were measured at 570 nm with a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
2-NBDG glucose uptake
HepG2 cells were routinely grown in MEM supplemented with 10 % (v/v) FBS, streptomycin (100 mg/mL), and penicillin (100 U/mL), in a humidified atmosphere of 95 % air–5 % CO2 at 37 °C. The insulin-resistant HepG2 cell model and glucose uptake assay were established according to a previous method (Jung et al. 2011; Liu et al. 2015; Xie et al. 2006) with slight modifications. Briefly, HepG2 cells were cultured in 96-well plates and in black, clear, flat-bottomed 96-well cell culture plates (Corning, Corning, NY, USA). After reaching 80 % confluence, the cells were treated with 10−6 M insulin for 24 h to induce insulin resistance as judged by a 64 % decrease in glucose uptake (data not shown). Then, different concentrations of fucosterol (12.5, 25, and 50 µM) or 10 µM metformin hydrochloride were added for 24 h and then incubated with 100 nM insulin for 30 min. After this incubation, 2-NBDG uptake by insulin-resistant HepG2 cells was measured. The cells were incubated with 40 μM 2-NBDG for 15–20 min., The cells were washed two to three times with ice-cold PBS to stop the reaction, and 2-NBDG fluorescence intensity was measured on a microplate reader (FL × 800; Bio-Tek Instruments Inc., Winooski, VT, USA) at 485 nm excitation and 528 nm emission. Five replicate wells were established, and each experiment was repeated three times.
Preparation of cell lysates and western blot analysis
Insulin-resistant HepG2 cells (density, 3 × 105 cells/well) in 6-well plates were treated with different concentrations of fucosterol for 24 h (Liu et al. 2015). After stimulation with 100 nM insulin for 30 min at 37 °C, the cells were washed three times with ice-cold PBS, collected, and lysed in sample buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 2.5 mM EDTA, 0.5 % NP-40, 1 mM PMSF, 1 mM DTT, 0.2 % aprotinin, 0.5 % leupeptin, 20 mM NaF, and 1 mM Na3VO4) on ice for 30 min. Insoluble material was removed by centrifugation at 25,000×g for 20 min. The protein concentrations were determined by the modified Bradford protein assay kit using BSA as the standard and stored at −80 °C until the Western blotting analyses. Protein samples from insulin-resistant HepG2 cells were separated by 10 % sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Burlington, MA, USA). The membranes were blocked with 5 % skim milk in Tris-buffered saline containing 0.1 % Tween 20 (TBST) for 2 h at room temperature and were then incubated with primary antibodies for 18 h at 4 °C. The following day, the membranes were washed in TBST for 30 min and probed with a secondary antibody for 3 h. The bands were detected using the Supersignal West Pico chemiluminescence substrate (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. The protein bands were visualized by X-ray film (Kodak, Rochester, NY, USA). Prestained blue protein markers were used to determine molecular weight. The bands were scanned using a scanner and quantitated using CS analyzer software (Atto Corp., Tokyo, Japan) in which the normal control was set to 1 (Kandasamy and Ashokkumar 2014). β-Actin antibody was used for normalization. Comparisons were made between mean values of bands within the same gel.
Molecular docking simulation of PTP1B inhibition
A docking study was performed to examine qualified binding poses of fucosterol against PTP1B. The X-ray crystallographic structure of a well known potent, selective PTP1B inhibitor [3-({5-[(n-acetyl-3-{4-[(carboxycarbonyl)(2-carboxyphenyl)amino]-1-naphthyl}-lalanyl)amino]pentyl} oxy)-2-naphthoic acid)], called compound 23 (Szczepankiewicz et al. 2003) and obtained from the RCSB Protein Data Bank (Berman et al. 2002) was determined using a linked-fragment strategy (PDB ID: 1NNY) at a resolution of 2.40 Å. The 515 heteroatoms were removed for the docking simulations; the protein was regarded as ligand-free, and water molecules were removed from the protein structure using Accelrys Discovery Studio 4.1 (DS, http://www.accelrys.com; Accelrys, Inc. San Diego, CA, USA). Polar hydrogen atoms were added to the protein using the Autodock 4.2 automated docking tool (Jones et al. 1997; Goodsell et al. 1996; Rarey et al. 1996). The 515 binding area of the protein was considered the most likely region for a docking simulation with respect to obtaining the best ligand binding results. The three-dimensional structure of the fucosterol constituents was obtained from Pub-Chem compound (NCBI). We used Autodock 4.2 for the docking simulation, which is an open-source program that significantly improves mean binding mode prediction accuracy compared to that of Autodock 4 (Trott and Olson 2010). Furthermore, the grid box size of 126 × 126 × 126 points with spacing of 0.375 Å between points was generated to cover almost the entire favorable protein binding site. The X, Y, and Z centers were 37.303, 30.97, and 33.501, respectively. The binding aspects of the PTP1B residues and their corresponding binding affinity scores were regarded as the best molecular interactions. Results were analyzed using UCSF Chimera (http://www.cgl.ucsf.edu/chimera/; Pettersen et al. 2004) and Lig Plot (v.1.4.5). Two-dimensional figures of the fucosterol–PTP1B interactions were groomed using Lig Plot (v.1.4.5). All docking simulations were performed using an Intel® Core ™ i5-2520 M CPU @ 2.50 GHz with Windows 8.1 and 64-bit operating system.
Statistical analysis
Data are expressed as the mean ± standard deviation (SD) of at least three separate experiments unless otherwise indicated. One-way analysis of variance and Student’s t test were used to detect differences (Systat Inc., Evanston, IL, USA). A P value < 0.05 was considered significant.
Results
Toxicity of fucosterol to HepG2 cells
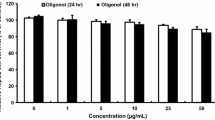
We initially examined toxicity of fucosterol to HepG2 cells. The HepG2 cells were treated with different concentrations of fucosterol for different time intervals, and cell viability was measured by the MTT assay. As shown in Fig. 2, up to 100 µM fucosterol for 24 did not reduce survival of HepG2 cells, but 48 h survival decreased at 200 µM fucosterol. Accordingly, further in vitro studies on the anti-diabetic activity of fucosterol were conducted with the 50, 25, and 12.5 µM non-toxic concentrations.

Cytotoxicity of fucosterol. HepG2 cells were incubated with 0–200 µM fucosterol for 24 or 48 h, and cell viability was measured by the MTT assay
Effect of fucosterol on glucose uptake
To examine the ability of fucosterol to increase glucose uptake, the 2-NBDG uptake assay was performed in insulin-resistant HepG2 cells. Fucosterol significantly increased insulin-stimulated uptake of 2-NBDG in a dose-dependent manner in insulin-resistant HepG2 cells at concentrations of 12.5, 25, and 50 μM (Fig. 3). The stimulatory effect of 50 µM fucosterol was greater than that of normal and metformin controls.

Effect of fucosterol on insulin-stimulated glucose uptake by insulin-resistant HepG2 cells. The glucose uptake assay was performed using the fluorescent d-glucose analogue 2-NBDG, and 10−6 M insulin was used for insulin resistance. The insulin-resistant HepG2 cells were treated with different concentrations of fucosterol or metformin for 24 h, and insulin-stimulated 2-NBDG uptake was measured. Values are mean ± SD. A p value <0.05, was considered significant compared with control
Effect of fucosterol on PTP1B expression level in insulin-resistant HepG2 cells
PTP1B negatively regulates insulin signaling, and its increased activity and expression are implicated in the pathogenesis of insulin resistance. To test whether PTP1B expression was affected by fucosterol, insulin-resistant HepG2 cells were incubated with selected concentrations of fucosterol for 24 h. As shown in Fig. 4, treating insulin-resistant HepG2 cells with 12.5, 25 and 50 μM, fucosterol decreased PTP1B expression level in a concentration-dependent manner. Interestingly, 50 µM fucosterol inhibited PTP1B expression more than the normal control group (P < 0.001). These results indicate that fucosterol improves insulin signaling by blocking PTP1B expression.

Effect of fucosterol on protein tyrosine phosphatase 1B (PTP1B) expression level in insulin-resistance HepG2 cells. Protein band intensities were quantified by densitometric analysis. Western blotting was performed as reported method. Upper panels display representative blots. Equal protein loading was ensured and normalized against β-actin levels. * P < 0.05, **P < 0.01, and ***P < 0.001 were determined by a t test compared with control
Effects of fucosterol on the IR/IRS-1/PI3K/Akt signaling pathway
To determine the molecular mechanism underlying the effects of fucosterol on insulin signaling, we determined the expression levels of proteins involved in the insulin signaling pathway by western blotting. Our results show that fucosterol dose-dependently decreased p-IRS-1 expression (Figs. 5, 6a) towards the levels observed in control insulin-sensitive cells. Fucosterol also significantly and dose-dependently increased the relative abundances of p-Akt (Figs. 5, 6b) and p-PI3K (Figs. 5, 6c) without affecting the expression levels of total Akt or PI3K (Fig. 5). Metformin was used in these experiments as a positive control. These findings indicate that fucosterol decreased p-IRS-1 (Ser307) expression levels and activated the downstream PI3K/Akt signaling pathway, and, therefore, enhanced insulin sensitivity in insulin-resistant HepG2 cells by stimulating glucose uptake.

Fucosterol reduced serine 307 phosphorylation of insulin receptor substrate 1 (IRS1), and restored phosphorylation of phosphatidylinositol-3-kinase (PI3K)/Akt, and extracellular regulated kinase1 (ERK1) in a dose-dependent manner. Western blotting was performed to determine total and phosphorylated protein expression levels. Protein band intensities were quantified by densitometric analysis. Equal protein loading was ensured and normalized against β-actin levels

Densitometric analysis of fucosterol on the levels of phosphorylated and total insulin receptor substrate 1 (IRS1)/phosphatidylinositol-3-kinase (PI3K)/Akt/extracellular regulated kinase1 (ERK1) in insulin-resistant HepG2 cells by using CS analyzer eng software. Values are expressed as ratios of pIRS-1/IRS-1, pPI3K/PI3K, p-Akt/Akt, and pERK1/ERK1. Mean ± standard deviations of three independent experiments are shown * P < 0.05, **P < 0.01, and ***P < 0.001 by a t test compared with control
Effects of fucosterol on phosphorylated and total levels of ERK1 in insulin-resistant HepG2 cells
To test the effects of fucosterol on ERK1 expression, insulin-resistant HepG2 cells were incubated with the same concentrations of fucosterol, and phosphorylated and total ERK 1 (Fig. 5) were evaluated in cell lysates by Western blotting. As shown in Fig. 6d, treating the insulin-resistant cells with any of the three concentrations of fucosterol increased the ERK1 phosphorylation level.
NF-κB inhibitory activity by fucosterol
NF-κB plays a primary role in insulin resistance and T2DM. Thus, in these experiments, we investigated the effect of fucosterol on NF-κB p65 protein levels using a NF-κB p65 antibody in a Western blot analysis. As shown in Fig. 7, NF-κB p65 protein expression increased significantly in the model group compared with that in the control group. The results also showed that fucosterol (12.5, 25, and 50 µM) decreased NF-κB p65 protein expression in insulin-resistant HepG2 cells in a dose-dependent manner.

Effect of fucosterol on nuclear factor kappa beta (NF-κB) expression level in insulin-resistant HepG2 cells. Protein band intensities were quantified by densitometric analysis. After Western blotting. Upper panels display representative blots. Equal protein loading was ensured and normalized against β-actin levels. * P < 0.05, and **P < 0.01 using a t test compared with controls
Fucosterol prevents caspase-3 activation
To test the effects of fucosterol on caspase-3 activation, insulin-resistant HepG2 cells were incubated with selected concentrations of fucosterol for 24 h. Figure 8 shows that fucosterol attenuated insulin-resistant cell death in a dose-dependent manner. Interestingly, 50 µM fucosterol inhibited activation of caspase-3 as in the normal control, whereas metformin did not inhibit caspase-3.

Inhibitory effects of fucosterol on caspase-3 activation in insulin-resistant HepG2 cells. Upper panels display representative Western blots. Protein band intensities were quantified by densitometric analysis. Equal protein loading was ensured and normalized against β-actin levels. **P < 0.01 using a t test compared with control
Molecular docking simulation studies
We next used molecular docking studies to obtain accurate predictions of the protein–ligand interaction geometries of fucosterol and PTP1B. Compound 23 (Szczepankiewicz et al. 2003) was used as a control ligand for validation in Autodock 4.2 (Fig. 9c). The docking scores of fucosterol with interacting residues as well as the number of hydrogen bonds formed between interacting residues and Vander Waals interacting force residues are shown in Table 1. Activity of fucosterol against PTP1B was correlated with binding energy and the number of hydrogen bonds formed at the active site. The binding affinity of fucosterol towards PTP1B was −8.39 kcal/mol, and Gln78, Ser80, Pro206, Ser205, Lys73, Pro210, Lys103, Gln102, His208, Gly209, Val211, Leu204, and Arg79 were identified as interacting residues (Fig. 9b). Fucosterol bound to PTP1B on a single selected hydrogen bond (Fig. 9a, b; Table 1). Specifically, fucosterol binding involved formation of a specific hydrogen bond with a GLU101-interacting oxygen atom group and the fucosterol hydroxyl group at position 3. Similarly, Gln78, Ser80, Pro206, Ser205, Gly209, Val211, Leu204, and Arg79 residues formed Vander Waals interactions with positions 22–29 of fucosterol, and the remaining residues formed Vander Waals interactions with other fucosterol positions, thereby strengthening the protein–ligand interaction between PTP1B and fucosterol.

Binding of fucosterol and compound 23 to the protein tyrosine phosphatase 1B (PTP1B) active site. The structural ligands were analyzed by Autodock 4.2 to depict the docked conformation of these compounds with PTP1B (PDB ID:1NNY) (a, b, c). Hydrogen bonding and hydrophobic interactions for the PTP1B–fucosterol (b) and PTP1B–compound 23 complexes (c)
Discussion
Prolonged hyperglycemia in patients with T2DM has been implicated in the pathogenesis of diabetic complications through several mechanisms, including insulin resistance, overexpression of PTP1B, and excessive oxidative stress, such as overproduction of superoxide, glycoxidation, and protein glycation. Among them, the PTP1B pathway has been proposed as a crucial target for developing drugs to treat diabetic complications (Laakso 1999). On the other hand, induction glucose uptake and glycogen accumulation as well stimulating insulin signaling are important anti-hyperglycemic mechanisms performed in hepatocytes (Shen et al. 2008). Insulin plays an important role in hepatic glycogen synthesis (Newgard et al. 2000). In the presence of high glucose, glycogen synthesis and levels are significantly downregulated in response to insulin stimulation in human HepG2 hepatoma cells (Lin and Lin 2008). Impaired glucose uptake leads to high circulating glucose levels; therefore, a strategy to treat T2DM is to stimulate glucose uptake by skeletal muscle, liver, and adipose tissue, where most plasma glucose is consumed. High-concentration insulin can causes insulin resistance in HepG2 cells, which show an insensitive response to glucose uptake and insulin signaling (Xie et al. 2006). We checked the glucose uptake effect of fucosterol in insulin-resistant HepG2 cells after 24 h incubation with 10−6 M insulin. Fucosterol improved insulin-provoked glucose uptake in insulin-resistant HepG2 cells in a dose-dependent manner. Western blotting was used to evaluate the potential mechanism of fucosterol to activate the insulin signaling pathway in insulin-resistant HepG2 cells. The biological effect of insulin is initiated by insulin binding to the α-subunit of the IR and activating intrinsic tyrosine kinase activity of the receptor β-subunit. Activation of the IR leads to tyrosine phosphorylation of IRS (White 2002), phosphorylated IRS activates PI3K (Cross et al. 1995; Franke et al. 2003), and the activated PI3K phosphorylates Ser/Thr kinase Akt. On the other hand, activating Akt promotes translocation of the intracellular glucose transporter 4 to the plasma membrane, resulting in enhanced glucose uptake. PTP1B is a key negative regulator of insulin signaling cascades (Byon et al. 1998), and IRβ and IRS-1 are two major PTP1B substrates. Some studies have reported that a PTP1B inhibitor increases tyrosine phosphorylation of the IR, and activates downstream insulin signaling molecules, such as IRS-1, Akt, and ERK1 (Takada et al. 2012; Maeda et al. 2014). In the present study, the effects of fucosterol on PTP1B expression and phosphorylated and total IRS, PI3K, and Akt in insulin-resistant HepG2 cells were evaluated. Fucosterol decreased PTP1B expression and Ser307 of p-IRS-1 in a dose-dependent manner and increased phosphorylation of PI3K, and Akt significantly (Figs. 4–6a–c) compared to the normal and positive controls. Additionally, ERK1, a subfamily of mitogen-activated protein kinases, is a downstream insulin signaling kinase. In this study, fucosterol increased ERK1 phosphorylation in insulin-resistant HepG2 cells (Figs. 5, 6d). Overall, these data reveal that fucosterol decreased PTP1B expression leading to increased phosphorylation of PI3K, Akt, and ERK1 in insulin-resistant HepG2 cells, which activated the IRS-1 and PI3K/Akt signaling pathways in these cell lines. Furthermore, nuclear factor-κB is a nuclear transcription factor found in all cell types and is involved in cellular responses to stimuli. NF-κB plays a primary role in insulin resistance and T2DM (Patel and Santani 2009). Inhibiting this transcription factor can have deleterious effects, leading to the development of insulin resistance and T2DM (Patel and Santani 2009). Fucosterol dose-dependently inhibited NF-kB (Fig. 7). Furthermore, caspase-3 is activated in apoptotic cells by the extrinsic (death ligand) and intrinsic (mitochondrial) pathways. Hepatic insulin resistance is associated with increased apoptosis after activation of the caspase family, including caspase-3. Inhibiting caspase-3 suppressed apoptosis induced by a high insulin concentration. Our results show that fucosterol inhibited activation of caspase-3 in insulin-resistant HepG2 cells (Fig. 8).
We also studied the fucosterol binding mode, which exhibited potent PTP1B inhibitory activity. A detailed binding mode analysis with a docking simulation showed that the inhibitors can be stabilized by the simultaneous establishment of hydrogen bonds as well Van der Waals interactions with the important PTP1B predicted residues, which defines the putative binding site. The fucosterol structural study showed that PTP1B inhibitory activity was strongly affected by the hydroxyl group at position 3, which interestingly increases proximity to the GLU101 residues through a hydrogen bond. On the other hand, other residues formed Van der Waals interactions with fucosterol resulting in strong binding energy and affinity to inhibit PTP1B, indicating that these inhibitors may reduce mobility toward a more rigid PTP1B conformation. Our previous studies reported kinetics and molecular docking studies of an anti-diabetic complication inhibitor fucosterol from edible brown algae Eisenia bicyclis and E. stolonifera (Jung et al. 2013). In particular, Lee et al. (2004) revealed that fucosterol has anti-diabetics effects when a dose of 30 mg/kg administered orally to streptozotocin-induced diabetic rats. Although previous report have shown that fucosterol possess anti-diabetic activities, no study has explained the fucosterol mechanism against PTP1B by evaluating glucose uptake with a molecular docking simulation. Thus, we investigated the possible mechanism of fucosterol, which activates insulin signaling by inhibiting PTP1B activity and enhancing insulin sensitivity. In summary, fucosterol exerted potent and efficacious anti-diabetic effects by inhibiting PTP1B and activating IRS-1 and PI3K/Akt signaling pathways in insulin-resistant HepG2 cells. These beneficial effects were mediated by stimulating glucose uptake, decreasing PTP1B.
References
Abdul QA, Choi RJ, Jung HA, Choi JS (2016) Health benefit of fucosterol from marine algae. J Sci Food Agric 96:1856–1866
Barrow C, Shahidi F (2008) Marine nutraceuticals and functional foods. CRC Press, New York, pp 185–187
Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C (2002) The protein data bank. Acta Crystallogr D Biol Crystallogr 58:899–907
Byon JCH, Kusari AB, Kusari J (1998) Protein-tyrosine phosphatase-1B acts as a negative regulator of insulin signal transduction. Mol Cell Biochem 182:101–108
Chandini SK, Ganeshan P, Suresh PV, Bhaskar N (2008) Seaweeds as a source of nutritionally beneficial compounds-A review. J Food Sci Technol 45:1–13
Clampit JE, Meuth JL, Smith HT, Reilly RM, Jirousek MR, Trevillyan JM, Rondinone CM (2003) Reduction of protein-tyrosine phosphatase-1B increases insulin signaling in FAO hepatoma cells. Biochem Biophys Res Commun 300:261–267
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–789
Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C (2003) PI3K/Akt and apoptosis: size matters. Oncogene 22:8983–8998
Gonzalez-Rodriguez A, Escribano O, Alba J, Rondinone CM, Benito M, Valverde AM (2007) Levels of protein tyrosine phosphatase 1B determine susceptibility to apoptosis in serum-deprived hepatocytes. J Cell Physiol 212:76–88
Goodsell DS, Morris GM, Olson AJ (1996) Automated docking of flexible ligands: applications of autodock. J Mol Recognit 9:1–5
He RJ, Yu ZH, Zhang RY, Zhang ZY (2014) Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin 35:1227–1246
Hwang SW, Jang JM, Lim SS (2012) Isolation of fucosterol from pelvetia siliquosa by high-speed countercurrent chromatography. Fish Aquat Sci 15:191–195
Johnson TO, Ermolieff J, Jirousek MR (2002) Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat Rev Drug Discov 1:696–709
Jones G, Willett P, Glen RC, Leach AR, Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. J Mol Biol 267:727–748
Jung DW, Ha HH, Zheng X, Chang YT, Williams DR (2011) Novel use of fluorescent glucose analogues to identify a new class of triazine-based insulin mimetics possessing useful secondary effects. Mol Bio Syst 7:346–358
Jung HA, Islam MN, Lee CM, Oh SH, Lee S, Jung JH, Choi JS (2013) Kinetics and molecular docking studies of an anti-diabetic complication inhibitor fucosterol from edible brown algae eisenia bicyclis and ecklonia stolonifera. Chem Biol Interact 206:55–62
Jung HA, Jung HJ, Jeong HY, Kwon HJ, Kim MS, Choi JS (2014) Anti-adipogenic activity of the edible brown alga ecklonia stolonifera and its constituent fucosterol in 3T3-L1 adipocytes. Arch Pharm Res 37:713–720
Kandasamy N, Ashokkumar N (2014) Protective effect of bioflavonoid myricetin enhances carbohydrate metabolic enzymes and insulin signaling molecules in streptozotocin-cadmium induced diabetic nephrotoxic rats. Toxicol Appl Pharmacol 279:173–185
Klover PJ, Mooney RA (2004) Hepatocytes: critical for glucose homeostasis. Int J Biochem Cell Biol 36:753–758
Laakso M (1999) Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes 48:937–942
Lee YS, Shin KH, Kim BK, Lee S (2004) Anti-diabetic activities of fucosterol from pelvetia siliquosa. Arch Pharm Res 27:1120–1122
Leng Y, Karlsson HKR, Zierath JR (2004) Insulin signaling defects in type 2 diabetes. Rev Endocr Metab Disord 5:111–117
Li YX, Kim SK (2011) Utilization of seaweed derived ingredients as potential antioxidants and functional ingredients in the food industry: an overview. Food Sci Biotechnol 20:1461–1466
Lin CL, Lin JK (2008) Epigallocatechin gallate (EGCG) attenuates high glucose-induced insulin signaling blockade in human HepG2 hepatoma cells. Mol Nutr Food Res 52:930–939
Liu Z-Q, Liua T, Chena C, Li M-Y, Wang Z-Y, Chen R-S, Wei G-X, Wang X-Y, Luo D-Q (2015) Fumosorinone, a novel PTP1B inhibitor, activates insulin signaling in insulin-resistance HepG2 cells and shows anti-diabetic effect in diabetic KKAy mice. Toxicol Appl Pharmacol 285:61–70
Maeda A, Kai K, Ishii M, Ishii T, Akagawa M (2014) Safranal, a novel protein tyrosine phosphatase 1B inhibitor, activates insulin signaling in C2C12 myotubes and improves glucose tolerance in diabetic KK-Ay mice. Mol Nutr Food Res 58:1177–1189
Moller DE (2001) New drug targets for type 2 diabetes and the metabolic syndrome. Nature 414:821–827
Mossman T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxic assays. J Immunol Methods 65:55–63
Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR (2000) Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes 49:1967–1977
Patel S, Santani D (2009) Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharmacol Rep 61:595–603
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Rarey M, Kramer B, Lengauer T, Klebe G (1996) A fast flexible docking method using an incremental construction algorithm. J Mol Biol 261:470–489
Saltiel AR (2001) New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104:517–529
Saltiel AR, Pessin JE (2002) Insulin signaling pathways in time and space. Trends Cell Biol 12:65–71
Sanchez-Machado DI, Lopez-Hernandez J, Paseiro-Losada P, Lopez-Cervantes J (2004) An HPLC method for the quantification of sterols in edible seaweeds. Biomed Chromatogr 18:183–190
Sangwan V, Paliouras GN, Cheng A, Dube N, Tremblay ML, Park M (2006) Protein-tyrosine phosphatase 1B deficiency protects against fas-induced hepatic failure. J Biol Chem 281:221–228
Shen SC, Cheng FC, Wu NJ (2008) Effect of guava (Psidium guajava Linn.) leaf soluble solids on glucose metabolism in type 2 diabetic rats. Phytother Res 22:1458–1464
Song H, Zhang Z, Wang L (2008) Small interference RNA against PTP-1B reduces hypoxia/reoxygenation induced apoptosis of rat cardiomyocytes. Apoptosis 13:383–393
Szczepankiewicz BG, Liu G, Hajduk PJ, Abad-Zapatero C, Pei Z, Xin Z, Lubben TH, Trevillyan JM, Stashko MA, Ballaron SJ, Liang H, Huang F, Hutchins CW, Fesik SW, Jirousek MR (2003) Discovery of a potent, selective protein tyrosine phosphatase 1B inhibitor using a linked-fragment strategy. J Am Chem Soc 125:4087–4096
Takada M, Sumi M, Maeda A, Watanabe F, Kamiya T, Ishii T, Nakano M, Akagawa M (2012) Pyrroloquinoline quinone, a novel protein tyrosine phosphatase 1B inhibitor, activates insulin signaling in C2C12 myotubes and improves impaired glucose tolerance in diabetic KK-A(y) mice. Biochem Biophys Res Commun 428:315–320
Tonks NK (2003) PTP1B: from the sidelines to the front lines. FEBS Lett 546:140–148
Trott O, Olson AJ (2010) Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
White MF (2002) IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 283:413–422
Wu X, Hoffstedt J, Deeb W, Singh R, Sedkova N, Zilbering A, Zhu L, Park PK, Arner P, Goldstein BJ (2001) Depot-specific variation in protein-tyrosine phosphatase activities in human omental and subcutaneous adipose tissue: a potential contribution to differential insulin sensitivity. J Clin Endocrinol Metab 86:5973–5980
Xie W, Wang W, Su H, Xing D, Pan Y, Du L (2006) Effect of ethanolic extracts of Ananas comosus L. leaves on insulin sensitivity in rats and HepG2. Comp Biochem Physiol C 143:429–435
Zabolotny JM, Haj FG, Kim YB, Kim HJ, Shulman GI, Kim JK, Neel BG, Kahn BB (2004) Transgenic overexpression of protein tyrosine phosphatase 1B in muscle causes insulin resistance, but overexpression with leukocyte antigen-related phosphatase does not additively impair insulin action. J Biol Chem 279:24844–24851
Zhang S, Zhang ZY (2007) PTP1B as a drug target: recent developments in PTP1B inhibitor discovery. Drug Discov Today 12:373–381
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (2012R1A6A1028677).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest.
Additional information
Hyun Ah Jung and Himanshu Kumar Bhakta have contributed equally to this study.
Rights and permissions
About this article

Cite this article
Jung, H.A., Bhakta, H.K., Min, BS. et al. Fucosterol activates the insulin signaling pathway in insulin resistant HepG2 cells via inhibiting PTP1B. Arch. Pharm. Res. 39, 1454–1464 (2016). https://doi.org/10.1007/s12272-016-0819-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0819-4