
Abstract
MALDI mass spectrometry imaging (MSI) provides a technology platform that allows the accurate visualization of unlabeled small molecules within the two-dimensional spaces of tissue samples. MSI has proven to be a powerful tool-box concept in the development of new drugs. MSI allows unlabeled drug compounds and drug metabolites to be detected and identified and quantified according to their mass-to-charge ratios (m/z) at high resolution in complex tissue environments. Such drug characterization in situ, by both spatial and temporal behaviors within tissue compartments, provide new understandings of the dynamic processes impacting drug uptake and metabolism at the local sites targeted by therapy. Further, MSI in combination with histology and immunohistochemistry, provides the added value of defining the context of cell biology present at the sites of drug localization thus providing invaluable information relating to treatment efficacy. In this report we provide mass spectrometry imaging data within various cancers such as malignant melanoma in patients administered with vemurafenib, a protein kinase inhibitor that is targeting BRAF mutated proteins and that has shown significant efficacy in restraining disease progression. We also provide an overview of other examples of the new generation of targeted drugs, and demonstrate the data on personalized medicine drugs localization within tumor compartments within in vivo models. In these cancer models we provide detailed data on drug and target protein co-localization of YCG185 and sunitinib. These drugs are targeting VEGFR2 within the angiogenesis mechanism. Our ability to resolve drug uptake at targeted sites of directed therapy provides important opportunities for increasing our understanding about the mode of action of drug activity within the environment of disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ever increasing demands in healthcare today posture high prospects and expectations onto the research community to invent and establish new solutions that can improve clinical outcome at lower cost. In response to these challenges, modern healthcare is looking for ways to treat patients that are both more efficient as well as more cost saving (Young et al. 2014; Fu et al. 2010; Dwyer 2011). It has to be taken into account that these improvements are expected to be managed without the necessary care that patients demand today. In these strives, large-scale patient repositories, biobanks with high quality of clinical materials are mandatory (Baker 2012; Marko-Varga et al. 2014). The introduction of regulatory directives are central to our research community in order to meet the demands from, e.g., cancer patients, that are expecting more safe drugs with lower mortalities and a fast onset of efficacy.
Together with the National Cancer Institute (NIH) and local clinicians and scientists there has been extensive progress made to work out and provide a protein biomarker discovery and validation strategy (Nilsson et al. 2015; Nishimura et al. 2014; Kato et al. 2011). These regulatory guidelines provide high-quality standardized, sensitive, specific, quantitative, and readily accessible protein, peptide, or other biomarkers of health, disease, response to therapy into the approval processes of regulatory agencies, such as the Food and Drug Administration (FDA) in USA and European Medicines Agency (EMA) in Europe. These developments are vital for healthcare professionals to improve diagnosis, detect cancer at an early stage, identify the likelihood of cancer recurrence, and stratify stages with differential survivals.
There is a growing trend in developing biorepositories around the focus of large population-based studies that address both active and silent non-symptomatic disease. Logistically these studies generate large numbers of clinical samples and practically place increasing demand upon healthcare systems to provide uniform sample handling, processing, storage, and documentation of both the sample and the subject as well to ensure that safeguards exist to protect the rights of the study subjects for deciding upon the fates of their samples. There has also been outlines with practical information to the potential users of biorepositories about some of the current developments in both the methodology of sample acquisition and in the regulatory environment governing their use (Marko-Varga et al. 2014).
Today, many millions of clinical samples are acquired every day for use in diagnostic tests that support clinical decision making. Worldwide, it is estimated that over one billion clinical samples are assembled into so called biobanks (Baker 2012; Marko-Varga et al. 2014). The preservation of patient samples is an important undertaking as the healthcare costs are increasing exponentially.
The link in-between disease pathology and samples representing these various phenotypes, on one hand and the characterization of drug compounds on the other in order to build a mechanistic understanding at a molecular level is at the heart of the FDA requests for drug approvals. Key to the process of developing new drugs is the need to deliver a complete package of information regarding both the effects of the drug uptake both within the targeted compartments of disease but also at sites throughout the body that may be effected by the pharmacological activities of the drug. The potentially beneficial or toxic effects of such drug therapy are determined by the unique chemical and pharmacological properties of each given drug form. Scientists can test these properties in the laboratory and in experimental models but until recently with the introduction of mass spectrometry imaging (MSI) it has not been possible to measure the unlabeled drug directly within a tissue or organ. In this respect, MSI is a powerful technology platform that we have developed in both pharma industry and academia that has proven to be of great value in the design of new drugs but also in measuring their efficacy and consequent effect at the sites of targeted therapy. We were the first to show drug localization in lung cancer, malignant melanoma, and chronic obstructive pulmonary disease (COPD) patients in joint studies with pharmaceutical industry (Marko-Varga et al. 2014; Nilsson et al. 2015; Nishimura et al. 2014; Kato et al. 2011; Fehniger et al. 2014). Already today, many major pharmaceutical manufacturers have included MSI in their development programs and are reporting on these findings in applications for new drug approval.
In this overview, we present studies in various solid tumors where tissues known to contain kinase mutations as determined by DNA sequencing and other protein receptors that are key cancer regulators have been co-localized with small molecule drug inhibitors using MSI.
Applying mass spectrometry imaging to characterize drugs
Absorption, distribution, metabolism and excretion (ADME) pharmacokinetic properties along with possible drug toxicity and drug metabolism, are key regulatory considerations that the FDA and the EMA demand in order to approve new drug entities, where animal models are commonly used (Nilsson et al. 2010). The mass spectrometry imaging technology is currently utilized in the drug discovery as well as the drug development process within the pharmaceutical industry (Chaurand et al. 2008; Willems et al. 2010). With regard to drug characterization, the MSI technology has been utilized in industry to investigate drug distribution in the main organs.
Nowadays, the matrix-assisted laser desorption ionization (MALDI) MSI is the most common mass spectrometry technology applied within pharma and biotech industry as well as national research centers and academia. MALDI-MSI studies on the clinical status and as risk assessments with respect to disease developments, e.g., cancer states of tumor genesis (Fehniger et al. 2011), and drug responders, as well as long-term survival (Marko-Varga et al. 2012; Sugihara et al. 2014). In addition to our own melanoma studies, studies undertaken by Caprioli et al., was first to present MALDI-MSI data where they used metastatic tumors, identifying calcyclin (Hardesty and Caprioli 2008; Lazova et al. 2012).
At the Center of Excellence in Lund (http://www.CEBMMS.se) we provide a dedicated activity in drug mass spectrometry imaging that apply MALDI Orbitrap mass spectrometers that provide high mass accuracy. MfSI has widespread usage throughout the community with academic clinical and commercial applications. Compared to other methods, which localize drugs in situ, such as autoradiography (ARG) and positron emission tomography (PET), optical fluorescence, MALDI-MSI is label-free and can be applied to active parent drugs and their metabolites within any tissue environment. Using this technology, we have recently provided evidence showing the exact tissue compartment localization of small drug molecules administered to patients.
Drug metabolism and pharmacokinetics (DMPK)
In drug discovery, and development, the investigation of the localization of drug is essential for the understanding of ADME, pharmacokinetics/pharmacodynamics (PK/PD) and toxicokinetics/toxicodynamics (TK/TD). This information can facilitate the decision making for the: (a) GO or (b) NO-GO developments of new drugs.
A desired pharmacological activity is generated by a proper distribution of a drug to a target site within the body. In addition, undesired localizations and accumulation of a drug or its metabolites in non-targeted site(s) can lead to toxicological effects (Pellegatti and Pagliarusco 2011). The distribution of a drug and its metabolites may not be homogeneous in a target tissue and is dependent on the physicochemical properties, tissue affinity and the transporter protein substrate profile of each molecular entity. Although the free drug hypothesis, stating that the unbound concentration of a drug in plasma is in equilibrium with that in tissue, is valid for many drugs, this is not true for drugs and metabolites that are dependent on transporters for their distribution. Therefore, the plasma concentration of drugs and their metabolites does commonly not reflect the levels which are present at the target site and consequently, may not be useful for understanding the efficacy or toxicity of the drug (Mouton et al. 2008; Langer and Muller 2004). It is the ultimate objective of DMPK studies to provide adequate information of drug distribution throughout the body and its metabolite profiles generated in various organs, and studied kinetically over time.
Generally, liquid chromatography coupled to tandem mass spectrometry (LC/MS/MS) is conventionally used for investigating drug levels in the target tissue. However, this method is limited in their usefulness due to the homogenization step of the tissue, which results in a loss of information on the spatial localization of the drug and its metabolites at a single cell resolution (Mouton et al. 2008; Lanao and Fraile 2005).
Autoradiography is traditionally used to determine the localization of the drug in the tissue. This quantitative methodology provides the kinetics associated with drug metabolism and elimination in preclinical studies. However, the synthesis of the radiolabeled drug is an expensive and frequently time-consuming process. Moreover, it can take several weeks of exposure, to develop radiographic images of sufficient sensitivity. In addition, as only the radioactivity of the label is measured, hence the drug cannot be distinguished from its metabolites (Solon et al. 2010).
MSI is an effective technology that makes it possible to determine the detailed distribution of drug in tissue as well as its metabolites without isotope labeling. Consequently, quantitative MSI was rapidly applied to evaluate the distribution of drug compounds (Solon et al. 2010; Nilsson et al. 2010; Koeniger et al. 2011; Takai et al. 2012; Lietz et al. 2013). Recently, high resolution MSI provided a significant advantage of autoradiography from the aspect of the drug metabolites (Fehniger et al. 2011; Jirasko et al. 2014; Liu et al. 2013; Buck et al. 2015) and demonstrated the distribution of a confirmed drug metabolite that possibly lead to the drug toxicity (Török et al. 2015). The technology is today making important contributions in fields such as PK screening (Castellino et al. 2013), novel pro-drug concepts (Nilsson et al. 2010; Fehniger et al. 2011), drug delivery systems (DDS) by micellar nanoparticle formulation (Swales et al. 2014), biomarkers for PD (Yasunaga et al. 2013; Shariatgorji et al. 2014; Cobice et al. 2013) as well as proteomics (Goto et al. 2014; Ye et al. 2014). MSI ultimately leads to a better understanding of ADME, PK/PD and TK/TD. Therefore, MSI in many drug discovery-, and development projects is a crucial technology for decision making. DMPK researchers have desired information of detailed distributions and accurate levels of the drug and its metabolite in target tissue for a long time.
Drug imaging studies
There are currently few therapeutic options for patients with various forms of cancer, and new insights into the pathogenesis of these often lethal diseases are urgently needed. Patients in feasibility studies in combination with disease models in rodents has been the strategy of our research teams for a number of years (Fehniger et al. 2011; Marko-Varga et al. 2012; Sugihara et al. 2014; Török et al. 2015). Further, the correlation in-between a given rodent model with administered drug and the ability to utilize controlled administrations to tissue sections from organs isolated with the intact tumor environment is a valuable tool for drug characterizations. Our team was able to prove that these in vivo and ex vivo drug models hold great promise for the assessment of novel drug candidates (Nilsson et al. 2010; Fehniger et al. 2011; Marko-Varga et al. 2012; Sugihara et al. 2014).
Malignant melanoma
Melanoma in general is a disease with unfavorable prognosis and has one of the highest incidence rates globally. In Sweden approximately 2800 new malignant melanoma (MM) patients are diagnosed every year and almost 500 patients die of disseminated melanoma disease annually. The 5-year survival rate in metastatic melanoma is around 5 % and the median survival is only 6–10 months. At the time of diagnosis 10–15 % of the patients are diagnosed with disseminated disease and hence a poor prognosis. In contrast to most other malignancies, MM is also common in young people. There are no blood or tissue biomarkers currently available for early detection, identifying disease progression or monitoring treatment of MM. Melanoma is a highly lethal malignancy with few effective therapies.
Recently, the FDA approved several drugs, including vemurafenib, dabrafenib, and trametinib, for use in treating MM patients. A key target of current therapy is the BRAF protein kinase, the mutated forms of which can occur in MM. The valine to glutamic acid mutation at amino acid 600 within the BRAF protein is the key targeted site, to which the drugs bind and affect tumor growth. Vemurafenib, a kinase inhibitor (KI), binds to the mutated BRAF site, leading to an interruption of proliferation signaling (Sugihara et al. 2014). The drug has a functional effect causing programmed cell death of MM cells. Vemurafenib also has effect against the rarer V600K mutation, one of several other BRAF protein kinase mutations (Table 1).
To examine the localization of BRAF V600E protein in the MM tissues, we conducted immunohistochemistry with a monoclonal antibody against BRAF V600E (Sugihara et al. 2014). The distribution of melanoma cells, containing the BRAF V600E mutation was homogeneous. Similar to a recent study, we used MM tissues known to contain the BRAF mutation status by DNA sequencing.
Exome sequencing and copy number analysis was applied to define the genomic aberrations in a prospectively accrued clinical cohort of stage III and IV patients. These patients showed sporadic tumor developments in lymph nodes that were isolated by surgery. Detailed analysis of these metastatic lymph nodes identified substantial heterogeneity with non-silent mutations. We and others were able to define significantly mutated genes, reaffirming known mutations from these patients (Tsao et al. 2004; Balch et al. 2009; Welinder et al. 2013; Harbst et al. 2014).
There are two major pathways that form the basis for MM drug studies:
(1) PI3K/Akt/mTOR pathway
(2) Ras-Raf-MEK-ERK pathway
Vemurafenib acts through the blocking of BRAF target with a specific mode-of-drug-action mechanism and this KI has been extensively investigated in clinical studies (Chapman et al. 2011; Bollag et al. 2010).
Drug localization in melanoma tumors
By mass spectrometry imaging, the vemurafenib drug compound was identified at m/z 490.078, as shown in the MALDI image of vemurafenib in Fig. 1. The drug compound with an ion of m/z 490.078 was clearly detected (see Fig. 1a) within BRAF V600E expressed region in the MM tissue, while it was not measurable in patients with wild type BRAF (see Fig. 1b).

Vemurafenib is identified at m/z 490.078 within the tumor regions of the tissue, where the upper MSI image is generated from a a BRAF positive and, b a BRAF negative tumor region
In order to validate the identity of m/z 490.078 in the tissue sections as being the vemurafenib precursor ion, we simultaneously analyzed the tissue in MS/MS mode as well, monitoring the fragmentation process of vemurafenib, as previously described (Sugihara et al. 2014). The patterns of precursor and product ions show strong consistency and coincident overlap with the image of the vemurafenib precursor ion (as shown in Fig. 2). For reference purposes, this same section was later histologically stained (H&E), in order to provide details of the micro-anatomy present in the sample (data not shown).

a Extracted Precursor ion image by a MALDI Orbitrap mass spectrometer platform of vemurafenib at m/z 490.078. b Extracted precursor ion image by a MALDI Orbitrap mass spectrometer platform of vemurafenib fragment, MS1 at m/z 383.1. c Extracted Precursor ion image by a MALDI Orbitrap mass spectrometer platform of vemurafenib fragment, MS2 at 262.1. d Overlay of the extracted precursor ion image MALDI Orbitrap mass spectrometer platform of vemurafenib precursor ion, and its MS1 and MS2 fragment ion images, respectively
These high spatial resolution MS images (in Fig. 2) revealed that the drug indeed localized within the BRAF-positive tumors. These extracted ion maps of vemurafenib at m/z 490.078 (see Fig. 2a), and its two corresponding fragment ions at m/z 383.1 and 262.1, respectively, determined from the MS/MS spectra have given strong evidences that the drug is highly concentrated in the tumor areas (see Fig. 2b, c). The distribution coincide well with the area, where BRAF V600E is highly up-regulated, as reported on previously (Sugihara et al. 2014). In this experiment, we observed the co-localization between drug compound and the respective two fragment ions (see Fig. 2d). The detailed evidence of the BRAF V600E mutation and the drug specific binding in crystal structure analysis was also presented recently (Bollag et al. 2010; Sosman et al. 2012).
Angiogenesis cancer disease models: xenograft models
Distribution of anti-angiogenic small molecule YCG185
The development of new anti-angiogenic agents has been considered as an efficient strategy for the treatment of cancer and other diseases related with angiogenesis. However, the clinical impact of development of suppressing angiogenesis remains controversial. Therefore, we focused on elucidating the PK/PD properties with identifying the localization of the anti-angiogenic agent YCG185 and its metabolites with the corresponding target protein on xenograft tumor tissues using MALDI-MSI. In previous studies, the compounds were also identified as in vitro and in vivo anti-angiogenic agents. We optimized the assay to provide high signal response of YCG185 and its metabolites on tissue samples using MALDI-MSI. In the present study, we performed immunofluorescence staining for detecting VEGFR2, the target candidate of YCG185, and determined the correlation of localization between YCG185 and VEGFR2 (data not shown). This study can serve as a systemic approach for understanding the pharmacokinetic properties of an unlabeled anti-angiogenic agent. YCG185 was identified by MALDI-MS with a an annotated spectrum in MS mode at m/z 369.21 (see Fig. 3). The histology image capture shown in Fig. 3, also provide evidence on the co-localization of the tumor region within the tissue by histology, and the MALDI-MSI. In addition, immunofluorescence staining could provide novel insights for validating interaction of drug and its targets within the tumor tissues.

Resulting outline from experimental settings with YCG185 providing a characteristic MS spectrum at m/z 369.21. The resulting image capture generated from the MS spectrum is shown together with the histology image (H&E), pointing to the localization of the drug binding area within the tissue
Distribution of anti-angiogenic small molecule sunitinib
The inhibition of tumor vessel growth by anti-angiogenic agents is one of the most intensively investigated fields of translational and clinical oncology. Despite of promising in vitro studies with angiogenic receptor tyrosine kinase inhibitors (RTKIs), both the in vivo experimental models and the clinical experiences were controversial (Jayson et al. 2012; Rapisarda and Melillo 2012; Loges et al. 2010; Moreno Garcia et al. 2012; Abe and Kamai 2013; Amir et al. 2009). Therefore, investigating the PK/PD properties of these compounds is a pivotal issue in furthering the understanding necessary for the development of future drug therapy. As reported in a recent paper, we were able to study the absorption, distribution, metabolism, elimination (ADME) properties of the RTKI sunitinib (Török et al. 2015). This drug compound was evaluated in a subcutaneous syngeneic murine tumor model of colorectal cancer using MALDI-MSI. After 2 weeks of treatment, the tumors and other organs were sectioned and analyzed to localize sunitinib, its fragment ions, and its metabolites within specified tissue microenvironments. The expression patterns of the main angiogenic receptor, which is one of sunitinib’s principle target receptors (VEGFR-2), was also studied by immunofluorescent analysis within the same tumors.
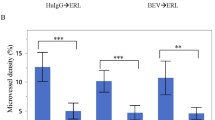
We were able to detect sunitinib in mice with established tumors, showing biological effect by significantly impairing further tumor growth. The precursor ion of sunitinib (m/z 399.218) and its fragment ions (m/z 326.1 and 283.1) were demonstrated by MALDI-MS in blood, tumor, liver and kidney tissues (Fig. 4a). Three peaks of major metabolites of the drug (m/z 371.188, 397.203 and 415.214) were detected both in the blood samples and in tissue sections, while other minor metabolites were detected only in the blood (Török et al. 2015). There were intratumor areas where the signal intensity of drug correlated with the histological density of VEGFR-2 (Fig. 4c). However, we also observed that within tumors, sunitinib treatment resulted in decreased expression of VEGFR-2 compared to tumors developing in non-treated mice (Török et al. 2015) (Fig. 4b).

MSI data of the distribution of sunitinib in a subcutaneously growing C26 tumor. a Signal intensity of m/z 399.218 normalized to total ion current. b Immunolabeling of VEGFR-2 in sunitinib treated and control tumors. c Overlap of the distribution of sunitinib (grey) and VEGFR-2 (green)
To our knowledge, this was the first study that demonstrated MALDI-MSI as a versatile and simple platform to outline the pharmacokinetic properties of an anti-angiogenic TKI, such as sunitinib (Török et al. 2015). This recent study supports the role of MALDI-MSI in the ADME characterization of anti-angiogenic drug candidates in preclinical drug development. The combination of two highly sensitive methods of micro-environment analysis, MALDI-MSI and immunohistochemistry, allows new insights into the molecular action and efficacy of drug compounds at the sites of targeted intervention (Török et al. 2015).
Conclusions
As a vision for the future we strongly believe that advances in MS, especially in structural MS will change the understanding of proteins to the benefit of society. Although the use of MS measurements to address questions of importance in biomedical MS is a relatively recent practice, there have already been major advancements in standard care procedures using MS, for example identification of lethal pathogens in clinical samples in hospitals around the world, thus saving lives every day. We foresee that the interactive expansion of our Center in Lund, the CEBMMS network will drive opportunities for both education and training, thus increasing the workforce in specialists capable of addressing the future applications of protein science both in industry and academia.
Targeted therapies with small molecular inhibitors for MM are developing from bench to bedside. Until 2013 June, three kinase inhibitors for MM have already approved by FDA. Further study will be needed to improve the prognosis for MM patients. Combination of targeting agents against different signaling pathways may provide additional benefits and warrant further clinical studies. In addition, novel agents and improved patient selection by characterization of the molecular targets in individual tumors show great promise and should be incorporated into future studies. The ability to characterize drugs within their targeted tissue environments holds great promise for furthering our understanding of drug actions and thus support the future development of more effective treatments of malignant melanoma.
It is also expected that the HUPO initiative, Chromosome based Human Protein Project (http://www.c-hpp.org) will provide an annotation of unknown proteins coded by the genome that will even further develop the protein target generated drug concept, and make pharmaceutical industry more prone to identify powerful key regulating proteins in disease that novel drug principles can be developed from (Nilsson et al. 2015; Paik et al. 2014; Marko-Varga et al. 2013; Paik et al. 2012a, b; Legrain et al. 2011).
References
Abe, H., and T. Kamai. 2013. Recent advances in the treatment of metastatic renal cell carcinoma. International Journal of Urolology 20(10): 944–955.
Amir, E., L. Mandoky, F. Blackhall, N. Thatcher, W. Klepetko, H.J. Ankersmit, M.A. Reza Hoda, G. Ostoros, M. Dank, and B. Dome. 2009. Antivascular agents for non-small-cell lung cancer: current status and future directions. Expert Opinion on Investigational Drugs 18(11): 1667–1686.
Baker, M. 2012. Biorepositories: Building better biobanks. Nature 486(7401): 141–146.
Balch, C.M., J.E. Gershenwald, S.J. Soong, J.F. Thompson, M.B. Atkins, D.R. Byrd, A.C. Buzaid, A.J. Cochran, D.G. Coit, S. Ding, A.M. Eggermont, K.T. Flaherty, P.A. Gimotty, J.M. Kirkwood, K.M. McMasters, M.C.Jr. Mihm, D.L. Morton, M.I. Ross, A.J. Sober, and V.K. Sondak. 2009. Final version of 2009 AJCC melanoma staging and classification. Journal of Clinical Oncology 27(36): 6199–6206.
Bollag, G., P. Hirth, J. Tsai, J. Zhang, P.N. Ibrahim, H. Cho, W. Spevak, C. Zhang, Y. Zhan, G. Habets, E.A. Burton, B. Wong, G. Tsang, B.L. West, B. Powell, R. Shellooe, A. Marimuthu, H. Nguyen, K.Y. Zhang, D.R. Artis, J. Schlessinger, F. Su, B. Higgins, R. Iyer, K. D’Andrea, A. Koehler, M. Stumm, P.S. Lin, R.J. Lee, J. Grippo, I. Puzanov, K.B. Kim, A. Ribas, G.A. McArthur, J.A. Sosman, P.B. Chapman, K.T. Flaherty, X. Xu, K.L. Nathanson, and K. Nolop. 2010. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467(7315): 596–599.
Buck, A., S. Halbritter, C. Spath, A. Feuchtinger, M. Aichler, H. Zitzelsberger, K.P. Janssen, and A. Walch. 2015. Distribution and quantification of irinotecan and its active metabolite SN-38 in colon cancer murine model systems using MALDI MSI. Analytical and Bioanalytical Chemistry 407(8): 2107–2116.
Castellino, S., M.R. Groseclose, J. Sigafoos, D. Wagner, M. de Serres, J.W. Polli, E. Romach, J. Myer, and B. Hamilton. 2013. Central nervous system disposition and metabolism of Fosdevirine (GSK2248761), a non-nucleoside reverse transcriptase inhibitor: An LC-MS and Matrix-assisted laser desorption/ionization imaging MS investigation into central nervous system toxicity. Chemical Research in Toxicology 26(2): 241–251.
Chapman, P.B., A. Hauschild, C. Robert, J.B. Haanen, P. Ascierto, J. Larkin, R. Dummer, C. Garbe, A. Testori, M. Maio, M. Hogg, P. Lorigan, C. Lebbe, T. Jouary, D. Schadendorf, A. Ribas, S.J. O’Day, J.A. Sosman, J.M. Kirkwood, A.M. Eggermont, B. Dreno, K. Nolop, J. Li, B. Nelson, J. Hou, R.J. Lee, K.T. Flaherty, G.A. McArthur, and BRISM-3 Study Group. 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New England Journal of Medicine 364(26): 2507–2516.
Chaurand, P., M.A. Rahman, T. Hunt, J.A. Mobley, G. Gu, J.C. Latham, R.M. Caprioli, and S. Kasper. 2008. Monitoring mouse prostate development by profiling and imaging mass spectrometry. Molecular and Cellular Proteomics 7(2): 411–423.
Cobice, D.F., C.L. Mackay, R.J. Goodwin, A. McBride, P.R. Langridge-Smith, S.P. Webster, B.R. Walker, and R. Andrew. 2013. Mass spectrometry imaging for dissecting steroid intracrinology within target tissues. Analytical Chemistry 85(23): 11576–11584.
Dwyer, D. 2011. Experiences of registered nurses as managers and leaders in residential aged care facilities: A systematic review. International Journal of Evidence Based Healthcare 9(4): 388–402.
Fehniger, T.E., A. Vegvari, M. Rezeli, K. Prikk, P. Ross, M. Dahlback, G. Edula, R. Sepper, and G. Marko-Varga. 2011. Direct demonstration of tissue uptake of an inhaled drug: Proof-of-principle study using matrix-assisted laser desorption ionization mass spectrometry imaging. Analalytical Chemistry 83(21): 8329–8336.
Fehniger, T.E., E.S. Boja, H. Rodriguez, M.S. Baker, and G. Marko-Varga. 2014. Four areas of engagement requiring strengthening in modern proteomics today. Journal of Proteome Research 13(12): 5310–5318.
Fu, Y., D. Kedziorek, and D.L. Kraitchman. 2010. Recent developments and future challenges on imaging for stem cell research. Journal of Cardiovascular and Translational Research 3(1): 24–29.
Goto, T., N. Terada, T. Inoue, K. Nakayama, Y. Okada, T. Yoshikawa, Y. Miyazaki, M. Uegaki, S. Sumiyoshi, T. Kobayashi, T. Kamba, K. Yoshimura, and O. Ogawa. 2014. The expression profile of phosphatidylinositol in high spatial resolution imaging mass spectrometry as a potential biomarker for prostate cancer. PLoS ONE 9(2): e90242.
Harbst, K., M. Lauss, H. Cirenajwis, C. Winter, J. Howlin, T. Törngren, A. Kvist, B. Nodin, E. Olsson, J. Häkkinen, K. Jirström, J. Staaf, L. Lundgren, H. Olsson, C. Ingvar, S.K. Gruvberger-Saal, L.H. Saal, and G. Jönsson. 2014. Molecular and genetic diversity in the metastatic process of melanoma. Journal of Pathology 233(1): 39–50.
Hardesty, W.M., and R.M. Caprioli. 2008. In situ molecular imaging of proteins in tissues using mass spectrometry. Analytical and Bioanalytical Chemistry 391(3): 899–903.
Jayson, G.C., D.J. Hicklin, and L.M. Ellis. 2012. Antiangiogenic therapy—evolving view based on clinical trial results. Nature Reviews Clinical Oncology 9(5): 297–303.
Jirasko, R., M. Holcapek, M. Kunes, and A. Svatos. 2014. Distribution study of atorvastatin and its metabolites in rat tissues using combined information from UHPLC/MS and MALDI-Orbitrap-MS imaging. Analytical and Bioanalytical Chemistry 406(19): 4601–4610.
Kato, H., T. Nishimura, N. Ikeda, T. Yamada, T. Kondo, N. Saijo, K. Nishio, J. Fujimoto, M. Nomura, Y. Oda, B. Lindmark, J. Maniwa, H. Hibino, M. Unno, T. Ito, Y. Sawa, H. Tojo, S. Egawa, G. Edula, M. Lopez, M. Wigmore, N. Inase, Y. Yoshizawa, F. Nomura, and G. Marko-Varga. 2011. Developments for a growing Japanese patient population: Facilitating new technologies for future health care. Journal of Proteomics 74(6): 759–764.
Koeniger, S.L., N. Talaty, Y. Luo, D. Ready, M. Voorbach, T. Seifert, S. Cepa, J.A. Fagerland, J. Bouska, W. Buck, R.W. Johnson, and S. Spanton. 2011. A quantitation method for mass spectrometry imaging. Rapid Communications in Mass Spectrometry 25(4): 503–510.
Lanao, J.M., and M.A. Fraile. 2005. Drug tissue distribution: Study methods and therapeutic implications. Current Pharmaceutical Design 11(29): 3829–3845.
Langer, O., and M. Muller. 2004. Methods to assess tissue-specific distribution and metabolism of drugs. Current Drug Metabolism 5(6): 463–481.
Lazova, R., E.H. Seeley, M. Keenan, R. Gueorguieva, and R.M. Caprioli. 2012. Imaging mass spectrometry—A new and promising method to differentiate Spitz nevi from Spitzoid malignant melanomas. American Journal of Dermatopathology 34(1): 82–90.
Legrain, P., R. Aebersold, A. Archakov, A. Bairoch, K. Bala, L. Beretta, J. Bergeron, C.H. Borchers, G.L. Corthals, C.E. Costello, E.W. Deutsch, B. Domon, W. Hancock, F. He, D. Hochstrasser, G. Marko-Varga, G.H. Salekdeh, S. Sechi, M. Snyder, S. Srivastava, M. Uhlén, C.H. Wu, T. Yamamoto, Y.K. Paik, and G.S. Omenn. 2011. The human proteome project: current state and future direction. Molecular and Cellular Proteomics 10(7): M111 009993.
Lietz, C.B., E. Gemperline, and L. Li. 2013. Qualitative and quantitative mass spectrometry imaging of drugs and metabolites. Advanced Drug Delivery Reveiws 65(8): 1074–1085.
Liu, X., J.L. Ide, I. Norton, M.A. Marchionni, M.C. Ebling, L.Y. Wang, E. Davis, C.M. Sauvageot, S. Kesari, K.A. Kellersberger, M.L. Easterling, S. Santagata, D.D. Stuart, J. Alberta, J.N. Agar, C.D. Stiles, and N.Y. Agar. 2013. Molecular imaging of drug transit through the blood–brain barrier with MALDI mass spectrometry imaging. Scientific Reports 3: 2859.
Loges, S., T. Schmidt, and P. Carmeliet. 2010. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer 1(1): 12–25.
Marko-Varga, G., A. Vegvari, M. Rezeli, K. Prikk, P. Ross, M. Dahlback, G. Edula, R. Sepper, and G. Marko-Varga. 2012. Understanding drug uptake and binding within targeted disease micro-environments in patients: a new tool for translational medicine. Clinical and Translational Medicine 1(1): 8.
Marko-Varga, G., G.S. Omenn, Y.K. Paik, and W.S. Hancock. 2013. A first step toward completion of a genome-wide characterization of the human proteome. Journal of Proteome Research 12(1): 1–5.
Marko-Varga, G., M.S. Baker, E.S. Boja, H. Rodriguez, and T.E. Fehniger. 2014. Biorepository regulatory frameworks: Building parallel resources that both promote scientific investigation and protect human subjects. Journal of Proteome Research 13(12): 5319–5324.
Moreno Garcia, V., B. Basu, L.R. Molife, and S.B. Kaye. 2012. Combining antiangiogenics to overcome resistance: rationale and clinical experience. Clinical Cancer Research 18(14): 3750–3761.
Mouton, J.W., U. Theuretzbacher, W.A. Craig, P.M. Tulkens, H. Derendorf, and O. Cars. 2008. Tissue concentrations: Do we ever learn? Journal of Antimicrobial Chemotherapy 61(2): 235–237.
Nilsson, A., T.E. Fehniger, L. Gustavsson, M. Andersson, K. Kenne, G. Marko-Varga, and P.E. Andrén. 2010. Fine mapping the spatial distribution and concentration of unlabeled drugs within tissue micro-compartments using imaging mass spectrometry. PLoS ONE 5(7): e11411.
Nilsson, C.L., E. Mostovenko, C.F. Lichti, K. Ruggles, D. Fenyo, K.R. Rosenbloom, W.S. Hancock, Y.K. Paik, G.S. Omenn, J. LaBaer, R.A. Kroes, M. Uhlén, S. Hober, A. Vegvari, P.E. Andrén, E.P. Sulman, F.F. Lang, M. Fuentes, E. Carlsohn, M.R. Emmett, J.R. Moskal, F.S. Berven, T.E. Fehniger, and G. Marko-Varga. 2015. Use of ENCODE resources to characterize novel proteoforms and missing proteins in the human proteome. Journal of Proteome Research 14(2): 603–608.
Nishimura, T., T. Kawamura, Y. Sugihara, Y. Bando, S. Sakamoto, M. Nomura, N. Ikeda, T. Ohira, J. Fujimoto, H. Tojo, T. Hamakubo, T. Kodama, R. Andersson, T.E. Fehniger, H. Kato, and G. Marko-Varga. 2014. Clinical initiatives linking Japanese and Swedish healthcare resources on cancer studies utilizing Biobank Repositories. Clinical and Translational Medicine 3(1): 61.
Paik, Y.K., S.K. Jeong, G.S. Omenn, M. Uhlen, S. Hanash, S.Y. Cho, H.J. Lee, K. Na, E.Y. Choi, F. Yan, F. Zhang, Y. Zhang, M. Snyder, Y. Cheng, R. Chen, G. Marko-Varga, E.W. Deutsch, H. Kim, J.Y. Kwon, R. Aebersold, A. Bairoch, A.D. Taylor, K.Y. Kim, E.Y. Lee, D. Hochstrasser, P. Legrain, and W.S. Hancock. 2012a. The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nature Biotechnology 30(3): 221–223.
Paik, Y.K., G.S. Omenn, M. Uhlen, S. Hanash, G. Marko-Varga, R. Aebersold, A. Bairoch, T. Yamamoto, P. Legrain, H.J. Lee, K. Na, S.K. Jeong, F. He, P.A. Binz, T. Nishimura, P. Keown, M.S. Baker, J.S. Yoo, J. Garin, A. Archakov, J. Bergeron, G.H. Salekdeh, and W.S. Hancock. 2012b. Standard guidelines for the chromosome-centric human proteome project. Journal of Proteome Research 11(4): 2005–2013.
Paik, Y.K., G.S. Omenn, V. Thongboonkerd, G. Marko-Varga, and W.S. Hancock. 2014. Genome-wide proteomics, Chromosome-Centric Human Proteome Project (C-HPP), part II. Journal of Proteome Research 13(1): 1–4.
Pellegatti, M., and S. Pagliarusco. 2011. Drug and metabolite concentrations in tissues in relationship to tissue adverse findings: A review. Expert Opinion on Drug Metabolism and Toxicology 7(2): 137–146.
Rapisarda, A., and G. Melillo. 2012. Overcoming disappointing results with antiangiogenic therapy by targeting hypoxia. Nature Reviews Clinical Oncology 9(7): 378–390.
Shariatgorji, M., A. Nilsson, R.J. Goodwin, P. Kallback, N. Schintu, X. Zhang, A.R. Crossman, E. Bezard, P. Svenningsson, and P.E. Andren. 2014. Direct targeted quantitative molecular imaging of neurotransmitters in brain tissue sections. Neuron 84(4): 697–707.
Solon, E.G., A. Schweitzer, M. Stoeckli, and B. Prideaux. 2010. Autoradiography, MALDI-MS, and SIMS-MS imaging in pharmaceutical discovery and development. American Association of Pharmaceutical Scientists Journal 12(1): 11–26.
Sosman, J.A., K.B. Kim, L. Schuchter, R. Gonzalez, A.C. Pavlick, J.S. Weber, G.A. McArthur, T.E. Hutson, S.J. Moschos, K.T. Flaherty, P. Hersey, R. Kefford, D. Lawrence, I. Puzanov, K.D. Lewis, R.K. Amaravadi, B. Chmielowski, H.J. Lawrence, Y. Shyr, F. Ye, J. Li, K.B. Nolop, R.J. Lee, A.K. Joe, and A. Ribas. 2012. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. New England Journal of Medicine 366(8): 707–714.
Sugihara, Y., A. Vegvari, C. Welinder, G. Jonsson, C. Ingvar, L. Lundgren, H. Olsson, T. Breslin, E. Wieslander, T. Laurell, M. Rezeli, B. Jansson, T. Nishimura, T.E. Fehniger, B. Baldetorp, and G. Marko-Varga. 2014. A new look at drugs targeting malignant melanoma—An application for mass spectrometry imaging. Proteomics 14(17–18): 1963–1970.
Swales, J.G., J.W. Tucker, N. Strittmatter, A. Nilsson, D. Cobice, M.R. Clench, C.L. Mackay, P.E. Andren, Z. Takats, P.J. Webborn, and R.J. Goodwin. 2014. Mass spectrometry imaging of cassette-dosed drugs for higher throughput pharmacokinetic and biodistribution analysis. Analytical Chemistry 86(16): 8473–8480.
Takai, N., Y. Tanaka, K. Inazawa, and H. Saji. 2012. Quantitative analysis of pharmaceutical drug distribution in multiple organs by imaging mass spectrometry. Rapid Communications in Mass Spectrometry 26(13): 1549–1556.
Török, S., Á. Végvári, M. Rezeli, T.E. Fehniger, J. Tóvári, S. Paku, V. Laszlo, B. Hegedus, A. Rozsas, B. Dome, and G. Marko-Varga. 2015. Localization of sunitinib, its metabolites and its target receptors in tumour-bearing mice: A MALDI-MS imaging study. British Journal of Pharmacology 172(4): 1148–1163.
Tsao, H., M.B. Atkins, and A.J. Sober. 2004. Management of cutaneous melanoma. New England Journal of Medicine 351(10): 998–1012.
Welinder, C., G. Jonsson, C. Ingvar, L. Lundgren, H. Olsson, T. Breslin, A. Vegvari, T. Laurell, M. Rezeli, B. Jansson, B. Baldetorp, and G. Marko-Varga. 2013. Establishing a Southern Swedish Malignant Melanoma OMICS and biobank clinical capability. Clinical and Translational Medicine 2(1): 7.
Willems, S.M., A. van Remoortere, R. van Zeijl, A.M. Deelder, L.A. McDonnell, and P.C. Hogendoorn. 2010. Imaging mass spectrometry of myxoid sarcomas identifies proteins and lipids specific to tumour type and grade, and reveals biochemical intratumour heterogeneity. Journal of Pathology 222(4): 400–409.
Yasunaga, M., M. Furuta, K. Ogata, Y. Koga, Y. Yamamoto, M. Takigahira, and Y. Matsumura. 2013. The significance of microscopic mass spectrometry with high resolution in the visualisation of drug distribution. Scientific Reports 3: 3050.
Ye, H., R. Mandal, A. Catherman, P.M. Thomas, N.L. Kelleher, C. Ikonomidou, and L. Li. 2014. Top-down proteomics with mass spectrometry imaging: A pilot study towards discovery of biomarkers for neurodevelopmental disorders. PLoS ONE 9(4): e92831.
Young, T., A. Rohwer, J. Volmink, and M. Clarke. 2014. What are the effects of teaching evidence-based health care (EBHC)? Overview of systematic reviews. PLoS One 9(1): e86706.
Acknowledgments
This work was supported by grants from Mrs. Berta Kamprad Foundation, Swedish Academy of Pharmaceutical Sciences, Swedish Research Council, the Swedish Foundation for Strategic Research, Vinnova, Ingabritt & Arne Lundbergs forskningsstiftelse, Fundacion Federico SA, the Crafoord Foundation and from the National Research Foundation of Korea (NRF) funded by the Korean Government (2009-0083522 and 2012M3A9D1054520), and the Brain Korea 21 Plus Project, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kwon, H.J., Kim, Y., Sugihara, Y. et al. Drug compound characterization by mass spectrometry imaging in cancer tissue. Arch. Pharm. Res. 38, 1718–1727 (2015). https://doi.org/10.1007/s12272-015-0627-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-015-0627-2