
Abstract
The concept of epigenetics is now providing the mechanisms by which cells transfer their new environmental-change-induced phenotypes to their daughter cells. However, how extracellular or cytoplasmic environmental cues are connected to the nuclear epigenome remains incompletely understood. Recently emerging evidence suggests that epigenetic changes are correlated with metabolic changes via chromatin remodeling. As many human complex diseases including cancer harbor both epigenetic changes and metabolic dysregulation, understanding the molecular processes linking them has huge implications for disease pathogenesis and therapeutic intervention. In this review, the impacts of metabolic changes on cancer epigenetics are discussed, along with the current knowledge on cancer metabolism and epigenetics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Metabolism changes drastically in cancer cells and this change possibly constitutes the very origin of cancer (Galluzzi et al. 2013; Wu and Zhao 2013). This “metabolic theory of cancer” was first proposed by Nobel prize-winning German biochemist Otto Warburg in 1924 (Koppenol et al. 2011). However, changes in metabolites themselves are not inherited by daughter cells and how deranged metabolism is maintained in various cancer-cell microenvironments has not been elucidated. Given that cancer cells harbor mutations, copy-number changes, and DNA methylation changes, it is possible that all metabolic changes originate from existing heritable genetic and/or epigenetic changes (Vander Heiden 2013). Nevertheless, metabolic changes might be the driving force for cancer progression by facilitating heritable epigenetic changes that transmit the metabolic signatures of cancer. In fact, emerging evidence supports this theory. In the following review, the metabolic and epigenetic changes in cancer and the possibility of a metabolic impetus for heritable epigenetic changes in cancer cells are discussed. The goal is to “fill the gap” in the metabolic theory of cancer.
Metabolic changes in cancer
The connection between metabolism and cancer-cell growth or survival is an area of growing interest. Nutrient utilization changes drastically to meet the large biosynthetic demands associated with cell growth and division when cancer cells receive proliferative signals. These changes include increased ATP production and synthesis of raw biomaterials, including lipids, proteins, and nucleic acids, through reorganization of intracellular metabolism from catabolic mitochondrial oxidative phosphorylation (OXPHOS) to glycolysis and other anabolic pathways (Marelli-Berg et al. 2012). Altered glucose metabolism from OXPHOS to lactate production due to increased biomass demands (Warburg Effect) is a metabolic hallmark of cancer cells (Jang et al. 2013). The significant role of the Warburg effect driving cell proliferation is suggested by: (1) the high rate of glycolysis and the glucose turnover to lactic acid for ATP production, and (2) increased lipid and nucleic acid biosynthesis via several biosynthetic pathways stimulated by accumulating glycolytic intermediates, including citrate (Ganapathy-Kanniappan and Geschwind 2013). Citrate is a substrate for three enzymes, including ATP citrate lyase, acetyl-CoA carboxylase, and fatty acid synthase (Table 1), and the rates of these enzyme reactions increase in cancer. Moreover, these enzymes are highly expressed and serve oncogenic functions in many human cancers.
In addition to the altered balance between glucose fermentation and OXPHOS, mutations or expression-level changes in metabolic enzymes, such as succinate dehydrogenase (SDH), fumarate hydratase, pyruvate kinase, and isocitrate dehydrogenase-1/2 (IDH1 and IDH2), are linked to certain types of tumorigenesis (Wu and Zhao 2013). Additionally, key oncogenes or tumor-suppressor genes including c-myc and TP53 are important metabolic regulators, and mutations or expression-level changes in these regulators alters metabolism in cancer cells (Wu and Zhao 2013; Marelli-Berg et al. 2012) (Table 1).
Epigenetic changes in cancer
Epigenetic modifications, such as DNA methylation, post-translational histone modifications, nucleosome positioning, and non-coding RNA expression (Cho et al. 2007), have tremendous impacts on local and global gene activities and are essential for maintenance of chromatin structure and genomic stability (Sadikovic et al. 2008). Microenvironment-mediated epigenetic perturbations can lead to altered gene function and play important roles in tumorigenes is through inappropriate gene silencing and/or activation (Shankar et al. 2013; Herceg and Hainaut 2007).
Among epigenetic modifications, DNA methylation, particularly promoter CpG island methylation, has been studied extensively in cancer. Cancer cells show characteristic global DNA hypomethylation of oncogenes (Wilson et al. 2007; Feinberg and Vogelstein 1983; Ehrlich 2009) and promoter CpG island hypermethylation in tumor suppressors genes (Esteller 2002; Kloten et al. 2013; Radpour et al. 2011; Ehrlich et al. 2002). Promoter CpG island methylation-affected genes in cancer are involved in several important cellular pathways such as DNA repair (hMLH1, MGMT, WRN, and BRCA1), vitamin response (RARB2 and CRBP1), Ras signaling (RASSFIA and NOREIA), cell-cycle control (p16INK4a, p15INK4b, and RB), the p53 network (p14ARF, TP73, and HIC-1), and apoptosis (TMS1, DAPK1, WIF-1, and SFRP1) (Lahtz and Pfeifer 2011; Jin and Robertson 2013; Esteller 2007). Global hypomethylation occurs in various malignancies, including breast, lung, bladder, and liver cancers (Di et al. 2011; Portela and Esteller 2010; Aporntewan et al. 2011) and hypermethylation of CpG islands has been identified in the HOXA2 (Li et al. 2013b), GATA2 (Li et al. 2013a), TGFB1 (Khin et al. 2011) and PAX5 (Liu et al. 2011) genes and affects their expression. Hypermethylation of miRNA promoters is another important epigenetic change in cancer, leading to overall miRNA downregulation in cancer cells (Suzuki et al. 2012, 2013). In particular, silencing of miR-148, miR-34b/c, or miR-9 by promoter hypermethylation occurs in cancer cells (Lujambio et al. 2008; Suzuki et al. 2013). Thus the changes in DNA methylation in specific genes or miRNA are major changes in cancer and contribute significantly to carcinogenesis or cancer progression.
Changes in modifications of the core histones H2A, H2B, H3, and H4 have also been reported in various cancer types (Podlaha et al. 2014). A plethora of reversible histone covalent post-translational modifications, including acetylation, methylation, phosphorylation, ubiquitination, sumoylation, proline isomerization, and ADP ribosylation has been reported in various cancers (Strahl and Allis 2000; Keum et al. 2013; Cohen et al. 2011). Additionally, translocation, mutation, and deletion in histone acetyltransferases (HATs) or HAT-related genes contribute to the global imbalance of histone acetylation in several cancer types (colon, uterus, and lung) by either activating or inactivating their transcription (Portela and Esteller 2010). Aberrant histone modifications found in cancer can affect transcription (Consortium et al. 2007; Koch et al. 2007), recombination (Miao and Natarajan 2005), DNA repair and replication (Consortium et al. 2007, Groth et al. 2007), as well as chromosomal organization (Jenuwein and Allis 2001; Luger et al. 1997), thereby potentially contributing to carcinogenesis, along with aberrant DNA methylation.
In addition to DNA methylation and histone modifications, epigenetic components, such as nucleosomal positioning, long non-coding RNAs, and RNA interference pathways, change in cancer (Beckedorff et al. 2013; Kornienko et al. 2013). A detailed description of these changes in cancer is beyond the scope of this review, but the importance of these epigenetic changes has aided the development of new diagnostic and therapeutic modalities as well as understating the cancer biology. However, questions, such as: (1) what will cause epigenetic changes in cancer cells, (2) can epigenetic changes induce cancer progression, and (3) can modifications of epigenetic changes cure patients with cancer, remain to be answered.
Metabolic regulation of epigenetics in cancer
Epigenetic modifications of DNA and histones by various epigenetic enzymes, such as DNA methyltransferases (DNMTs) and histone methyltransferases (HMTs) require metabolites as co-factors or substrates (Lu and Thompson 2012). It has long been suspected that elevations or depletions of these metabolite levels beyond the normal range can influence epigenetic regulation and chromatin structure, thereby altering gene expression. In addition, analyses of cancers harboring SDH and IDH mutants have found abnormal metabolites including 2-hydroxyglutarate (2-HG) (Ward et al. 2010; Dang et al. 2010). Redundant 2-HG inhibits the TET family of dioxygenases, which is involved in DNA demethylation by oxidizing 5-methylcytosine (5mC) to 5-hydroxymethylcytosine, resulting in increased 5mC in SDH mutant cancers (Yang et al. 2013). This finding clearly demonstrates a close connection between metabolic rewiring and epigenetic alterations. Considering this close association and recent suggestions of epigenetic-change-based facilitation of genomic perturbation, epigenetic changes may be an important driver of tumor progression or initiation. In the following section we focus on the mechanism by which various metabolite alterations lead to epigenetic changes that may be associated with cancer development (Table 2).
S-adenosyl-l-methionine (SAM) and methylation reactions
SAM, produced by methionine adenosyltransferase (MAT or SAM synthetase) using ATP and methionine as substrates, is an important co-factor involved in methyl group transfer reactions (Markham and Pajares 2009). Additionally, it is involved in reactions related to epigenetic regulation such as methylation of DNA, RNA, and selective arginine or lysine residues of histones and non-histone transcriptional regulators including tumor-suppressor p53 and transcriptional factor TAF10 (Huang and Berger 2008). DNMTs and HMTs are the major enzymes involved in this process. After SAM donates its methyl group, it is transformed into S-adenosyl-homocysteine (SAH), which is a potent inhibitor of DNMTs and HMTs and the key metabolic determinant of methyltransferase reactions (Lu and Thompson 2012). The transformed SAH is converted to homocysteine and then to methionine or cysteine by accepting methyl groups from 5-methyl tetrahydrofolate and vitamin B12.
Changes in intracellular energy or multiple metabolic inputs can change SAM and SAH levels; thus, affecting DNA and histone methylation. A diet deficient in choline-methionine, which limits SAM availability, can produce liver cancer in rats (Ghoshal and Farber 1984) and deficiency of a methyl donor such as folate in humans can lead to global DNA hypomethylation, and affect the epigenome leading to tumorigenesis (Duthie 1999). These results suggest that changes in the intracellular levels of methyl donors, including SAM, can promote or initiate tumorigenesis via their effect on DNA methylation.
Acetyl coenzyme A (acetyl CoA)
Acetyl-CoA is the acetyl-group donor for acetylation reactions in histones and non-histone proteins. Acetyl-CoA is synthesized via two routes: (1) condensation of acetate and coenzyme A into acetyl-CoA by acetyl-CoA synthetase (ACS), or (2) generation of acetyl-CoA from pyruvate by the pyruvate dehydrogenase complex (PDH) in the tricarboxylic acid (TCA) cycle. Citrate, which is produced in the TCA cycle and exported from mitochondria, is the major source of nuclear and cytosolic acetyl-CoA, and its conversion from acetyl-CoA is facilitated by ATP-citrate lyase (ACL) in the cytoplasm or nucleus. In yeast cells, acetyl-CoA is synthesized mainly from condensation of acetate and coenzyme A by ACS1 or ACS2. Inactivation of the two ACS enzymes in yeast reduces histone acetylation and global transcription defects (Takahashi et al. 2006), whereas glucose supplementation during quiescent status increases the histone acetylation level dramatically (Friis et al. 2009). The acetyl-CoA level oscillates dynamically, with 10-fold variations, according to the growth phases and is correlated with H3K9 acetylation levels in genes differentially expressed during various growth phases (Cai et al. 2011; Lu and Thompson 2012). These findings suggest an intriguing role for acetyl-CoA as a link between cell metabolism and gene expression by modulating histone acetylation.
Excess acetyl-CoA production in the mitochondria of proliferating or tumor cells allows for an increased level in the cytoplasm and nucleus via export in the form of citrate into the cytosol, where it is converted back to acetyl-CoA by ACL (Munoz-Pinedo et al. 2012). An increased cytosolic or nuclear level of acetyl-CoA can affect histone acetylation reactions. Interestingly, ACL serves as a molecular link between cell metabolism and histone acetylation; histone acetylation increases in response to stimulation by growth factors and increased expression of genes, such as GLUT4, HK2, PFK1, and LDH, that regulate glucose metabolism (Wellen et al. 2009; Govardhan et al. 2011). A profound decrease in histone acetylation and in the expression of a selective subset of genes occurs when ACL activity is knocked down (Wellen et al. 2009). These results indicate that global and gene-specific control of transcription can be intertwined with the metabolic status of cells via acetyl-CoA.
The importance of histone acetylation in cancer has been recognized since the first observation that sodium butyrate, a histone deacetylase (HDAC) inhibitor, reverses the morphology of transformed cancer cells (Singh et al. 2011; Glozak and Seto 2007). HDAC inhibitors, such as vorinostat and romidepsin, have been approved for treating cutaneous T cell lymphoma, and many other new inhibitors are in clinical trials (Mack 2010). The exact mechanism of action of the HDAC inhibitors is not clear, although epigenetic pathways via modulation of histone acetylation have been proposed (Glozak and Seto 2007). Cyclin-dependent kinase inhibitor p21, a regulator of p53 tumor-suppressor activity, is one of the best studied targets of HDAC inhibitor-mediated de-repression. HDAC inhibitors induce p21 expression, which coincides with hyperacetylation of histones H3 and H4 in the promoter region (Sambucetti et al. 1999). Many non-histone HDAC targets and their potential applications in cancer have been reported (Choudhary et al. 2014). Therefore, it is plausible that deregulated acetyl-CoA levels affect derangement in histone and/or non-histone acetylation during tumorigenesis and tumor progression.
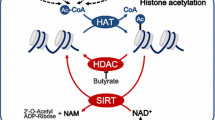
Nicotinamide adenine dinucleotide (NAD+)
NAD+ is as a coenzyme that captures electrons during glycolysis and the TCA cycle, and is critical for several reactions that directly or indirectly modulate chromatin dynamics, DNA repair, and transcription. NAD+ also acts as an enzyme cofactor, such as sirtuins [NAD+-dependent class III as histone deacetylases (HDACs)] and poly (ADP-ribose) polymerase (Ying 2008), which have important roles in the DNA damage response, epigenetic regulation of chromatin structure, and gene expression.
NAD+ is hydrolyzed to nicotinamide and O-acetyl-ribose during sirtuin-mediated deacetylation reactions. The former is a potent inhibitor of sirtuin HDAC activity, whereas the latter is a novel signaling molecule related to calcium homeostasis and other pathways (Sauve et al. 2006). Changes in the NAD+/NADH ratio due to metabolic stress affects sirtuin activities (Sauve et al. 2006). SIRT1, the most well-studied sirtuin, represses glycolysis by deacetylating histones at the promoters of several genes, such as forkhead box class O, CRTC2, STAT3, TP53, and peroxisome proliferator-activated receptor gamma coactivator-1α (Saunders and Verdin 2007). The role of sirtuins in cancer is complex and multifaceted, because sirtuins act as both oncogenes and tumor suppressors. NAD+ molecules are reduced to NADH during each glycolysis and TCA cycle round, which decreases the NAD+/NADH ratio, and downregulates overall sirtuin activity. Reduced sirtuin activity in cancer cells is correlated with histone hyperacetylation and chromatin decondensation, which stimulates the expression of genes related to cell proliferation (Hitchler and Domann 2009). Thus, metabolism and availability of NAD+ can affect the genome and cellular physiology in multiple ways, including global and local histone modifications.
Flavin adenine dinucleotide (FAD)
FAD is a redox coenzyme derived from the vitamin riboflavin (vitamin B2) and an electron acceptor in mitochondrial respiratory chain complex II. FAD exists in two different redox forms of oxidized FAD and reduced FADH2 (Teperino et al. 2010). Furthermore, FAD is an important cofactor in histone demethylation reactions with the histone demethylase LSD1. Covalent methyl groups in DNA or histones are relatively stable, but they can be removed by demethylases (Shi and Whetstine 2007). LSD1 is the first identified histone demethylase that uses FAD and releases formaldehyde as a byproduct. Histone demethylation by LSD1 modifies chromatin structure and gene transcription and contributes to proliferation and metastasis of lung and colon cancers through epigenetic modifications (Lv et al. 2012b; Ding et al. 2013). Correspondingly, FAD availability originates from the activities of other flavin-linked dehydrogenases and oxidases in the fatty acid β-oxidation and the TCA cycle pathways, and is an important determining factor in epigenetic regulation of energy-expenditure genes by LSD1 (Hino et al. 2012). Thus, cellular redox or energy status can influence LSD1 activity and change target gene transcription via its epigenetic modulation. Other oxidoreductase enzymes that use FAD as a cofactor include SDH (complex II), acyl-CoA dehydrogenase, α–ketoglutarate dehydrogenase, and a component of the PDH complex, the enzymatic activity of which can be influenced by intracellular FAD level (Teperino et al. 2010).
α-Ketoglutarate (α-KG) and 2-hydroxyglutarate (2-HG)
α-KG, also called oxo-glutarate, is an important biological compound and a key intermediate in the TCA cycle. It is produced either from isocitrate by isocitrate dehydrogenase (IDH), or from glutamine and glutamate by enzymes, such as glutaminase 1 and glutamate dehydrogenase (Oermann et al. 2012). α-KG levels in cancer cells are elevated due to the increased glutamine utilization. α-KG in the nucleus can be a substrate for α-KG-dependent methyl dioxygenases, such as ten eleven translocation (TET) and Jumonji-C domain-containing HDM (JHDM) (Teperino et al. 2010). TET converts methylated cytosine to 5-hydroxy methylcytosine, which is altered to unmethylated cytosine by hydroxymethylcytosine glycosylase; thus, activating target genes epigenetically or protecting promoters from aberrant DNA methylation (Putiri et al. 2014; Klug et al. 2013). JHDM removes repressive histone methylation marks, such as H3K9me3 and H3K27me3; thus, activating differentiation-related genes during progenitor cell differentiation (Lu and Thompson 2012). Therefore, the reduced availability of α-KG due to a depressed TCA cycle decreases the activity of α-KG-dependent proteins important for epigenetic regulation by hydroxymethylation.
IDH1 and IDH2 are not TCA-cycle enzymes but produce α-KG from isocitrate using NADP+ as a cofactor instead of NAD+ (Reitman and Yan 2010). Mutant forms of IDH1 and IDH2 in cancer produce an abnormal oncometabolite, 2-hydroxyglutarate (2-HG), from α-KG (Cairns and Mak 2013). Since 2-HG is structurally similar to α-KG, it competes with α-KG for binding to several classes of α-KG-dependent enzymes (Ye et al. 2013) and inhibits DNA demethylases, such as TET2 and JHDM, which serves to permanently silence differentiation-related genes by DNA and histone hypermethylation (Lu and Thompson 2012). Thereby, 2-HG induces differentiation-arrest and expansion of progenitor cells; thus, facilitating tumor development. 2-HG also inhibits histone demethylases, including lysine-specific demethylase 4C, lysine-specific demethylase 7A, and lysine-specific demethylase 4A (Schulze and Harris 2012), causing increased histone methylation and inhibited expression of lineage-specific differentiation-related genes (Xu et al. 2011). These reports demonstrate how deranged cancer metabolites such as 2-HG facilitate tumorigenesis by modifying enzymatic activities during the epigenetic process.
Transmission of metabolic phenotypes to daughter cells
Most changes on cancer-cell metabolite levels are transient because cancer cells migrate into a neighboring area where the tumor microenvironment is different from the original site, and the metabolic state of the cancer cells is altered. However, most tumor cell metabolic phenotypes persist and are transmitted to daughter cells (Pattabiraman and Weinberg 2014). Genetic and epigenetic changes in cancer explain all metabolic phenotypes when they originate from pre-existing inheritable changes. Metabolic stress has been suggested to change epigenetic mechanisms (Lu and Thompson 2012). In the following section, the mode of epigenetic inheritance and its possible link in the inheritance of newly acquired metabolic phenotypes during cell division will be discussed.
Inheritance of DNA methylation during replication
DNA methylation is transmitted with high fidelity, and the process is facilitated by DNA methyltransferase 1 (DNMT1), which shows high affinity for hemi-methylated DNA (Holliday and Ho 2002). However, the mechanism for high-fidelity transmission of DNA methylation is unclear. Many fork-related replication proteins are involved in DNA replication to link information from the two strands and ensure high fidelity replication of DNA sequences. One of the fork replication proteins is proliferating cell nuclear antigen (PCNA), a DNA processivity factor (Probst et al. 2009). In addition to the role of PCNA in DNA replication, a function in the inheritance of DNA methylation has also been suggested, as D. melanogaster harboring a PCNA mutation show position-effect variegation related to transcriptional silencing due to translocation of a specific gene to the heterochromatin regions, followed by DNA methylation (Henderson et al. 1994).
Another fork replication protein, NP95 (also known as ICBP90 and UHRF1) product, interacts with DNMT1 during transmission of epigenetic marks, and preferentially binds to hemi-methylated DNA. Deleting NP95 causes methylation defects similar to those from loss of DNMT1 (Sharif et al. 2007; Bostick et al. 2007). The NP95 product binds to the hemi-methylated DNA form made from semiconservative DNA replication. Then, the associated DNMT1 in the replication fork methylates the newly synthesized unmethylated daughter strand, producing methylated daughter DNA in both strands (Probst et al. 2009).
Although several molecular mechanisms have been revealed, the DNA methylation process and the dynamics of reversible DNA methylation are not fully understood. Therefore, further data on the basic mechanisms of the DNA methylation and demethylation processes are needed before the entire translational process for input of the metabolic signature into the epigenome can be elucidated.
Inheritance of histone modifications
Parental nucleosomes are disrupted at the replication fork during replication; thus, the histone modifications must be correctly reassembled during or after DNA replication for transmission of histone information to daughter cells (Huang et al. 2013; Zhu and Reinberg 2011). However, the histone-modification process is much less clear than the DNA methylation and demethylation processes. In contrast to the DNA methylation inheritance mechanism, the template for histone modification is lacking. In addition, some marks, including histone acetylation and phosphorylation are unstable (Chestier and Yaniv 1979), and these modifications are not considered heritable modifications.
Several mechanisms to transmit histone modifications during cell replication have been reported. Histone variant-specific chaperones are involved in disrupting and reassembling nucleosomes during DNA replication. The H3-H4 chaperone ASF1 binds to newly synthesized or recycled histone dimers and transfers them to another histone chaperone, CAF1 (Mello et al. 2002). CAF1 is recruited to the replication fork by interacting with PCNA, and mediates the histone-modification process by interacting with other PCNA-associated histone modifiers, such as HDACs and lysine methyltransferases.
The self-reinforcing loop model has been proposed as the mechanism for transmitting H3 K9 histone marks to daughter cells. In the model, parental marks are recognized by the chromatin-binding protein, heterochromatin protein 1 (HP1), which recruits a chromatin modifier, such as SUV39H1 (Bannister et al. 2001; Lachner et al. 2001); thus, ensuring methylation of surrounding histones near the parental histone marks. A similar self-reinforcing loop during replication has been proposed for H3K27me3 marks, in which PRC2 instead of HP1 binds to its own methylation sites, ensuring methylation of surrounding histones near the parental histone marks (Hansen et al. 2008). These self-reinforcing loop models apply only to repetitive regions (in which long arrays of nucleosomes carry the same histone marks) where the disrupted parental histones from the replication forks and the newly synthesized histones are randomly incorporated into the daughter strands. However, the transmission of histone modifications in other parts of the genome is not clear.
The histone modification process cannot be completed during the S phase. Although H3K9me2 modification of newly deposited histones is almost complete shortly after the S phase, the levels of H3K9me3 and H3K27me3 in S-phase cells are only about 70 % of those in the histones of the mother strands, and the levels fully recover only in the G1 phase (Xu et al. 2012). The histone modifications during the S phase indicate the presence of histone-modification mechanisms that are independent of DNA replication forks, although the relevant information revealed remains insufficient.
Correlating metabolic stress-induced changes with heritable DNA and histone marks
Heritable information should ideally be precisely duplicated during cell division, but when or how metabolic stress-induced changes are translated into heritable epigenetic marks is not fully understood. As we discussed in a previous section, changes in intracellular SAM levels can affect DNA and histone methylation, particularly during the S phase of the cell cycle, when most DNA and histone modifications occur (Nelson et al. 2002). Even after the S phase, H3K9me3 and H3K27me3 progress; thus, intracellular SAM-level changes beyond the S phase can also affect histone methylation status.
In contrast to histone methylation, histone acetylation is a transient modification and cannot constitute a heritable modification, suggesting that intracellular acetyl-CoA or NAD+ level changes can affect the histone acetylation level but cannot induce heritable histone modifications. However, changes in acetylated histones can affect the expression of genes associated with acetyl-modified histones and could induce heritable changes indirectly via those gene-expression changes (Verdone et al. 2005).
Most microenvironments near tumor cells change continually and consistently; thus, metabolic stress can be transient and may not be translated into an epigenetic change. However, persistent metabolic stress can be induced from mutations at well-known oncogenes or tumor-suppressor genes. For example, p53 is a tumor suppressor that plays important roles in cell growth, apoptosis, cell cycle, autophagy, DNA damage and repair, reactive oxygen species (ROS) regulation, and metabolism (Zhang et al. 2010; Rai et al. 2011). p53 is not only an important stress sensor for intracellular ROS level, hypoxia, and DNA damage (Horn and Vousden 2007; Vousden and Prives 2009), but also a key regulator of both glycolysis and OXPHOS (Vousden and Ryan 2009). Reduced nutrient or energy levels activate AMPK and decrease AKT-mTOR pathway activities, both of which activate p53 (Vousden and Ryan 2009). Induced p53 can change intracellular metabolism by regulating glucose transporter expression or the levels of enzymes in the glycolytic pathway (Schwartzenberg-Bar-Yoseph et al. 2004; Bensaad et al. 2006; Mathupala et al. 2001). p53 mutations or inactivation found in various cancer cells can disrupt energy balance and metabolism under stressed conditions, and the metabolic changes affect epigenomic changes and tumor progression.
Another example is amplification of c-myc found in many different cancers, which plays important roles regulating cell proliferation and metastasis. Activating c-myc increases levels of glucose transporters, glycolytic enzymes, and glutaminase (Wu and Zhao 2013). Activating c-myc by amplification in cancer cells induces a deranged response to the tumor microenvironment, which leads to epigenetic derangement and tumor progression.
Is there any direct evidence for the induction of epigenetic change by metabolic stress? A direct answer to this question is difficult because mutations in oncogenes or tumor-suppressor genes, such as TP53 and c-myc, affect various signaling pathways as well as metabolism and because each specific metabolite change most often accompanies other functional or signaling pathway changes. However, we can estimate the effect of metabolism on epigenetic changes indirectly by observing IDH1 and IDH2 mutants, because these mutations induce relatively pure metabolic changes. For example, abnormal depletion of the TCA-cycle constituent α-ketoglutarate occurs at the expense of a marked increase in 2-HG. Interestingly, human gastrointestinal stromal tumor (GIST) cells and paragangliomas harboring these IDH mutations have abnormal DNA and histone methylation patterns (Figueroa et al. 2010; Killian et al. 2013; Letouze et al. 2013). An animal model with an IDH1 mutation reproduces this abnormal methylation pattern, suggesting that metabolic change can affect epigenetic phenotypes (Sasaki et al. 2012). The mechanism of this abnormal methylation pattern is competitive inhibition of α-KG-dependent dioxygenases (Xu et al. 2011) and inhibition of histone lysine methylases (Chowdhury et al. 2011). Mutations in fumarate hydratase, another TCA-cycle enzyme, also show hypermethylation patterns, suggesting that different TCA-cycle lesions result in similar epigenetic outcomes (Letouze et al. 2013). These results clearly demonstrate a link between metabolic deregulation and heritable epigenetic abnormalities. This correlation could confirm the mechanism for tumor heterogeneity, which currently is suspected to be a major reason for drug resistance and cancer progression, because the metabolic effects could be dependent on the various microenvironments in different areas (Meacham and Morrison 2013; Easwaran et al. 2014). Therefore, revealing the underlying mechanisms linking metabolism and the epigenome could help solve problems related to drug resistance and cancer progression.
Targeting metabolic regulators affecting the epigenome: a new therapeutic approach
Epigenetic alterations have emerged as promising candidates for developing specific markers for cancer detection, diagnosis, and progression. Most epigenetic alterations are largely mediated by chemical modifications in DNA bases or histones (Paschos and Allday 2010) and are potentially reversible as they are controlled by epigenetic enzymes. Thus, investigating epigenomic profiles, including DNA methylation, histone marks, chromatin conformation, and miRNAs is important due to the implications for developing novel target agents that reverse transcriptional abnormalities by modulating the cancer epigenome (Boumber and Issa 2011; Campbell and Tummino 2014). Studies have yielded new promising compounds targeting the epigenetic modifiers or enzymes. Most importantly, DNA methyltransferase inhibitors (decitabine, 5-azacitidine, and its deoxy derivatives for myelodysplastic syndromes) and HDAC inhibitors (romidepsin and vorinostat for T-cell lymphoma) have been successfully developed as epigenetic drugs that have anti-tumor activities in vitro and in vivo (Boumber and Issa 2011; Ghoshal and Bai 2007; Connolly and Stearns 2012) and received FDA approval.
Recent recognition of the correlation between metabolic and epigenetic changes can provide novel strategies to improve the effect of epigenetic modifiers or to decrease side effects on normal cells when cancer-specific metabolism affecting the epigenome is utilized. Table 3 summarizes the metabolic targets that can affect epigenetic modifications. In addition, understanding the transformation mechanism of metabolic signals into heritable epigenetic information may provide another novel strategy for reversing epigenetic abnormalities in human diseases including cancer.
Conclusion
In this review, we have summarized current knowledge on cancer cell metabolism and its impacts on epigenetic modulation or inheritance (Fig. 1). Alterations in finely tuned metabolic equilibrium via disturbed metabolites can change enzyme activities related to epigenetic modifications, which can lead to heritable epigenetic changes. Hence, two conjectures can be proposed about the role of metabolism in cancer initiation and progression. First, tumor-associated gene mutations, such as TP53 or c-myc, can lead to metabolic changes, resulting in new metabolic status and cancer progression. Alternatively, metabolic changes initiated by environmental factors, which are usually transient, can modify the epigenome and initiate cancer development. These two processes can potentiate each other and speed up malignant transformation or cancer progression. As many complex human diseases including cancers harbor both epigenetic aberrations and metabolic dysregulation, understanding the molecular processes linking them has huge implications for disease pathogenesis and therapeutic intervention.

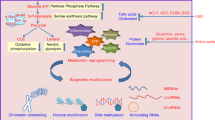
Possible mechanisms of metabolic control over epigenetic modulation and their inheritance. Metabolic pathways and metabolites that can affect the epigenome are shown. Important metabolites that can affect the epigenome include citrate, α-ketoglutarate (α-KG), 2-hydroxyglutarate (2-HG) and acetyl-CoA carboxylase (acetyl-CoA) (from the TCA cycle), S-adenosyl-l-methionine (SAM) (from the methionine cycle), N-acetylglucosamine (GlcNAc) (from the hexosamine biosynthetic pathway), and NAD+/NADH (from glycolysis). The epigenetic enzymes modulated by metabolites are shown. DNA methyltransferase (DNMT), ten-eleven translocation protein (TET), and methyl CpG binding proteins are responsible for DNA modifications, whereas histone acetyltransferases, deacetylases, methyltransferases and demethylases are responsible for histone modifications. Changes in epigenetic enzyme activities affect the level of epigenetic modifications; thereby, changing the epigenomic and expression profiles. The schematic structure of the molecules involved in DNA or histone epigenome for inheritance modifications are also shown. Proliferating cell nuclear antigen (PCNA) recruits enzymes, such as histone deacetylases (HDACs) or Lys methyltransferase SET8, chromatin remodelers (Williams syndrome transcription factor (WSTF)–SNF2H and chromatin assembly factor 1 (CAF1) at the replication forks, and induce changes in DNA methylation and histone modifications on newly synthesized daughter strands
References
Alisch, R.S., B.G. Barwick, P. Chopra, L.K. Myrick, G.A. Satten, K.N. Conneely, and S.T. Warren. 2012. Age-associated DNA methylation in pediatric populations. Genome Research 22(4): 623–632. doi:10.1101/gr.125187.111.
Aporntewan, C., C. Phokaew, J. Piriyapongsa, C. Ngamphiw, C. Ittiwut, S. Tongsima, and A. Mutirangura. 2011. Hypomethylation of intragenic LINE-1 represses transcription in cancer cells through AGO2. PLoS One 6(3): e17934. doi:10.1371/journal.pone.0017934.
Arlauckas, S.P., A.V. Popov, and E.J. Delikatny. 2014. Direct inhibition of choline kinase by a near-infrared fluorescent carbocyanine. Molecular Cancer Therapeutics 13(9): 2149–2158. doi:10.1158/1535-7163.MCT-14-0085.
Banerjee, S., D. Kong, Z. Wang, B. Bao, G.G. Hillman, and F.H. Sarkar. 2011. Attenuation of multi-targeted proliferation-linked signaling by 3,3′-diindolylmethane (DIM): From bench to clinic. Mutation Research 728(1–2): 47–66. doi:10.1016/j.mrrev.2011.06.001.
Bannister, A.J., P. Zegerman, J.F. Partridge, E.A. Miska, J.O. Thomas, R.C. Allshire, and T. Kouzarides. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410(6824): 120–124. doi:10.1038/35065138.
Beckedorff, F.C., M.S. Amaral, C. Deocesano-Pereira, and S. Verjovski-Almeida. 2013. Long non-coding RNAs and their implications in cancer epigenetics. Bioscience Reports. doi:10.1042/BSR20130054.
Behrooz, A., and F. Ismail-Beigi. 1997. Dual control of glut1 glucose transporter gene expression by hypoxia and by inhibition of oxidative phosphorylation. The Journal of Biological Chemistry 272(9): 5555–5562.
Bensaad, K., A. Tsuruta, M.A. Selak, M.N. Vidal, K. Nakano, R. Bartrons, E. Gottlieb, and K.H. Vousden. 2006. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126(1): 107–120. doi:10.1016/j.cell.2006.05.036.
Berg, J.M., J.L. Tymoczko, and L. Stryer. 2002. Biochemistry. In The citric acid cycle is a source of biosynthetic precursors, 5th edn. New York: W H Freeman.
Boocock, D.J., G.E. Faust, K.R. Patel, A.M. Schinas, V.A. Brown, M.P. Ducharme, T.D. Booth, J.A. Crowell, M. Perloff, A.J. Gescher, W.P. Steward, and D.E. Brenner. 2007. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiology, Biomarkers & Prevention 16(6): 1246–1252. doi:10.1158/1055-9965.EPI-07-0022.
Bostick, M., J.K. Kim, P.O. Esteve, A. Clark, S. Pradhan, and S.E. Jacobsen. 2007. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317(5845): 1760–1764. doi:10.1126/science.1147939.
Boumber, Y., and J.P. Issa. 2011. Epigenetics in cancer: What’s the future? Oncology (Williston Park) 25(3): 220–226, 228.
Cai, L., B.M. Sutter, B. Li, and B.P. Tu. 2011. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Molecular Cell 42(4): 426–437. doi:10.1016/j.molcel.2011.05.004.
Cairns, R.A., and T.W. Mak. 2013. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discovery 3(7): 730–741. doi:10.1158/2159-8290.CD-13-0083.
Campbell, R.M., and P.J. Tummino. 2014. Cancer epigenetics drug discovery and development: The challenge of hitting the mark. The Journal of Clinical Investigation 124(1): 64–69. doi:10.1172/JCI71605.
Canto, C., A.A. Sauve, and P. Bai. 2013. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Molecular Aspects of Medicine 34(6): 1168–1201. doi:10.1016/j.mam.2013.01.004.
Chen, D., S.B. Wan, H. Yang, J. Yuan, T.H. Chan, and Q.P. Dou. 2011. EGCG, green tea polyphenols and their synthetic analogs and prodrugs for human cancer prevention and treatment. Advances in Clinical Chemistry 53: 155–177.
Carter, L.G., J.A. D’Orazio, and K.J. Pearson. 2014. Resveratrol and cancer: Focus on in vivo evidence. Endocrine-Related Cancer 21(3): R209–R225. doi:10.1530/ERC-13-0171.
Chajes, V., M. Cambot, K. Moreau, G.M. Lenoir, and V. Joulin. 2006. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Research 66(10): 5287–5294. doi:10.1158/0008-5472.CAN-05-1489.
Chen, Y., W. Shu, W. Chen, Q. Wu, H. Liu, and G. Cui. 2007. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic & Clinical Pharmacology & Toxicology 101(6): 427–433. doi:10.1111/j.1742-7843.2007.00142.x.
Cheng, A.L., C.H. Hsu, J.K. Lin, M.M. Hsu, Y.F. Ho, T.S. Shen, J.Y. Ko, J.T. Lin, B.R. Lin, W. Ming-Shiang, H.S. Yu, S.H. Jee, G.S. Chen, T.M. Chen, C.A. Chen, M.K. Lai, Y.S. Pu, M.H. Pan, Y.J. Wang, C.C. Tsai, and C.Y. Hsieh. 2001. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Research 21(4B): 2895–2900.
Chestier, A., and M. Yaniv. 1979. Rapid turnover of acetyl groups in the four core histones of simian virus 40 minichromosomes. Proceedings of the National Academy of Sciences of the United States of America 76(1): 46–50.
Cho, H.S., J.H. Park, and Y.J. Kim. 2007. Epigenomics: Novel aspect of genomic regulation. Journal of Biochemistry and Molecular Biology 40(2): 151–155.
Choi, S.W., and S. Friso. 2010. Epigenetics: A new bridge between nutrition and health. Advances in Nutrition 1(1): 8–16. doi:10.3945/an.110.1004.
Choudhary, C., B.T. Weinert, Y. Nishida, E. Verdin, and M. Mann. 2014. The growing landscape of lysine acetylation links metabolism and cell signalling. Nature Reviews Molecular Cell Biology 15(8): 536–550. doi:10.1038/nrm3841.
Chowdhury, R., K.K. Yeoh, Y.M. Tian, L. Hillringhaus, E.A. Bagg, N.R. Rose, I.K. Leung, X.S. Li, E.C. Woon, M. Yang, M.A. McDonough, O.N. King, I.J. Clifton, R.J. Klose, T.D. Claridge, P.J. Ratcliffe, C.J. Schofield, and A. Kawamura. 2011. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Reports 12(5): 463–469. doi:10.1038/embor.2011.43.
Chua, B.T., D. Gallego-Ortega, A. Ramirez de Molina, A. Ullrich, J.C. Lacal, and J. Downward. 2009. Regulation of Akt(ser473) phosphorylation by choline kinase in breast carcinoma cells. Molecular Cancer 8: 131. doi:10.1186/1476-4598-8-131.
Cohen, I., E. Poreba, K. Kamieniarz, and R. Schneider. 2011. Histone modifiers in cancer: Friends or foes? Genes & Cancer 2(6): 631–647. doi:10.1177/1947601911417176.
Connolly, R., and V. Stearns. 2012. Epigenetics as a therapeutic target in breast cancer. Journal of Mammary Gland Biology and Neoplasia 17(3–4): 191–204. doi:10.1007/s10911-012-9263-3.
Consortium, Encode Project, E. Birney, J.A. Stamatoyannopoulos, A. Dutta, R. Guigo, T.R. Gingeras, E.H. Margulies, Z. Weng, M. Snyder, E.T. Dermitzakis, R.E. Thurman, M.S. Kuehn, C.M. Taylor, S. Neph, C.M. Koch, S. Asthana, A. Malhotra, I. Adzhubei, J.A. Greenbaum, R.M. Andrews, P. Flicek, P.J. Boyle, H. Cao, N.P. Carter, G.K. Clelland, S. Davis, N. Day, P. Dhami, S.C. Dillon, M.O. Dorschner, H. Fiegler, P.G. Giresi, J. Goldy, M. Hawrylycz, A. Haydock, R. Humbert, K.D. James, B.E. Johnson, E.M. Johnson, T.T. Frum, E.R. Rosenzweig, N. Karnani, K. Lee, G.C. Lefebvre, P.A. Navas, F. Neri, S.C. Parker, P.J. Sabo, R. Sandstrom, A. Shafer, D. Vetrie, M. Weaver, S. Wilcox, M. Yu, F.S. Collins, J. Dekker, J.D. Lieb, T.D. Tullius, G.E. Crawford, S. Sunyaev, W.S. Noble, I. Dunham, F. Denoeud, A. Reymond, P. Kapranov, J. Rozowsky, D. Zheng, R. Castelo, A. Frankish, J. Harrow, S. Ghosh, A. Sandelin, I.L. Hofacker, R. Baertsch, D. Keefe, S. Dike, J. Cheng, H.A. Hirsch, E.A. Sekinger, J. Lagarde, J.F. Abril, A. Shahab, C. Flamm, C. Fried, J. Hackermuller, J. Hertel, M. Lindemeyer, K. Missal, A. Tanzer, S. Washietl, J. Korbel, O. Emanuelsson, J.S. Pedersen, N. Holroyd, R. Taylor, D. Swarbreck, N. Matthews, M.C. Dickson, D.J. Thomas, M.T. Weirauch, J. Gilbert, J. Drenkow, I. Bell, X. Zhao, K.G. Srinivasan, W.K. Sung, H.S. Ooi, K.P. Chiu, S. Foissac, T. Alioto, M. Brent, L. Pachter, M.L. Tress, A. Valencia, S.W. Choo, C.Y. Choo, C. Ucla, C. Manzano, C. Wyss, E. Cheung, T.G. Clark, J.B. Brown, M. Ganesh, S. Patel, H. Tammana, J. Chrast, C.N. Henrichsen, C. Kai, J. Kawai, U. Nagalakshmi, J. Wu, Z. Lian, J. Lian, P. Newburger, X. Zhang, P. Bickel, J.S. Mattick, P. Carninci, Y. Hayashizaki, S. Weissman, T. Hubbard, R.M. Myers, J. Rogers, P.F. Stadler, T.M. Lowe, C.L. Wei, Y. Ruan, K. Struhl, M. Gerstein, S.E. Antonarakis, Y. Fu, E.D. Green, U. Karaoz, A. Siepel, J. Taylor, L.A. Liefer, K.A. Wetterstrand, P.J. Good, E.A. Feingold, M.S. Guyer, G.M. Cooper, G. Asimenos, C.N. Dewey, M. Hou, S. Nikolaev, J.I. Montoya-Burgos, A. Loytynoja, S. Whelan, F. Pardi, T. Massingham, H. Huang, N.R. Zhang, I. Holmes, J.C. Mullikin, A. Ureta-Vidal, B. Paten, M. Seringhaus, D. Church, K. Rosenbloom, W.J. Kent, E.A. Stone, Nisc Comparative Sequencing Program, Center Baylor College of Medicine Human Genome Sequencing, Center Washington University Genome Sequencing, Institute Broad, Institute Children’s Hospital Oakland Research, S. Batzoglou, N. Goldman, R.C. Hardison, D. Haussler, W. Miller, A. Sidow, N.D. Trinklein, Z.D. Zhang, L. Barrera, R. Stuart, D.C. King, A. Ameur, S. Enroth, M.C. Bieda, J. Kim, A.A. Bhinge, N. Jiang, J. Liu, F. Yao, V.B. Vega, C.W. Lee, P. Ng, A. Shahab, A. Yang, Z. Moqtaderi, Z. Zhu, X. Xu, S. Squazzo, M.J. Oberley, D. Inman, M.A. Singer, T.A. Richmond, K.J. Munn, A. Rada-Iglesias, O. Wallerman, J. Komorowski, J.C. Fowler, P. Couttet, A.W. Bruce, O.M. Dovey, P.D. Ellis, C.F. Langford, D.A. Nix, G. Euskirchen, S. Hartman, E. Urban, P. Kraus, S. Van Calcar, N. Heintzman, T.H. Kim, K. Wang, C. Qu, G. Hon, R. Luna, C.K. Glass, M.G. Rosenfeld, S.F. Aldred, S.J. Cooper, J.M. Lin, H.P. Shulha, X. Zhang, M. Xu, J.N. Haidar, Y. Yu, Y. Ruan, V.R. Iyer, rd Green, C. Wadelius, P.J. Farnham, B. Ren, R.A. Harte, A.S. Hinrichs, H. Trumbower, H. Clawson, J. Hillman-Jackson, A.S. Zweig, K. Smith, A. Thakkapallayil, G. Barber, R.M. Kuhn, D. Karolchik, L. Armengol, C.P. Bird, P.I. de Bakker, A.D. Kern, N. Lopez-Bigas, J.D. Martin, B.E. Stranger, A. Woodroffe, E. Davydov, A. Dimas, E. Eyras, I.B. Hallgrimsdottir, J. Huppert, M.C. Zody, G.R. Abecasis, X. Estivill, G.G. Bouffard, X. Guan, N.F. Hansen, J.R. Idol, V.V. Maduro, B. Maskeri, J.C. McDowell, M. Park, P.J. Thomas, A.C. Young, R.W. Blakesley, D.M. Muzny, E. Sodergren, D.A. Wheeler, K.C. Worley, H. Jiang, G.M. Weinstock, R.A. Gibbs, T. Graves, R. Fulton, E.R. Mardis, R.K. Wilson, M. Clamp, J. Cuff, S. Gnerre, D.B. Jaffe, J.L. Chang, K. Lindblad-Toh, E.S. Lander, M. Koriabine, M. Nefedov, K. Osoegawa, Y. Yoshinaga, B. Zhu, and P.J. de Jong. 2007. Identification and analysis of functional elements in 1 % of the human genome by the ENCODE pilot project. Nature 447(7146): 799–816. doi:10.1038/nature05874.
Dang, C.V., B.C. Lewis, C. Dolde, G. Dang, and H. Shim. 1997. Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. Journal of Bioenergetics and Biomembranes 29(4): 345–354.
Dang, C.V., and G.L. Semenza. 1999. Oncogenic alterations of metabolism. Trends in Biochemical Sciences 24(2): 68–72.
Dang, L., D.W. White, S. Gross, B.D. Bennett, M.A. Bittinger, E.M. Driggers, V.R. Fantin, H.G. Jang, S. Jin, M.C. Keenan, K.M. Marks, R.M. Prins, P.S. Ward, K.E. Yen, L.M. Liau, J.D. Rabinowitz, L.C. Cantley, C.B. Thompson, M.G. Vander Heiden, and S.M. Su. 2010. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 465(7300): 966. doi:10.1038/nature09132.
Davis, C.D., and E.O. Uthus. 2002. Dietary selenite and azadeoxycytidine treatments affect dimethylhydrazine-induced aberrant crypt formation in rat colon and DNA methylation in HT-29 cells. Journal of Nutrition 132(2): 292–297.
Davis, C.D., and E.O. Uthus. 2003. Dietary folate and selenium affect dimethylhydrazine-induced aberrant crypt formation, global DNA methylation and one-carbon metabolism in rats. Journal of Nutrition 133(9): 2907–2914.
Deberardinis, R.J., J.J. Lum, and C.B. Thompson. 2006. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. The Journal of Biological Chemistry 281(49): 37372–37380. doi:10.1074/jbc.M608372200.
Di, J.Z., X.D. Han, W.Y. Gu, Y. Wang, Q. Zheng, P. Zhang, H.M. Wu, and Z.Z. Zhu. 2011. Association of hypomethylation of LINE-1 repetitive element in blood leukocyte DNA with an increased risk of hepatocellular carcinoma. Journal of Zhejiang University Science B 12(10): 805–811. doi:10.1631/jzus.B1000422.
Ding, J., Z.M. Zhang, Y. Xia, G.Q. Liao, Y. Pan, S. Liu, Y. Zhang, and Z.S. Yan. 2013. LSD1-mediated epigenetic modification contributes to proliferation and metastasis of colon cancer. British Journal of Cancer 109(4): 994–1003. doi:10.1038/bjc.2013.364.
Ding, W., M. Mouzaki, H. You, J.C. Laird, J. Mato, S.C. Lu, and C.B. Rountree. 2009. CD133+ liver cancer stem cells from methionine adenosyl transferase 1A-deficient mice demonstrate resistance to transforming growth factor (TGF)-beta-induced apoptosis. Hepatology 49(4): 1277–1286. doi:10.1002/hep.22743.
Duthie, G.G. 1999. Determination of activity of antioxidants in human subjects. Proceedings of the Nutrition Society 58(4): 1015–1024.
Duthie, S.J. 2011. Folate and cancer: How DNA damage, repair and methylation impact on colon carcinogenesis. Journal of Inherited Metabolic Disease 34(1): 101–109. doi:10.1007/s10545-010-9128-0.
Easwaran, H., H.C. Tsai, and S.B. Baylin. 2014. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Molecular Cell 54(5): 716–727. doi:10.1016/j.molcel.2014.05.015.
Ehrlich, M. 2009. DNA hypomethylation in cancer cells. Epigenomics 1(2): 239–259. doi:10.2217/epi.09.33.
Ehrlich, M., G. Jiang, E. Fiala, J.S. Dome, M.C. Yu, T.I. Long, B. Youn, O.S. Sohn, M. Widschwendter, G.E. Tomlinson, M. Chintagumpala, M. Champagne, D. Parham, G. Liang, K. Malik, and P.W. Laird. 2002. Hypomethylation and hypermethylation of DNA in Wilms tumors. Oncogene 21(43): 6694–6702. doi:10.1038/sj.onc.1205890.
Esteller, M. 2002. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 21(35): 5427–5440. doi:10.1038/sj.onc.1205600.
Esteller, M. 2007. Epigenetic gene silencing in cancer: The DNA hypermethylome. Human Molecular Genetics. doi:10.1093/hmg/ddm018.
Fares, F., N. Azzam, B. Appel, B. Fares, and A. Stein. 2010. The potential efficacy of 3,3′-diindolylmethane in prevention of prostate cancer development. European Journal of Cancer Prevention 19(3): 199–203. doi:10.1097/CEJ.0b013e328333fbce.
Feinberg, A.P., and B. Vogelstein. 1983. Hypomethylation of ras oncogenes in primary human cancers. Biochemical and Biophysical Research Communications 111(1): 47–54.
Figueroa, M.E., O. Abdel-Wahab, C. Lu, P.S. Ward, J. Patel, A. Shih, Y. Li, N. Bhagwat, A. Vasanthakumar, H.F. Fernandez, M.S. Tallman, Z. Sun, K. Wolniak, J.K. Peeters, W. Liu, S.E. Choe, V.R. Fantin, E. Paietta, B. Lowenberg, J.D. Licht, L.A. Godley, R. Delwel, P.J. Valk, C.B. Thompson, R.L. Levine, and A. Melnick. 2010. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18(6): 553–567. doi:10.1016/j.ccr.2010.11.015.
Friis, R.M., B.P. Wu, S.N. Reinke, D.J. Hockman, B.D. Sykes, and M.C. Schultz. 2009. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Research 37(12): 3969–3980. doi:10.1093/nar/gkp270.
Fukao, T., T. Hosono, S. Misawa, T. Seki, and T. Ariga. 2004. The effects of allyl sulfides on the induction of phase II detoxification enzymes and liver injury by carbon tetrachloride. Food and Chemical Toxicology 42(5): 743–749. doi:10.1016/j.fct.2003.12.010.
Galluzzi, L., O. Kepp, M.G. Vander Heiden, and G. Kroemer. 2013. Metabolic targets for cancer therapy. Nature Reviews Drug Discovery 12(11): 829–846. doi:10.1038/nrd4145.
Ganapathy-Kanniappan, S., and J.F. Geschwind. 2013. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Molecular Cancer 12: 152. doi:10.1186/1476-4598-12-152.
Gerhäuser, Clarissa. 2012. Cancer cell metabolism, epigenetics and the potential influence of dietary components—A perspective. Biomedical Research 23(1): 1–21.
Ghoshal, A.K., and E. Farber. 1984. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis 5(10): 1367–1370.
Ghoshal, K., and S. Bai. 2007. DNA methyltransferases as targets for cancer therapy. Drugs Today (Barc) 43(6): 395–422. doi:10.1358/dot.2007.43.6.1062666.
Glozak, M.A., and E. Seto. 2007. Histone deacetylases and cancer. Oncogene 26(37): 5420–5432. doi:10.1038/sj.onc.1210610.
Gossell-Williams, M., H. Fletcher, N. McFarlane-Anderson, A. Jacob, J. Patel, and S. Zeisel. 2005. Dietary intake of choline and plasma choline concentrations in pregnant women in Jamaica. West Indian Medical Journal 54(6): 355–359.
Govardhan, K.S., K. Ramyasri, D. Kethora, Y. Ravishekar, and M. Prasenjit. 2011. Harnessing impaired energy metabolism in cancer cell: small molecule- mediated ways to regulate tumorigenesis. Anticancer Agents in Medicinal Chemistry 11(3): 272–279.
Groth, A., W. Rocha, A. Verreault, and G. Almouzni. 2007. Chromatin challenges during DNA replication and repair. Cell 128(4): 721–733. doi:10.1016/j.cell.2007.01.030.
Guo, X.P., X.Y. Zhang, and S.D. Zhang. 1991. [Clinical trial on the effects of shikonin mixture on later stage lung cancer]. Zhong Xi Yi Jie He Za Zhi 11(10): 580, 598–599.
Hansen, K.H., A.P. Bracken, D. Pasini, N. Dietrich, S.S. Gehani, A. Monrad, J. Rappsilber, M. Lerdrup, and K. Helin. 2008. A model for transmission of the H3K27me3 epigenetic mark. Nature Cell Biology 10(11): 1291–1300. doi:10.1038/ncb1787.
Henderson, D.S., S.S. Banga, T.A. Grigliatti, and J.B. Boyd. 1994. Mutagen sensitivity and suppression of position-effect variegation result from mutations in mus209, the Drosophila gene encoding PCNA. EMBO Journal 13(6): 1450–1459.
Herceg, Z., and P. Hainaut. 2007. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Molecular Oncology 1(1): 26–41. doi:10.1016/j.molonc.2007.01.004.
Herr, I., V. Lozanovski, P. Houben, P. Schemmer, and M.W. Buchler. 2013. Sulforaphane and related mustard oils in focus of cancer prevention and therapy. Wiener Medizinische Wochenschrift 163(3–4): 80–88. doi:10.1007/s10354-012-0163-3.
Hino, S., A. Sakamoto, K. Nagaoka, K. Anan, Y. Wang, S. Mimasu, T. Umehara, S. Yokoyama, K. Kosai, and M. Nakao. 2012. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nature Communications 3: 758. doi:10.1038/ncomms1755.
Hitchler, M.J., and F.E. Domann. 2009. Metabolic defects provide a spark for the epigenetic switch in cancer. Free Radical Biology & Medicine 47(2): 115–127. doi:10.1016/j.freeradbiomed.2009.04.010.
Holliday, R., and T. Ho. 2002. DNA methylation and epigenetic inheritance. Methods 27(2): 179–183.
Horn, H.F., and K.H. Vousden. 2007. Coping with stress: Multiple ways to activate p53. Oncogene 26(9): 1306–1316. doi:10.1038/sj.onc.1210263.
Houtkooper, R.H., and J. Auwerx. 2012. Exploring the therapeutic space around NAD+. The Journal of Cell Biology 199(2): 205–209. doi:10.1083/jcb.201207019.
Howells, L.M., D.P. Berry, P.J. Elliott, E.W. Jacobson, E. Hoffmann, B. Hegarty, K. Brown, W.P. Steward, and A.J. Gescher. 2011. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases—Safety, pharmacokinetics, and pharmacodynamics. Cancer Prevention Research (Philadelphia, PA) 4(9): 1419–1425. doi:10.1158/1940-6207.CAPR-11-0148.
Huang, C., M. Xu, and B. Zhu. 2013. Epigenetic inheritance mediated by histone lysine methylation: Maintaining transcriptional states without the precise restoration of marks? Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 368(1609): 20110332. doi:10.1098/rstb.2011.0332.
Huang, J., and S.L. Berger. 2008. The emerging field of dynamic lysine methylation of non-histone proteins. Current Opinion in Genetics and Development 18(2): 152–158. doi:10.1016/j.gde.2008.01.012.
Huang, J., C. Plass, and C. Gerhauser. 2011. Cancer chemoprevention by targeting the epigenome. Current Drug Targets 12(13): 1925–1956.
Huang, S. 2002. Histone methyltransferases, diet nutrients and tumour suppressors. Nature Reviews Cancer 2(6): 469–476. doi:10.1038/nrc819.
Hui, E.P., A.T. Chan, F. Pezzella, H. Turley, K.F. To, T.C. Poon, B. Zee, F. Mo, P.M. Teo, D.P. Huang, K.C. Gatter, P.J. Johnson, and A.L. Harris. 2002. Coexpression of hypoxia-inducible factors 1alpha and 2alpha, carbonic anhydrase IX, and vascular endothelial growth factor in nasopharyngeal carcinoma and relationship to survival. Clinical Cancer Research 8(8): 2595–2604.
Hustad, S., M.C. McKinley, H. McNulty, J. Schneede, J.J. Strain, J.M. Scott, and P.M. Ueland. 2002. Riboflavin, flavin mononucleotide, and flavin adenine dinucleotide in human plasma and erythrocytes at baseline and after low-dose riboflavin supplementation. Clinical Chemistry 48(9): 1571–1577.
Jang, M., S.S. Kim, and J. Lee. 2013. Cancer cell metabolism: Implications for therapeutic targets. Experimental & Molecular Medicine 45: e45. doi:10.1038/emm.2013.85.
Jenuwein, T., and C.D. Allis. 2001. Translating the histone code. Science 293(5532): 1074–1080. doi:10.1126/science.1063127.
Jin, B., and K.D. Robertson. 2013. DNA methyltransferases, DNA damage repair, and cancer. Advances in Experimental Medicine & Biology 754: 3–29. doi:10.1007/978-1-4419-9967-2_1.
Jo, H.J., J.D. Song, K.M. Kim, Y.H. Cho, K.H. Kim, and Y.C. Park. 2008. Diallyl disulfide induces reversible G2/M phase arrest on a p53-independent mechanism in human colon cancer HCT-116 cells. Oncology Reports 19(1): 275–280.
Kaluz, S., M. Kaluzova, S.Y. Liao, M. Lerman, and E.J. Stanbridge. 2009. Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: A one transcription factor (HIF-1) show? Biochimica et Biophysica Acta 1795(2): 162–172. doi:10.1016/j.bbcan.2009.01.001.
Keum, Y.S., H.G. Kim, A.M. Bode, Y.J. Surh, and Z. Dong. 2013. UVB-induced COX-2 expression requires histone H3 phosphorylation at Ser10 and Ser28. Oncogene 32(4): 444–452. doi:10.1038/onc.2012.71.
Khin, S.S., R. Kitazawa, T. Kondo, Y. Idei, M. Fujimoto, R. Haraguchi, K. Mori, and S. Kitazawa. 2011. Epigenetic alteration by DNA promoter hypermethylation of genes related to transforming growth factor-beta (TGF-beta) signaling in cancer. Cancers (Basel) 3(1): 982–993. doi:10.3390/cancers3010982.
Kihira, Y., N. Yamano, Y. Izawa-Ishizawa, K. Ishizawa, Y. Ikeda, K. Tsuchiya, T. Tamaki, and S. Tomita. 2011. Basic fibroblast growth factor regulates glucose metabolism through glucose transporter 1 induced by hypoxia-inducible factor-1alpha in adipocytes. The International Journal of Biochemistry & Cell Biology 43(11): 1602–1611. doi:10.1016/j.biocel.2011.07.009.
Killian, J.K., S.Y. Kim, M. Miettinen, C. Smith, M. Merino, M. Tsokos, M. Quezado, W.I. Smith Jr, M.S. Jahromi, P. Xekouki, E. Szarek, R.L. Walker, J. Lasota, M. Raffeld, B. Klotzle, Z. Wang, L. Jones, Y. Zhu, Y. Wang, J.J. Waterfall, M.J. O’Sullivan, M. Bibikova, K. Pacak, C. Stratakis, K.A. Janeway, J.D. Schiffman, J.B. Fan, L. Helman, and P.S. Meltzer. 2013. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discovery 3(6): 648–657. doi:10.1158/2159-8290.CD-13-0092.
Klein, E.A., I.M. Thompson Jr, C.M. Tangen, J.J. Crowley, M.S. Lucia, P.J. Goodman, L.M. Minasian, L.G. Ford, H.L. Parnes, J.M. Gaziano, D.D. Karp, M.M. Lieber, P.J. Walther, L. Klotz, J.K. Parsons, J.L. Chin, A.K. Darke, S.M. Lippman, G.E. Goodman, F.L. Meyskens Jr, and L.H. Baker. 2011. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 306(14): 1549–1556. doi:10.1001/jama.2011.1437.
Kloten, V., B. Becker, K. Winner, M.G. Schrauder, P.A. Fasching, T. Anzeneder, J. Veeck, A. Hartmann, R. Knuchel, and E. Dahl. 2013. Promoter hypermethylation of the tumor-suppressor genes ITIH5, DKK3, and RASSF1A as novel biomarkers for blood-based breast cancer screening. Breast Cancer Research 15(1): R4. doi:10.1186/bcr3375.
Klug, M., S. Schmidhofer, C. Gebhard, R. Andreesen, and M. Rehli. 2013. 5-Hydroxymethylcytosine is an essential intermediate of active DNA demethylation processes in primary human monocytes. Genome Biology 14(5): R46. doi:10.1186/gb-2013-14-5-r46.
Koch, C.M., R.M. Andrews, P. Flicek, S.C. Dillon, U. Karaoz, G.K. Clelland, S. Wilcox, D.M. Beare, J.C. Fowler, P. Couttet, K.D. James, G.C. Lefebvre, A.W. Bruce, O.M. Dovey, P.D. Ellis, P. Dhami, C.F. Langford, Z. Weng, E. Birney, N.P. Carter, D. Vetrie, and I. Dunham. 2007. The landscape of histone modifications across 1 % of the human genome in five human cell lines. Genome Research 17(6): 691–707. doi:10.1101/gr.5704207.
Kohn, A.D., S.A. Summers, M.J. Birnbaum, and R.A. Roth. 1996. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. The Journal of Biological Chemistry 271(49): 31372–31378.
Koppenol, W.H., P.L. Bounds, and C.V. Dang. 2011. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature Reviews. Cancer 11(5): 325–337. doi:10.1038/nrc3038.
Kornienko, A.E., P.M. Guenzl, D.P. Barlow, and F.M. Pauler. 2013. Gene regulation by the act of long non-coding RNA transcription. BMC Biology 11: 59. doi:10.1186/1741-7007-11-59.
Koukourakis, M.I., A. Giatromanolaki, E. Sivridis, G. Bougioukas, V. Didilis, K.C. Gatter, A.L. Harris, and Tumour and Group Angiogenesis Research. 2003. Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. British Journal of Cancer 89(5): 877–885. doi:10.1038/sj.bjc.6601205.
Lachner, M., D. O’Carroll, S. Rea, K. Mechtler, and T. Jenuwein. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410(6824): 116–120. doi:10.1038/35065132.
Lahtz, C., and G.P. Pfeifer. 2011. Epigenetic changes of DNA repair genes in cancer. Journal of Molecular Cell Biology 3(1): 51–58. doi:10.1093/jmcb/mjq053.
Lamprecht, S.A., and M. Lipkin. 2003. Chemoprevention of colon cancer by calcium, vitamin D and folate: Molecular mechanisms. Nature Reviews. Cancer 3(8): 601–614. doi:10.1038/nrc1144.
Laplante, M., and D.M. Sabatini. 2009. An emerging role of mTOR in lipid biosynthesis. Current Biology 19(22): R1046–R1052. doi:10.1016/j.cub.2009.09.058.
Lea, M.A., M. Rasheed, V.M. Randolph, F. Khan, A. Shareef, and C. desBordes. 2002. Induction of histone acetylation and inhibition of growth of mouse erythroleukemia cells by S-allylmercaptocysteine. Nutrition and Cancer 43(1): 90–102. doi:10.1207/S15327914NC431_11.
Lee, W.J., J.Y. Shim, and B.T. Zhu. 2005. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Molecular Pharmacology 68(4): 1018–1030. doi:10.1124/mol.104.008367.
Letouze, E., C. Martinelli, C. Loriot, N. Burnichon, N. Abermil, C. Ottolenghi, M. Janin, M. Menara, A.T. Nguyen, P. Benit, A. Buffet, C. Marcaillou, J. Bertherat, L. Amar, P. Rustin, A. De Reynies, A.P. Gimenez-Roqueplo, and J. Favier. 2013. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23(6): 739–752. doi:10.1016/j.ccr.2013.04.018.
Li, B., Q. Lu, Z.G. Song, L. Yang, H. Jin, Z.G. Li, T.J. Zhao, Y.F. Bai, J. Zhu, H.Z. Chen, and Z.Y. Xu. 2013a. Functional analysis of DNA methylation in lung cancer. European Review for Medical and Pharmacological Sciences 17(9): 1191–1197.
Li, H.P., C.C. Peng, I.C. Chung, M.Y. Huang, S.T. Huang, C.C. Chen, K.P. Chang, C.L. Hsu, and Y.S. Chang. 2013b. Aberrantly hypermethylated Homeobox A2 derepresses metalloproteinase-9 through TBP and promotes invasion in Nasopharyngeal carcinoma. Oncotarget 4(11): 2154–2165.
Li, Y., X. Li, and B. Guo. 2010. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Research 70(2): 646–654. doi:10.1158/0008-5472.CAN-09-1924.
Liu, W., K. Glunde, Z.M. Bhujwalla, V. Raman, A. Sharma, and J.M. Phang. 2012. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Research 72(14): 3677–3686. doi:10.1158/0008-5472.CAN-12-0080.
Liu, W., X. Li, E.S. Chu, M.Y. Go, L. Xu, G. Zhao, L. Li, N. Dai, J. Si, Q. Tao, J.J. Sung, and J. Yu. 2011. Paired box gene 5 is a novel tumor suppressor in hepatocellular carcinoma through interaction with p53 signaling pathway. Hepatology 53(3): 843–853. doi:10.1002/hep.24124.
Liu, W., and J.M. Phang. 2012. Proline dehydrogenase (oxidase) in cancer. Biofactors 38(6): 398–406. doi:10.1002/biof.1036.
Logan, R.F., M.J. Grainge, V.C. Shepherd, N.C. Armitage, K.R. Muir, and ukCAP Trial Group. 2008. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology 134(1): 29–38. doi:10.1053/j.gastro.2007.10.014.
Lozanovski, V.J., P. Houben, U. Hinz, T. Hackert, I. Herr, and P. Schemmer. 2014. Pilot study evaluating broccoli sprouts in advanced pancreatic cancer (POUDER trial)—Study protocol for a randomized controlled trial. Trials 15: 204. doi:10.1186/1745-6215-15-204.
Lu, C., and C.B. Thompson. 2012. Metabolic regulation of epigenetics. Cell Metabolism 16(1): 9–17. doi:10.1016/j.cmet.2012.06.001.
Luger, K., A.W. Mader, R.K. Richmond, D.F. Sargent, and T.J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389(6648): 251–260. doi:10.1038/38444.
Lujambio, A., G.A. Calin, A. Villanueva, S. Ropero, M. Sanchez-Cespedes, D. Blanco, L.M. Montuenga, S. Rossi, M.S. Nicoloso, W.J. Faller, W.M. Gallagher, S.A. Eccles, C.M. Croce, and M. Esteller. 2008. A microRNA DNA methylation signature for human cancer metastasis. Proceedings of the National Academy of Sciences of the United States of America 105(36): 13556–13561. doi:10.1073/pnas.0803055105.
Lukey, M.J., K.F. Wilson, and R.A. Cerione. 2013. Therapeutic strategies impacting cancer cell glutamine metabolism. Future Medicinal Chemistry 5(14): 1685–1700. doi:10.4155/fmc.13.130.
Lv, C., X. Yang, B. Yu, Q. Ma, B. Liu, and Y. Liu. 2012a. Blocking the Na+/H+ exchanger 1 with cariporide (HOE642) reduces the hypoxia-induced invasion of human tongue squamous cell carcinoma. International Journal of Oral and Maxillofacial Surgery 41(10): 1206–1210. doi:10.1016/j.ijom.2012.03.001.
Lv, T., D. Yuan, X. Miao, Y. Lv, P. Zhan, X. Shen, and Y. Song. 2012b. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS One 7(4): e35065. doi:10.1371/journal.pone.0035065.
Ma, W., H.J. Sung, J.Y. Park, S. Matoba, and P.M. Hwang. 2007. A pivotal role for p53: Balancing aerobic respiration and glycolysis. Journal of Bioenergetics and Biomembranes 39(3): 243–246. doi:10.1007/s10863-007-9083-0.
Mack, G.S. 2010. To selectivity and beyond. Nature Biotechnology 28(12): 1259–1266. doi:10.1038/nbt.1724.
Maggio, A., A. Magli, T. Rancati, C. Fiorino, F. Valvo, G. Fellin, U. Ricardi, F. Munoz, D. Cosentino, L.F. Cazzaniga, R. Valdagni, and V. Vavassori. 2014. Daily sodium butyrate enema for the prevention of radiation proctitis in prostate cancer patients undergoing radical radiation therapy: Results of a multicenter randomized placebo-controlled dose-finding phase 2 study. International Journal of Radiation Oncology, Biology, Physics 89(3): 518–524. doi:10.1016/j.ijrobp.2014.03.018.
Majmundar, A.J., W.J. Wong, and M.C. Simon. 2010. Hypoxia-inducible factors and the response to hypoxic stress. Molecular Cell 40(2): 294–309. doi:10.1016/j.molcel.2010.09.022.
Marelli-Berg, F.M., H. Fu, and C. Mauro. 2012. Molecular mechanisms of metabolic reprogramming in proliferating cells: Implications for T-cell-mediated immunity. Immunology 136(4): 363–369. doi:10.1111/j.1365-2567.2012.03583.x.
Markham, G.D., and M.A. Pajares. 2009. Structure-function relationships in methionine adenosyltransferases. Cellular and Molecular Life Science 66(4): 636–648. doi:10.1007/s00018-008-8516-1.
Marshall, J.R., C.M. Tangen, W.A. Sakr, D.P. Wood Jr, D.L. Berry, E.A. Klein, S.M. Lippman, H.L. Parnes, D.S. Alberts, D.F. Jarrard, W.R. Lee, J.M. Gaziano, E.D. Crawford, B. Ely, M. Ray, W. Davis, L.M. Minasian, and I.M. Thompson Jr. 2011. Phase III trial of selenium to prevent prostate cancer in men with high-grade prostatic intraepithelial neoplasia: SWOG S9917. Cancer Prevention Research (Philadelphia, PA) 4(11): 1761–1769. doi:10.1158/1940-6207.CAPR-10-0343.
Mathupala, S.P., A. Rempel, and P.L. Pedersen. 2001. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. The Journal of Biological Chemistry 276(46): 43407–43412. doi:10.1074/jbc.M108181200.
Mazurek, S. 2011. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. The International Journal of Biochemistry & Cell Biology 43(7): 969–980. doi:10.1016/j.biocel.2010.02.005.
Meacham, C.E., and S.J. Morrison. 2013. Tumour heterogeneity and cancer cell plasticity. Nature 501(7467): 328–337. doi:10.1038/nature12624.
Mello, J.A., H.H. Sillje, D.M. Roche, D.B. Kirschner, E.A. Nigg, and G. Almouzni. 2002. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Reports 3(4): 329–334. doi:10.1093/embo-reports/kvf068.
Messing, E., J.R. Gee, D.R. Saltzstein, K. Kim, A. diSant’Agnese, J. Kolesar, L. Harris, A. Faerber, T. Havighurst, J.M. Young, M. Efros, R.H. Getzenberg, M.A. Wheeler, J. Tangrea, H. Parnes, M. House, J.E. Busby, R. Hohl, and H. Bailey. 2012. A phase 2 cancer chemoprevention biomarker trial of isoflavone G-2535 (genistein) in presurgical bladder cancer patients. Cancer Prevention Research (Philadelphia, PA) 5(4): 621–630. doi:10.1158/1940-6207.CAPR-11-0455.
Miao, F., and R. Natarajan. 2005. Mapping global histone methylation patterns in the coding regions of human genes. Molecular and Cellular Biology 25(11): 4650–4661. doi:10.1128/MCB.25.11.4650-4661.2005.
Mimeault, M., and S.K. Batra. 2013. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. Journal of Cellular and Molecular Medicine 17(1): 30–54. doi:10.1111/jcmm.12004.
Molinie, B., and P. Georgel. 2009. Genetic and epigenetic regulations of prostate cancer by genistein. Drug News & Perspectives 22(5): 247–254. doi:10.1358/dnp.2009.22.5.1378633.
Munoz-Pinedo, C., N. El Mjiyad, and J.E. Ricci. 2012. Cancer metabolism: Current perspectives and future directions. Cell Death & Disease 3: e248. doi:10.1038/cddis.2011.123.
Myzak, M.C., K. Hardin, R. Wang, R.H. Dashwood, and E. Ho. 2006. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 27(4): 811–819. doi:10.1093/carcin/bgi265.
Myzak, M.C., P.A. Karplus, F.L. Chung, and R.H. Dashwood. 2004. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Research 64(16): 5767–5774. doi:10.1158/0008-5472.CAN-04-1326.
Nelson, D.M., X. Ye, C. Hall, H. Santos, T. Ma, G.D. Kao, T.J. Yen, J.W. Harper, and P.D. Adams. 2002. Coupling of DNA synthesis and histone synthesis in S phase independent of cyclin/cdk2 activity. Molecular and Cellular Biology 22(21): 7459–7472.
Nian, H., B. Delage, E. Ho, and R.H. Dashwood. 2009. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: Studies with sulforaphane and garlic organosulfur compounds. Environmental and Molecular Mutagenesis 50(3): 213–221. doi:10.1002/em.20454.
Niculescu, M.D., C.N. Craciunescu, and S.H. Zeisel. 2006. Dietary choline deficiency alters global and gene-specific DNA methylation in the developing hippocampus of mouse fetal brains. FASEB Journal 20(1): 43–49. doi:10.1096/fj.05-4707com.
Oermann, E.K., J. Wu, K.L. Guan, and Y. Xiong. 2012. Alterations of metabolic genes and metabolites in cancer. Seminars in Cell & Developmental Biology 23(4): 370–380. doi:10.1016/j.semcdb.2012.01.013.
Pascale, R.M., M.M. Simile, M.R. De Miglio, and F. Feo. 2002. Chemoprevention of hepatocarcinogenesis: S-adenosyl-l-methionine. Alcohol 27(3): 193–198.
Paschos, K., and M.J. Allday. 2010. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends in Microbiology 18(10): 439–447. doi:10.1016/j.tim.2010.07.003.
Pattabiraman, D.R., and R.A. Weinberg. 2014. Tackling the cancer stem cells—What challenges do they pose? Nature Reviews Drug Discovery 13(7): 497–512. doi:10.1038/nrd4253.
Phang, J.M., S.P. Donald, J. Pandhare, and Y. Liu. 2008. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids 35(4): 681–690. doi:10.1007/s00726-008-0063-4.
Pledgie-Tracy, A., M.D. Sobolewski, and N.E. Davidson. 2007. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Molecular Cancer Therapeutics 6(3): 1013–1021. doi:10.1158/1535-7163.MCT-06-0494.
Podlaha, O., S. De, M. Gonen, and F. Michor. 2014. Histone modifications are associated with transcript isoform diversity in normal and cancer cells. PLoS Computational Biology 10(6): e1003611. doi:10.1371/journal.pcbi.1003611.
Portela, A., and M. Esteller. 2010. Epigenetic modifications and human disease. Nature Biotechnology 28(10): 1057–1068. doi:10.1038/nbt.1685.
Probst, A.V., E. Dunleavy, and G. Almouzni. 2009. Epigenetic inheritance during the cell cycle. Nature Reviews Molecular Cell Biology 10(3): 192–206. doi:10.1038/nrm2640.
Putiri, E.L., R.L. Tiedemann, J.J. Thompson, C. Liu, T. Ho, J.H. Choi, and K.D. Robertson. 2014. Distinct and overlapping control of 5-methylcytosine and 5-hydroxymethylcytosine by the TET proteins in human cancer cells. Genome Biology 15(6): R81. doi:10.1186/gb-2014-15-6-r81.
Radpour, R., Z. Barekati, C. Kohler, Q. Lv, N. Burki, C. Diesch, J. Bitzer, H. Zheng, S. Schmid, and X.Y. Zhong. 2011. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One 6(1): e16080. doi:10.1371/journal.pone.0016080.
Ragsdale, S.W. 2008. Catalysis of methyl group transfers involving tetrahydrofolate and B(12). Vitamins & Hormones 79: 293–324. doi:10.1016/S0083-6729(08)00410-X.
Rai, R., M. Tewari, M. Kumar, A.K. Singh, and H.S. Shukla. 2011. p53: Its alteration and gallbladder cancer. European Journal of Cancer Prevention 20(2): 77–85. doi:10.1097/CEJ.0b013e328341e371.
Ramani, K., J.M. Mato, and S.C. Lu. 2011. Role of methionine adenosyltransferase genes in hepatocarcinogenesis. Cancers (Basel) 3(2): 1480–1497. doi:10.3390/cancers3021480.
Reid, T., F. Valone, W. Lipera, D. Irwin, W. Paroly, R. Natale, S. Sreedharan, H. Keer, B. Lum, F. Scappaticci, and A. Bhatnagar. 2004. Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer. Lung Cancer 45(3): 381–386. doi:10.1016/j.lungcan.2004.03.002.
Reitman, Z.J., and H. Yan. 2010. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. Journal of the National Cancer Institute 102(13): 932–941. doi:10.1093/jnci/djq187.
Reuter, S., S.C. Gupta, B. Park, A. Goel, and B.B. Aggarwal. 2011. Epigenetic changes induced by curcumin and other natural compounds. Genes & Nutrition 6(2): 93–108. doi:10.1007/s12263-011-0222-1.
Riggs, M.G., R.G. Whittaker, J.R. Neumann, and V.M. Ingram. 1977. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 268(5619): 462–464.
Robey, R.B., and N. Hay. 2009. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Seminars in Cancer Biology 19(1): 25–31. doi:10.1016/j.semcancer.2008.11.010.
Sadikovic, B., K. Al-Romaih, J.A. Squire, and M. Zielenska. 2008. Cause and consequences of genetic and epigenetic alterations in human cancer. Current Genomics 9(6): 394–408. doi:10.2174/138920208785699580.
Sambucetti, L.C., D.D. Fischer, S. Zabludoff, P.O. Kwon, H. Chamberlin, N. Trogani, H. Xu, and D. Cohen. 1999. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. The Journal of Biological Chemistry 274(49): 34940–34947.
Sasaki, M., C.B. Knobbe, M. Itsumi, A.J. Elia, I.S. Harris, I.I. Chio, R.A. Cairns, S. McCracken, A. Wakeham, J. Haight, A.Y. Ten, B. Snow, T. Ueda, S. Inoue, K. Yamamoto, M. Ko, A. Rao, K.E. Yen, S.M. Su, and T.W. Mak. 2012. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes & Development 26(18): 2038–2049. doi:10.1101/gad.198200.112.
Saunders, L.R., and E. Verdin. 2007. Sirtuins: Critical regulators at the crossroads between cancer and aging. Oncogene 26(37): 5489–5504. doi:10.1038/sj.onc.1210616.
Sauve, A.A., C. Wolberger, V.L. Schramm, and J.D. Boeke. 2006. The biochemistry of sirtuins. Annual Review of Biochemistry 75: 435–465. doi:10.1146/annurev.biochem.74.082803.133500.
Schulze, A., and A.L. Harris. 2012. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491(7424): 364–373. doi:10.1038/nature11706.
Schwartzenberg-Bar-Yoseph, F., M. Armoni, and E. Karnieli. 2004. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Research 64(7): 2627–2633.
Sealy, L., and R. Chalkley. 1978. The effect of sodium butyrate on histone modification. Cell 14(1): 115–121.
Shankar, S., D. Kumar, and R.K. Srivastava. 2013. Epigenetic modifications by dietary phytochemicals: Implications for personalized nutrition. Pharmacology & Therapeutics 138(1): 1–17. doi:10.1016/j.pharmthera.2012.11.002.
Shapiro, T.A., J.W. Fahey, A.T. Dinkova-Kostova, W.D. Holtzclaw, K.K. Stephenson, K.L. Wade, L. Ye, and P. Talalay. 2006. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutrition and Cancer 55(1): 53–62. doi:10.1207/s15327914nc5501_7.
Sharif, J., M. Muto, S. Takebayashi, I. Suetake, A. Iwamatsu, T.A. Endo, J. Shinga, Y. Mizutani-Koseki, T. Toyoda, K. Okamura, S. Tajima, K. Mitsuya, M. Okano, and H. Koseki. 2007. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450(7171): 908–912. doi:10.1038/nature06397.
Sharma, R.A., S.A. Euden, S.L. Platton, D.N. Cooke, A. Shafayat, H.R. Hewitt, T.H. Marczylo, B. Morgan, D. Hemingway, S.M. Plummer, M. Pirmohamed, A.J. Gescher, and W.P. Steward. 2004. Phase I clinical trial of oral curcumin: Biomarkers of systemic activity and compliance. Clinical Cancer Research 10(20): 6847–6854. doi:10.1158/1078-0432.CCR-04-0744.
Shay, J.E., and M. Celeste Simon. 2012. Hypoxia-inducible factors: Crosstalk between inflammation and metabolism. Seminars in Cell & Developmental Biology 23(4): 389–394. doi:10.1016/j.semcdb.2012.04.004.
Shi, Y., and J.R. Whetstine. 2007. Dynamic regulation of histone lysine methylation by demethylases. Molecular Cell 25(1): 1–14. doi:10.1016/j.molcel.2006.12.010.
Shor, B., J.J. Gibbons, R.T. Abraham, and K. Yu. 2009. Targeting mTOR globally in cancer: Thinking beyond rapamycin. Cell Cycle 8(23): 3831–3837.
Singh-Gupta, V., S. Banerjee, C.K. Yunker, J.T. Rakowski, M.C. Joiner, A.A. Konski, F.H. Sarkar, and G.G. Hillman. 2012. B-DIM impairs radiation-induced survival pathways independently of androgen receptor expression and augments radiation efficacy in prostate cancer. Cancer Letters 318(1): 86–92. doi:10.1016/j.canlet.2011.12.006.
Singh, B.N., H. Zhou, J. Li, T. Tipton, B. Wang, G. Shao, E.N. Gilbert, Q. Li, and S.W. Jiang. 2011. Preclinical studies on histone deacetylase inhibitors as therapeutic reagents for endometrial and ovarian cancers. Future Oncology 7(12): 1415–1428. doi:10.2217/fon.11.124.
Singh, S., and B.B. Aggarwal. 1995. Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected]. The Journal of Biological Chemistry 270(42): 24995–25000.
Smolkova, K., L. Plecita-Hlavata, N. Bellance, G. Benard, R. Rossignol, and P. Jezek. 2011. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. The International Journal of Biochemistry & Cell Biology 43(7): 950–968. doi:10.1016/j.biocel.2010.05.003.
Soga, T. 2013. Cancer metabolism: Key players in metabolic reprogramming. Cancer Science 104(3): 275–281. doi:10.1111/cas.12085.
Stadtman, E.R., G.D. Novelli, and F. Lipmann. 1951. Coenzyme A function in and acetyl transfer by the phosphotransacetylase system. The Journal of Biological Chemistry 191(1): 365–376.
Steiner, C., S. Arnould, A. Scalbert, and C. Manach. 2008. Isoflavones and the prevention of breast and prostate cancer: New perspectives opened by nutrigenomics. The British Journal of Nutrition 99 E Suppl 1: ES78–ES108. doi:10.1017/S0007114508965788.
Strahl, B.D., and C.D. Allis. 2000. The language of covalent histone modifications. Nature 403(6765): 41–45. doi:10.1038/47412.
Sun, M., Z. Estrov, Y. Ji, K.R. Coombes, D.H. Harris, and R. Kurzrock. 2008. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Molecular Cancer Therapeutics 7(3): 464–473. doi:10.1158/1535-7163.MCT-07-2272.
Suzuki, H., R. Maruyama, E. Yamamoto, and M. Kai. 2012. DNA methylation and microRNA dysregulation in cancer. Molecular Oncology 6(6): 567–578. doi:10.1016/j.molonc.2012.07.007.
Suzuki, H., R. Maruyama, E. Yamamoto, and M. Kai. 2013. Epigenetic alteration and microRNA dysregulation in cancer. Frontiers in Genetics 4: 258. doi:10.3389/fgene.2013.00258.
Takahashi, H., J. M. McCaffery, R. A. Irizarry, and J. D. Boeke. 2006. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Molecular cell 23(2):207–217. doi: 10.1016/j.molcel.2006.05.040
Tamada, M., M. Suematsu, and H. Saya. 2012. Pyruvate kinase M2: Multiple faces for conferring benefits on cancer cells. Clinical Cancer Research 18(20): 5554–5561. doi:10.1158/1078-0432.CCR-12-0859.
Taylor, C.K., R.M. Levy, J.C. Elliott, and B.P. Burnett. 2009. The effect of genistein aglycone on cancer and cancer risk: A review of in vitro, preclinical, and clinical studies. Nutrition Reviews 67(7): 398–415. doi:10.1111/j.1753-4887.2009.00213.x.
Teperino, R., K. Schoonjans, and J. Auwerx. 2010. Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metabolism 12(4): 321–327. doi:10.1016/j.cmet.2010.09.004.
Tormos, K.V., and N.S. Chandel. 2010. Inter-connection between mitochondria and HIFs. Journal of Cellular and Molecular Medicine 14(4): 795–804. doi:10.1111/j.1582-4934.2010.01031.x.
Vander Heiden, M.G. 2013. Exploiting tumor metabolism: Challenges for clinical translation. The Journal of Clinical Investigation 123(9): 3648–3651. doi:10.1172/JCI72391.
Venkatesh, H.S., M.M. Chaumeil, C.S. Ward, D.A. Haas-Kogan, C.D. James, and S.M. Ronen. 2012. Reduced phosphocholine and hyperpolarized lactate provide magnetic resonance biomarkers of PI3K/Akt/mTOR inhibition in glioblastoma. Neuro Oncology 14(3): 315–325. doi:10.1093/neuonc/nor209.
Verdone, L., M. Caserta, and E. Di Mauro. 2005. Role of histone acetylation in the control of gene expression. Biochemistry and Cell Biology 83(3): 344–353. doi:10.1139/o05-041.
Vousden, K.H., and C. Prives. 2009. Blinded by the light: The growing complexity of p53. Cell 137(3): 413–431. doi:10.1016/j.cell.2009.04.037.
Vousden, K.H., and K.M. Ryan. 2009. p53 and metabolism. Nature Reviews Cancer 9(10): 691–700. doi:10.1038/nrc2715.
Wanka, C., D.P. Brucker, O. Bahr, M. Ronellenfitsch, M. Weller, J.P. Steinbach, and J. Rieger. 2012. Synthesis of cytochrome C oxidase 2: A p53-dependent metabolic regulator that promotes respiratory function and protects glioma and colon cancer cells from hypoxia-induced cell death. Oncogene 31(33): 3764–3776. doi:10.1038/onc.2011.530.
Ward, P.S., J. Patel, D.R. Wise, O. Abdel-Wahab, B.D. Bennett, H.A. Coller, J.R. Cross, V.R. Fantin, C.V. Hedvat, A.E. Perl, J.D. Rabinowitz, M. Carroll, S.M. Su, K.A. Sharp, R.L. Levine, and C.B. Thompson. 2010. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17(3): 225–234. doi:10.1016/j.ccr.2010.01.020.
Wellen, K.E., G. Hatzivassiliou, U.M. Sachdeva, T.V. Bui, J.R. Cross, and C.B. Thompson. 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324(5930): 1076–1080. doi:10.1126/science.1164097.
Wilson, A.S., B.E. Power, and P.L. Molloy. 2007. DNA hypomethylation and human diseases. Biochimica et Biophysica Acta 1775(1): 138–162. doi:10.1016/j.bbcan.2006.08.007.
Wise, D.R., P.S. Ward, J.E. Shay, J.R. Cross, J.J. Gruber, U.M. Sachdeva, J.M. Platt, R.G. DeMatteo, M.C. Simon, and C.B. Thompson. 2011. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proceedings of the National Academy of Sciences of the United States of America 108(49): 19611–19616. doi:10.1073/pnas.1117773108.
Wu, W., and S. Zhao. 2013. Metabolic changes in cancer: Beyond the Warburg effect. Acta Biochimica et Biophysica Sinica (Shanghai) 45(1): 18–26. doi:10.1093/abbs/gms104.
Xu, M., W. Wang, S. Chen, and B. Zhu. 2012. A model for mitotic inheritance of histone lysine methylation. EMBO Reports 13(1): 60–67. doi:10.1038/embor.2011.206.
Xu, W., H. Yang, Y. Liu, Y. Yang, P. Wang, S.H. Kim, S. Ito, C. Yang, P. Wang, M.T. Xiao, L.X. Liu, W.Q. Jiang, J. Liu, J.Y. Zhang, B. Wang, S. Frye, Y. Zhang, Y.H. Xu, Q.Y. Lei, K.L. Guan, S.M. Zhao, and Y. Xiong. 2011. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19(1): 17–30. doi:10.1016/j.ccr.2010.12.014.
Yang, C.S., X. Wang, G. Lu, and S.C. Picinich. 2009a. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nature Reviews Cancer 9(6): 429–439. doi:10.1038/nrc2641.
Yang, H., J.A. Zonder, and Q.P. Dou. 2009b. Clinical development of novel proteasome inhibitors for cancer treatment. Expert Opinion on Investigational Drugs 18(7): 957–971. doi:10.1517/13543780903002074.
Yang, M., T. Soga, and P.J. Pollard. 2013. Oncometabolites: linking altered metabolism with cancer. The Journal of Clinical Investigation 123(9): 3652–3658. doi:10.1172/JCI67228.
Yang, X., D. Wang, W. Dong, Z. Song, and K. Dou. 2010. Inhibition of Na(+)/H(+) exchanger 1 by 5-(N-ethyl-N-isopropyl) amiloride reduces hypoxia-induced hepatocellular carcinoma invasion and motility. Cancer Letters 295(2): 198–204. doi:10.1016/j.canlet.2010.03.001.
Ye, D., S. Ma, Y. Xiong, and K.L. Guan. 2013. R-2-hydroxyglutarate as the key effector of IDH mutations promoting oncogenesis. Cancer Cell 23(3): 274–276. doi:10.1016/j.ccr.2013.03.005.
Ying, W. 2008. NAD +/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxidants & Redox Signaling 10(2): 179–206. doi:10.1089/ars.2007.1672.
Yoon, S., M.Y. Lee, S.W. Park, J.S. Moon, Y.K. Koh, Y.H. Ahn, B.W. Park, and K.S. Kim. 2007. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. The Journal of Biological Chemistry 282(36): 26122–26131. doi:10.1074/jbc.M702854200.
Yu, R., J.J. Jiao, J.L. Duh, K. Gudehithlu, T.H. Tan, and A.N. Kong. 1997. Activation of mitogen-activated protein kinases by green tea polyphenols: Potential signaling pathways in the regulation of antioxidant-responsive element-mediated phase II enzyme gene expression. Carcinogenesis 18(2): 451–456.
Zhang, X.D., Z.H. Qin, and J. Wang. 2010. The role of p53 in cell metabolism. Acta Pharmacologica Sinica 31(9): 1208–1212. doi:10.1038/aps.2010.151.
Zhang, Y., and L. Tang. 2007. Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta Pharmacologica Sinica 28(9): 1343–1354. doi:10.1111/j.1745-7254.2007.00679.x.
Zhu, B., and D. Reinberg. 2011. Epigenetic inheritance: Uncontested? Cell Research 21(3): 435–441. doi:10.1038/cr.2011.26.
Acknowledgments
This study was supported by Grants to K.-M.H. from the National Cancer Center through the Korean Ministry of Health and Welfare (#1410674 and #1510121).
Conflict of interest
We declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chandra, V., Hong, KM. Effects of deranged metabolism on epigenetic changes in cancer. Arch. Pharm. Res. 38, 321–337 (2015). https://doi.org/10.1007/s12272-015-0561-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-015-0561-3