
Abstract
Remote ischemic conditioning (RIC) can be effectively applied for cardio-protection. Here, to clarify whether RIC exerts myocardial protection via aldehyde dehydrogenase 2 (ALDH2), we established a myocardial ischemia/reperfusion (I/R) model in C57BL/6 and ALDH2 knockout (ALDH2-KO) mice and treated them with RIC. Echocardiography and single-cell contraction experiments showed that RIC significantly improved myocardial function and alleviated I/R injury in C57BL/6 mice but did not exhibit its cardioprotective effects in ALDH2-KO mice. TUNEL, Evan’s blue/triphenyl tetrazolium chloride, and reactive oxygen species (ROS) assays showed that RIC’s effect on reducing myocardial cell apoptosis, myocardial infarction area, and ROS levels was insignificant in ALDH2-KO mice. Our results showed that RIC could increase ALDH2 protein levels, activate sirtuin 3 (SIRT3)/hypoxia-inducible factor 1-alpha (HIF1α), inhibit autophagy, and exert myocardial protection. This study revealed that RIC could exert myocardial protection via the ALDH2/SIRT3/HIF1α signaling pathway by reducing 4-HNE secretion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocardial ischemia/reperfusion (I/R) injury is common in the clinical treatment of cardiovascular diseases [1]. Reducing myocardial I/R injury after opening the aorta during cardiopulmonary bypass surgery and coronary stent implantation in coronary heart disease has consistently been the focus of general cardiovascular clinical attention [2]. Preconditioning and postconditioning can play a role in myocardial protection; however, they have low applicability and operability [3,4,5]. Remote ischemic conditioning (RIC) refers to the short-term myocardial I/R of another organ before post-ischemia reperfusion to activate the body’s endogenous protective mechanism [6, 7]. However, the specific mechanism of action of RIC remains unclear. Therefore, exploring the mechanism of RIC can provide a basis for the clinical application of RIC [5, 8].
Aldehyde dehydrogenase 2 (ALDH2), an essential isoenzyme in the ALDH family, is an endogenous cardioprotective factor in the mitochondria, closely related to cardiovascular disease occurrence. It is involved in the pathology of coronary heart disease, heart failure, cardiomyopathy, and several other physiological processes [9, 10]. It is distributed in various tissues and organs of the human body, primarily in the mitochondria of the human heart, brain, lung, liver, and kidney cells [10, 11]. The level of ALDH2 in the heart is much higher than that of other types of aldehyde dehydrogenases, and it has the most robust activity. Approximately 40% of the East Asian population has the ALDH2 deletion genotype closely related to myocardial infarction (MI) [12].
Recent studies have shown that a variety of preconditioning and postconditioning strategies can play a role in myocardial I/R injury via ALDH2, indicating that ALDH2 is crucial for pre and post-stimulation [11, 13]. In addition, research suggests that RIC alleviates myocardial I/R injury by upregulating ALDH2 expression levels via the PI3K/Akt or PI3K/mTOR pathway [14, 15]. However, there are numerous challenges (intervention time, intervention mode, secondary ischemic injury, and the complex clinical circumstances of cardiac patients) to be examined [16, 17]. In addition, the complex mechanism of RIC has not been elucidated, especially the mechanism of ALDH2 mediated cardioprotection. In this study, we investigated the role of ALDH2 in RIC-induced myocardial protection and related mechanisms by using ALDH2 knockout (ALDH2-KO) and wild-type (WT) mice to establish myocardial I/R models.
Materials and Methods
Animals
WT mice (C57BL/6 age 8–10 weeks) were obtained from Cavens Biogle Model Animal Research Co., Ltd. (Suzhou, China), and matched ALDH2-KO male mice were generated as described previously [18]. All protocols were approved by the Animal Care Ethics Committee of Fudan University and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The mice were kept under a 12:12-h light/dark cycle at a consistent temperature and humidity and given ad libitum access to food and water. A dose of analgesics was given if the animals appeared to be experiencing pain (based on criteria such as immobility and failure to eat). At the indicated intervals, the mice were euthanized by CO2/cervical dislocation, and tissues were subsequently harvested for analyses. Mice were randomly assigned to four groups of 20 each: sham, RIC, I/R, and RIC+ I/R groups.
Myocardial I/R Injury and RIC Treatment
To establish a mouse myocardial I/R injury model, we used 8–10-week-old mice. Anesthesia was maintained under 2% isoflurane induction (RWD Life Science Inc., Shenzhen, China). The limbs of the mice were fixed, the left chest of the mice was exposed, and the hair was shaved. We selected four or five intercostal openings on the left side of the mouse, separated the muscles, identified the strongest point of the apex, inserted the mosquito vascular clamp, opened the chest, and applied slight pressure on the right index finger to make the mouse heart jump out of the chest cavity. The left ventricular descending anterior descending branch was ligated with a 6-0 silk suture slipknot. After ligation, the heart was returned to the pericardial cavity, and the chest cavity was closed using two slip knots. The wound was sutured; the knot was loosened after waiting for 45 min and reperfusion was performed for 24 h to construct the myocardial I/R model. The detailed method was explained previously [19]. During the 45-min ischemic period, we used a 3 × 5 tourniquet latex tube to ligate mouse legs to block blood flow for 5 min and then released for 5 min three times for RIC treatment.
Echocardiography Analysis
Mice were anesthetized with isoflurane to evaluate cardiac function 24 h after myocardial I/R, and M-mode images were acquired using a Vevo 2100 high-frequency ultrasound system (VisualSonics, Toronto, ON, Canada). The data was averaged based on the measurements of at least six cardiac cycles, which included recording the left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) scores (n>6). The specific operation was performed as described previously [19, 20].
Single-Cell Systolic and Diastolic Function of Cardiomyocytes
Cardiomyocytes were isolated from the model mice using a previously described method [21]. The systolic and diastolic functions of primary cardiomyocytes were detected using the IonOptix™ system (IonOptix Corporation, Milton, MA, USA). The detection buffer, cardiomyocyte calcium buffer, comprised 130 mM NaCl, 5.4 mM KCl, 10 mM HEPES, 1.8 mM CaCl2, 0.5 mM MgCl2, and 10 mM glucose, pH 7.4. Two drops of the buffer were added to each slide. The contractile function of cardiomyocytes was evaluated by measuring the peak shortening (PS) and maximal velocity of shortening (-dL/dt) of cardiomyocytes. We measured and averaged the contractile function of 30 cardiomyocytes per mouse.
Evan’s Blue/Triphenyl Tetrazolium Chloride (TTC) Staining
After 24 h of reperfusion, the mice were anesthetized intraperitoneally with 2% sodium pentobarbital. The chest cavity was opened, the heart was exposed, the anterior coronary artery was retied, and 1% Evan’s blue was injected into the left atrial appendage to allow the heart to beat freely. The heart was then cut out, washed with PBS, and immediately frozen on dry ice. After 30 min, the heart was cut into 5-6 short-axis sections on average with a blade. Next, 1% 2,3,5-TTC was incubated at 37 °C in a water bath for 30 min. Each section was flattened and fixed with 4% paraformaldehyde for 2 h. The blue area impregnated with Evan’s blue was the non-ischemic area. The red area impregnated by TTC was the ischemic area. The white unstained area was the myocardial infarction site. The area at risk (AAR) included both white unstained area and red area (n>4). Image quantification was performed by segmenting the stained areas of each section using ImageJ software [19, 22]. Infarct size is expressed as the ratio of white unstained area to AAR and presented as a percentage.
TUNEL Assay
Myocardial tissues (4–8 mice per group, with two incisions at the connective tissue of myocardial infarction) were fixed with 4% paraformaldehyde and stained using the One Step TUNEL Apoptosis Assay Kit (Beyotime Biotechnology, China) according to previous reports [19].
Reactive Oxygen Species (ROS) Measurement
ROS production was evaluated by analyzing the fluorescence intensity resulting from dihydroethidium (DHE) staining (Invitrogen D11347). Briefly, frozen mice hearts were cut into 5-μm sections. The heart sections were stained 37 times with 5 μM DHE for 30 min followed by staining with DAPI for 10 min and examined using a fluorescence microscope (n>4).
Histological Analysis
The myocardial tissue was fixed with 4% paraformaldehyde (4–8 mice in each group, with two incisions at the connective tissue of myocardial infarction) 24 h after reperfusion. Macrophage infiltration was detected using anti-mouse F480 (ab25377; Abcam) according to a previously described method [20].
Electron Microscopy
Transmission electron microscopy was used to observe the ultrastructure of the cardiomyocytes (n=3). Briefly, the hearts of mice in each experimental group were perfused and fixed with tube-buffered formaldehyde-glutaraldehyde. The left ventricular myocardium was removed from the middle of the ventricle and cut into 1-mm3 pieces. The blocks were fixed overnight with a 10:1 liquid/tissue ratio at 4 °C. To further process the myocardial mass, it was incubated in 2% sucrose (pH 7.4), 1% OsO4, and 1.5% K3[Fe(CN)6]·3H2O buffer overnight at 22–24 °C. It was then dehydrated with graded ethanol and propylene oxide and finally encapsulated with Epon/Araldite. An RMC-MTXL ultramicrotome and diatom diamond knife were used to obtain sections. The images were obtained using a CM-120 transmission electron microscope (Philips, Netherlands). Each heart sample was observed in at least 10 fields.
Western Blot Analysis
Cardiac tissue (n≥4) at the infarct site was harvested in the RIPA lysis buffer containing 1 mM phenylmethanesulfonyl fluoride. Protein concentration was determined using the BCA protein assay kit (Bio-Rad, 5000006JA). Next, 20-μg, normalized protein samples were separated via 10% and 15% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Biotech Well). The membranes were blocked with 5% bovine serum albumin in TBST for 2 h and incubated with the following primary antibodies at 4 °C overnight: ALDH2 (ab133306; Abcam; 1:1000), hypoxia-inducible factor 1-alpha (HIF1α) (36169S; Cell Signaling Technology; 1:1000), 4-hydroxynonenal (4-HNE) (ab46545, Abcam; 1:3000), sirtuin 3 (5490S; Cell Signaling Technology; 1:1000), P62 (ab109012; Abcam; 1:10000), LC3B (ab192890; Abcam; 1:2000), caspase-3 (19245S; Cell Signaling Technology; 1:1000), cleaved caspase-3 (9664S; Cell Signaling Technology; 1:1000), Bax (14796S; Cell Signaling Technology; 1:1000), BCL-2 (3498S; Cell Signaling Technology; 1:1000), and β-actin (4970S; Cell Signaling Technology; 1:4000). The horseradish peroxidase-conjugated secondary antibody was allowed to stand at room temperature (24 °C) for 1.5 h. Detection was performed using Immobilon Western chemiluminescent HRP substrate (Millipore, Billerica, MA, USA). Gel images were captured using an Image Quant LAS 4000 Mini (n=6, GE Healthcare, Barrington, IL, USA).
Statistical Analysis
Data is expressed as the mean ± standard error of the mean. Statistical analyses were conducted using GraphPad Prism 5.01 software. The normality of the data distribution was tested using the Kolmogorov-Smirnov test. The Mann-Whitney-U test was used when the group data were not normally distributed or if the group variances were unequal. The homogeneity of variance test was performed using Levene’s test. Continuous data with normal distribution were assessed by one-way analysis of variance (ANOVA) with post hoc test or two-way ANOVA with post hoc test (Tukey-Kramer) as indicated [20].
Results
RIC Treatment Ameliorated Myocardial I/R Injury in Mice
To investigate the therapeutic potential of RIC in vivo, we used the myocardial I/R mouse model. Myocardial I/R injury was mimicked by coronary artery ligation for 45 min, followed by 24-h reperfusion with RIC. SFigure 1a shows representative echocardiograms illustrating the comparison of mouse hearts after surgery and those in the sham group 24 h after treatment with RIC. During echocardiography, the heart rates of the mice were similar in all groups (SFig. 1b). In the sham groups, it was evident that RIC treatment did not change the LVEF (SFig. 1c) or LVFS (SFig. 1d). After 24 h of reperfusion, echocardiographic parameters were significantly restored in mice treated with RIC compared with those in the control group, with LVEF values of I/R + RIC group (61.89 ± 4.120%, n=10) and I/R group (49.77 ± 3.399%, n=10), with p values <0.05 (SFig. 1c). The LVFS values were I/R + RIC group (34.73 ± 1.952%, n=10) and I/R group (21.00 ± 2.638%, n=10), with p <0.001 (SFig. 1d). After isolating the mouse cardiomyocytes in each group, we used the IonOptix™ Soft-Edge single-cell contractile function detection system to evaluate the functional improvement of RIC in cardiomyocytes after I/R at the cellular level. In terms of systolic function, PS and -dL/dt in failed cardiomyocytes, after I/R, were significantly lower than those in the sham group, and they were significantly improved after RIC treatment (SFig. 1e and 1f).
Myocardial infarction size was assessed via Evan’s blue/TTC staining, and a small infarction size was observed after RIC treatment, as shown by a reduced ratio of the white-to-red area (Fig. 1a and b). Furthermore, the results of apoptotic protein analysis showed that after myocardial I/R, the expression of myocardial apoptotic protein Bax and cleaved caspase-3 increased significantly (Fig. 1c, d, and g). Conversely, after RIC treatment, the protein levels of Bax and cleaved caspase-3 were significantly decreased (Fig. 1d, e, and g). TUNEL staining also showed that RIC significantly reduced cardiomyocyte apoptosis (Fig. 1f and h). In addition, we examined the ROS deposition levels in the frozen sections using DHE staining. RIC treatment significantly reduced postoperative injury after myocardial I/R (Fig. 2a and c). F480 results also showed that RIC significantly reduced macrophage infiltration in myocardial tissues (Fig. 2b and d).

RIC treatment ameliorates I/R injury in mice. Representative images of Evan’s blue dye and TTC staining (a). The ratio of risk area to left ventricular area in each group (AAR/LV, n>6, b). Change in infarction size induced by RIC or without RIC (IA/AAR, n>6, b). Western blot of Bax (n=6, d, c), caspase 3 (n=6, d, e), and cleaved caspase3 (n=6, d, g) levels in myocardium after I/R treatment and RIC treatment. Fluorescence imaging of cardiomyocyte apoptosis induced by I/R treatment with RIC. The cell nuclei were stained with DAPI (blue). Red represents apoptotic cardiomyocytes (n=6, Scale bar=200 μm, f, h). Data are depicted as the mean ± SEM. Statistical significance was determined by two-way ANOVA with a post hoc Holm-Sidak test; ns, not significant; *P<0.05; **P<0.001; ***P<0.001; ****P<0.0001

RIC protects myocardium from I/R injury through ALDH2/SIRT3 regulation of autophagy and reduction of 4-HNE levels in mice. Representative images of DHE-stained heart sections from mice 1 day after I/R. Scale bar =200 μm (a). Relative index of ROS fluorescence (n=4, c). Representation of myocardial macrophage infiltration (F480, scale bar=240 μm, b) and related statistics (n=4, d). Representative western blot of ALDH2, SIRT3, HIF1α, P62, LC3II, and 4-HNE levels in mice treated with RIC and control animals (e). Effects of RIC on ALDH2 (n=6, f), HIF1α (n=6, f), SIRT3 (n=6, g), P62 (n=6, g), LC3II (n=6, i), and 4-HNE (n=6, i) expression in I/R mouse model. Transmission electron microscopy (TEM) images of the left ventricle. Red arrows mark autophagosomes (h). Data are depicted as the mean ± SEM. Statistical significance was determined by two-way ANOVA with a post hoc Holm-Sidak test, *P<0.05, **P<0.001, ***P<0.001, ****P<0.0001
ALDH2/SIRT3-Based Regulation of Autophagy and Reduction of 4-HNE Levels in Mice
ALDH2 is an endogenous protective factor that plays an essential role in repairing myocardial I/R injury [12]. RIC treatment significantly promoted the protein levels of ALDH2 in both sham and I/R hearts (Fig. 2e and f). The protein levels of HIF1α and SIRT3 were similar to ALDH2, which was downregulated after I/R, and upregulated after RIC (Fig. 2f and g). In addition, RIC decreased myocardial autophagy after myocardial I/R (Fig. 2e and i). The electron microscopy results showed that the arrangement of the myocardium was disordered, and the number of autophagosomes increased after I/R (Fig. 2h). Post-RIC treatment, the arrangement of the myocardium was improved, with reduced number of myocardial autophagosomes (Fig. 2h). We also found that RIC attenuated 4-HNE induction by myocardial I/R injury, further reducing ROS levels and myocardial apoptosis (Fig. 2e and i). Hence, we speculate that RIC protects the myocardium from myocardial I/R injury via autophagy by regulating the ALDH2/SIRT3 signaling pathway and via attenuation of 4-HNE by the ALDH2/4-HNE signaling pathway in mice.
ALDH2 Deficiency Attenuates the Protective Effect of RIC on Myocardial I/R Injury in Mice
After 24 h of reperfusion, echocardiography examination revealed worse LV functions in ALDH2-KO mice than in the WT controls, presenting lower EF and FS values in ALDH2-KO mice (Fig. 3a, c, and d). Furthermore, we used the IonOptix™ Soft-Edge single-cell contractile function detection system to evaluate cardiac function. The study found that PS (Fig. 3e) and -dL/dt (Fig. 3f) in the ALDH2-KO group were worse than those in the WT group. In addition, RIC significantly improved I/R-induced myocardial dysfunction in the WT group but not in the ALKDH-KO group (Fig. 3).

ALDH2 deficiency attenuates the protective effect of RIC on I/R injury in mice. Representative images of echocardiography tracing in WT and ALDH2-KO groups after 24 h of reperfusion (a). Heart rate (BPM, n>6, b). Left ventricular ejection fraction (LVEF, n>6, c). Left ventricular fractional shortening (LVFS, n>6, d). Peak shortening (% cell lengthening, n>6, e). Maximal shortening velocity (-dl/dt, n>6, f). Data are depicted as the mean ± SEM. Statistical significance was determined by two-way ANOVA with a post hoc Holm-Sidak test; ns, not significant; *P<0.05; **P<0.001; ***P<0.001
Evan’s blue/TTC staining and TUNEL staining showed that in both the control and RIC groups, the area of myocardial infarction and myocardial apoptosis in the ALDH2-KO group increased significantly (Fig. 4). This further proved that the myocardial protective effect of RIC could be attenuated by ALDH2 deficiency (Fig. 5a–d). In addition, ROS and F480 staining showed that RIC could reduce ROS levels and myocardial inflammation after I/R in WT mice; however, the therapeutic effect was attenuated in the ALDH2-KO group (Fig. 5e–g). Furthermore, ROS and myocardial inflammation in the ALDH2-KO group increased significantly after I/R compared with that in the WT group (Fig. 5e–g).

RIC reduces myocardial infarction size and apoptosis after I/R was attenuated by ALDH2. Representative images of Evan’s blue dye and TTC staining (a). The ratio of risk area to left ventricular area in each group (AAR/LV, n=5–7, b) and change in infarction size induced by RIC or without RIC in the WT and KO groups (IA/AAR, n=5–7, b). Fluorescence imaging of cardiomyocyte apoptosis induced by I/R treatment with RIC. Cell nuclei were stained with DAPI (blue). Red represents apoptotic cardiomyocytes (n=6, Scale bar=200 μm, c, d). Data are depicted as the mean ± SEM. Statistical significance was determined by two-way ANOVA with a post hoc Holm-Sidak test; ns, not significant; *P<0.05; **P<0.001; ***P<0.001

RIC reduces ROS damage and inflammatory infiltration after I/R was attenuated by ALDH2. Representative western blot of caspase-3, cleaved caspase-3, and Bax in WT and ALDH2-KO mice treated with RIC (a). After I/R, RIC downregulated cleaved caspase-3 (n=4, b) and Bax (n=4, c) expression, but ALDH2 attenuated this effect. Representative images of DHE-stained heart sections from mice 1 day after I/R (n=4, Scale bar=200 μm, d, f). Representation of myocardial macrophage infiltration (F480, scale bar=240 μm, g) and related statistics (n=4, e). Data are depicted as the mean ± SEM. Statistical significance was determined by two-way ANOVA with a post hoc Holm-Sidak test; ns, not significant; *P<0.05; **P<0.001; ***P<0.001, ****P<0.0001
The above results show that RIC treatment reduced the area of myocardial infarction in the WT group and decreased myocardial apoptosis and ROS levels; however, there was no significant improvement in the ALDH2-KO group (Figs. 4 and 5). In summary, our results show that RIC can protect mouse myocardium from I/R injury; however, ALDH2 deficiency can attenuate this protective effect.
RIC Exhibits Cardioprotection After Myocardial I/R Injury
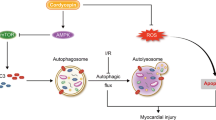
Western blotting revealed that RIC can cause transient ischemia in the lower limbs of mice, induce high protein levels of HIF1α, and exert myocardial protection (Fig. 6a and c). However, in the ALDH2-KO group, RIC did not induce a high protein level of HIF1α (Fig. 6a and c). SIRT3 is one of several nicotinamide adenine dinucleotide-dependent histone deacetylases that regulates various functions in mammals, including aging and metabolism [23, 24]. ALDH2 is a direct SIRT3 substrate, and its deacetylation increases acetaminophen toxic-metabolite binding and enzyme inactivation [25]. Under normal circumstances, the deletion of ALDH2 did not affect the protein levels of SIRT3 (Fig. 6a and d). However, after I/R in the WT group, the protein levels of SIRT3 were significantly reduced but significantly increased after RIC treatment (Fig. 6a and d). However, when ALDH2 was deficient, SIRT3 protein levels in the I/R group were significantly decreased, and RIC did not increase SIRT3 protein levels (Fig. 6a and d). In addition, we found that myocardial autophagy was significantly increased after I/R, and RIC inhibited myocardial autophagy (Fig. 6a, e, and f). However, when ALDH2 was deficient, the regulatory effect of RIC on autophagy disappeared (Fig. 6a, e, and f). Furthermore, RIC can reduce the induction of 4-HNE after myocardial I/R injury. However, the effect of RIC was attenuated by ALDH2 deletion (Fig. 6g). In summary, we speculate that RIC protects the myocardium from I/R injury via the regulation of autophagy by the ALDH2/SIRT3/HIF1α signaling pathway and attenuates 4-HNE via the ALDH2/4-HNE signaling pathway in mice (Fig. 6h).

RIC exhibits cardio-protection after I/R injury by eliminating 4-HNE and regulating autophagy through ALDH2/SIRT3/HIF1α signaling pathway. Representative western blot of ALDH2, SIRT3, HIF1α, P62, LC3II, and 4-HNE expression (a). In the I/R model, RIC-treatment of the WT group upregulated the expression of ALDH2 (n=4, b), SIRT3 (n=4, c), and HIF1α (n=4, d) and regulated autophagy-related proteins, P62 and LC3II (n=4, e, f). However, after ALDH2-KO, the effect of RIC was blocked. Furthermore, RIC reduced 4-HNE levels, but there was no significant difference in the ALDH2-KO group (n=4, g). RIC cardio-protection after I/R by eliminating 4-HNE and regulating autophagy through the ALDH2/SIRT3/HIF1α signaling pathway (h). Data are depicted as the mean ± SEM. Statistical significance was determined by two-way ANOVA with a post hoc Holm-Sidak test; ns, not significant; *P<0.05; **P<0.001; ***P<0.001; ***P<0.001
Discussion
To our understanding, this is the first study to demonstrate that RIC can inhibit autophagy via the ALDH2/SIRT3/HIF1α signaling pathway and attenuate 4-HNE, consequently, protecting the myocardium from I/R injury. RIC is feasible and straightforward and is expected to be a more promising myocardial protection measure in clinical applications. Using this strategy, we observed a significant improvement in myocardial function after I/R injury in mice. In addition, our study demonstrated that RIC could attenuate 4-HNE and regulate myocardial autophagy via the ALDH2/SIRT3/HIF1α pathway, reduce myocardial infarction area, and inhibit myocardial cell apoptosis. This study suggests that RIC plays a vital role in cardiac protection after myocardial I/R injury.
Myocardial I/R injury is widely used in the clinical treatment of cardiovascular diseases, such as aortic opening after cardiopulmonary bypass surgery and coronary artery stent implantation. RIC can initiate the endogenous protective mechanism of the body by temporary I/R of another organ before the onset of myocardial I/R. Previous studies have shown that RIC can reduce infarct size, protect myocardial function, and improve adverse cardiac remodeling in patients with MI [7, 26]. Furthermore, RIC can improve the inflammatory response after cerebral ischemia and reduce both the risk and symptoms of cerebral hemorrhage [27]. As an exogenous intervention, RIC is simple and easy to implement. In this study, a mouse myocardial I/R model was constructed and treated with RIC. It was found that RIC can significantly reduce the area of myocardial infarction and myocardial cell apoptosis and protect heart function.
RIC could promote the secretion of a variety of humoral factors and activate a variety of signal transduction pathways, consequently playing a role in myocardial protection [28, 29]. Numerous studies have shown that the humoral component of RIC includes endogenous opioids, endocannabinoids, adrenomedullin, as well as calcitonin gene-bound peptide, and miRNAs as a component of exosomes [29]. These mediators trigger cardioprotective signaling and mediate cardiac repair after I/R [28, 29]. A recent study revealed RIC mediated cardiovascular protection via regulation of plasma cytokines as well as changes in cell surface characteristics of monocytes [30]. In addition, RIC can also inhibit Rho-kinase [31], JNK activation [32], downregulate STAT3 phosphorylation [33], and ERK pathways [34] to attenuate I/R injury. It was worth noting that when we studied the cardioprotective effect of RIC on I/R, we found that RIC could significantly promote ALDH2 protein expression in the myocardium. However, when ALDH2 is deficient, the myocardial protection of RIC attenuated. This data indicates that ALDH2 plays an extremely important role in RIC mediated cardio-protection.
ALDH2 is an endogenous cardioprotective factor in the mitochondria and is involved in the pathophysiological processes of coronary heart disease, heart failure, cardiomyopathy, and several other diseases [35,36,37]. Ma et al. performed I/R treatment in WT mice, ALDH2-overexpressed mice, and ALDH2-KO mice and found that the area of myocardial infarction in ALDH2-overexpressed mice was significantly decreased, whereas that in ALDH2-KO mice was increased [38]. Our research shows that after I/R, ALDH2 protein levels were significantly decreased, and RIC significantly increased the protein levels of ALDH2. Furthermore, we found that RIC also upregulated SIRT3. SIRT3 is primarily located in the mitochondria and can reduce oxidative stress damage and the area of myocardial infarction by activating the anti-oxidative stress signaling pathway, thereby protecting the myocardium from reperfusion injury [39]. ALDH2 is a direct SIRT3 substrate, and its deacetylation increases acetaminophen toxic-metabolite binding and enzyme inactivation [25, 40]. Therefore, we believe that RIC can reduce myocardial I/R injury by promoting the protein levels of ALDH2/SIRT3, thereby exerting myocardial protection.
Myocardial I/R injury stabilizes HIF1α, the primary regulator of the transcriptional response initiated by hypoxia [31], and HIF2α [41]. Previous research has shown that remote ischemic preconditioning (RIPC) increases plasma IL-10 levels and decreases myocardial infarct size in WT mice but not in HIF1α-deficient mice [42]. However, another study revealed that RIPC-induced cardioprotection was preserved in partially HIF1α-deficient mice and in rats pretreated with cadmium (HIF-1α inhibitors) [43]. Two studies presented controversial conclusions. However, the role of HIF-1α in RIC remains unclear. Our results reveal that RIC can increase the protein levels of HIF-1α and participate in RIC-induced cardioprotection. ALDH2 can regulate mitochondrial fission and smooth muscle cell proliferation via the HIF1α signal pathway [44, 45]. Thus, ALDH2 acts as an endogenous cardiac protective factor in the mitochondria and can exert myocardial protection by regulating autophagy [46].
Mitochondria are the main source of ROS in cells, and when ROS exceed their antioxidant capacity, they lead to fatty acid oxidation, a process known as lipid peroxidation [47]. 4-HNE is the most abundant lipid peroxidation product and forms adducts with proteins, which affects its biological function and destroys intracellular homeostasis [48]. The level of plasma 4-HNE was increased in patients with heart failure, which was negatively correlated with cardiac function [49]. ALDH2 is a mitochondrial enzyme that metabolizes ethanol and toxic aldehydes, such as 4-HNE [36]. In this study, we found that myocardial I/R injury leads to excessive 4-HNE levels and has serious consequences in cardiac dysfunction after I/R. The absence of ALDH2 significantly increased the protein levels of 4-HNE, which aggravated myocardial I/R injury, further revealing that ALDH2 could play a role in promoting 4-HNE metabolism. Conversely, RIC can upregulate ALDH2 protein levels, clear excessive 4-HNE levels, protect the myocardium, and reduce ROS levels.
Notably, 40% of the East Asian population and 8% of the global population carry the ALDH2 mutation, which is caused by the replacement of glutamate with lysine at amino acid 487 and results in only 15% of the catalytic activity of the WT ALDH2 [50,51,52]. Our present study indicates that RIC can protect the myocardium from I/R damage via the ALDH2/SIRT3/HIF1α pathway and by decreasing 4-HNE levels. Furthermore, RIC is highly operable, simple, easy to implement, and significant for clinical transformation. Therefore, it is expected to become a myocardial protection measure with more clinical application prospects.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code Availability
Not applicable.
Abbreviations
- RIC:
-
Remote ischemic conditioning
- ALDH2:
-
Aldehyde dehydrogenase 2
- I/R:
-
Ischemia/reperfusion
- ROS:
-
Reactive oxygen species
- SIRT3:
-
Sirtuin 3
- HIF1α:
-
Hypoxia-inducible factor 1-alpha
- 4-HNE:
-
4-hydroxynonenal
- ALDH2-KO:
-
ALDH2 knockout
- TTC:
-
1% 2,3,5-triphenyl tetrazolium chloride
- PS:
-
Peak shortening
- -dL/dt:
-
Maximal velocity of shortening
- DHE:
-
Dihydroethidium
- LVFS:
-
Left ventricular fractional shortening
- LVEF:
-
Left ventricular ejection fraction
- AAR:
-
Area at risk
- WT:
-
Wild-type
References
Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. 2020;17:773–89.
Davidson SM, Ferdinandy P, Andreadou I, et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J Am Coll Cardiol. 2019;73:89–99.
Hirschhäuser C, Lissoni A, Görge PM, et al. Connexin 43 phosphorylation by casein kinase 1 is essential for the cardioprotection by ischemic preconditioning. Basic Res Cardiol. 2021;116:21.
Hong DM, Lee EH, Kim HJ, et al. Does remote ischaemic preconditioning with postconditioning improve clinical outcomes of patients undergoing cardiac surgery? Remote Ischaemic Preconditioning with Postconditioning Outcome Trial. Eur Heart J. 2014;35:176–83.
Wei X, Wu B, Zhao J, et al. Myocardial hypertrophic preconditioning attenuates cardiomyocyte hypertrophy and slows progression to heart failure through upregulation of S100A8/A9. Circulation. 2015;131:1506–17.
Skyschally A, Gent S, Amanakis G, et al. Across-species transfer of protection by remote ischemic preconditioning with species-specific myocardial signal transduction by reperfusion injury salvage kinase and survival activating factor enhancement pathways. Circ Res. 2015;117:279–88.
Amanakis G, Kleinbongard P, Heusch G, et al. Attenuation of ST-segment elevation after ischemic conditioning maneuvers reflects cardioprotection online. Basic Res Cardiol. 2019;114:22.
Zarbock A, Schmidt C, Van Aken H, et al. Effect of remote ischemic preconditioning on kidney injury among high-risk patients undergoing cardiac surgery: a randomized clinical trial. JAMA. 2015;313:2133–41.
Chen CH, Ferreira JC, Gross ER, et al. Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev. 2014;94:1–34.
Gross ER, Zambelli VO, Small BA, et al. A personalized medicine approach for Asian Americans with the aldehyde dehydrogenase 2*2 variant. Annu Rev Pharmacol Toxicol. 2015;55:107–27.
Ma LL, Ding ZW, Yin PP, et al. Hypertrophic preconditioning cardioprotection after myocardial ischaemia/reperfusion injury involves ALDH2-dependent metabolism modulation. Redox Biol. 2021;43:101960.
Chen CH, Budas GR, Churchill EN, et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–5.
Ueta CB, Campos JC, Albuquerque RPE, et al. Cardioprotection induced by a brief exposure to acetaldehyde: role of aldehyde dehydrogenase 2. Cardiovasc Res. 2018;114:1006–15.
Zhang ZX, Li H, He JS, et al. Remote ischemic postconditioning alleviates myocardial ischemia/reperfusion injury by up-regulating ALDH2. Eur Rev Med Pharmacol Sci. 2018;22:6475–84.
Yu Y, Jia XJ, Zong QF, et al. Remote ischemic postconditioning protects the heart by upregulating ALDH2 expression levels through the PI3K/Akt signaling pathway. Mol Med Rep. 2014;10:536–42.
Li CY, Ma W, Liu KP, et al. Advances in intervention methods and brain protection mechanisms of in situ and remote ischemic postconditioning. Metab Brain Dis. 2021;36:53–65.
Joseph B, Pandit V, Zangbar B, et al. Secondary brain injury in trauma patients: the effects of remote ischemic conditioning. J Trauma Acute Care Surg. 2015;78:698–703.
Kitagawa K, Kawamoto T, Kunugita N, et al. Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 2000;476:306–11.
Sun X, Gao R, Li W, et al. Alda-1 treatment promotes the therapeutic effect of mitochondrial transplantation for myocardial ischemia-reperfusion injury. Bioact Mater. 2021;6:2058–69.
Gao RF, Li X, Xiang HY, et al. The covalent NLRP3-inflammasome inhibitor Oridonin relieves myocardial infarction induced myocardial fibrosis and cardiac remodeling in mice. Int Immunopharmacol. 2021;90:107133.
Ackers-Johnson M, Li PY, Holmes AP, et al. A simplified, Langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ Res. 2016;119:909–20.
Fan Q, Tao R, Zhang H, et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. 2019;139:663–78.
Si Y, Bao H, Han L, et al. Dexmedetomidine attenuation of renal ischaemia-reperfusion injury requires sirtuin 3 activation. Br J Anaesth. 2018;121:1260–71.
Wang Z, Sun R, Wang G, et al. SIRT3-mediated deacetylation of PRDX3 alleviates mitochondrial oxidative damage and apoptosis induced by intestinal ischemia/reperfusion injury. Redox Biol. 2020;28:101343.
Lu Z, Bourdi M, Li JH, et al. SIRT3-dependent deacetylation exacerbates acetaminophen hepatotoxicity. EMBO Rep. 2011;12:840–6.
Stiermaier T, Jensen JO, Rommel KP, et al. Combined intrahospital remote ischemic perconditioning and postconditioning improves clinical outcome in ST-elevation myocardial infarction. Circ Res. 2019;124:1482–91.
Vaibhav K, Braun M, Khan MB, et al. Remote ischemic post-conditioning promotes hematoma resolution via AMPK-dependent immune regulation. J Exp Med. 2018;215:2636–54.
Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674–99.
Tsibulnikov SY, Maslov LN, Gorbunov AS, et al. A review of humoral factors in remote preconditioning of the heart. J Cardiovasc Pharmacol Ther. 2019;24:403–21.
Hummitzsch L, Zitta K, Fritze L, et al. Effects of remote ischemic preconditioning (RIPC) and chronic remote ischemic preconditioning (cRIPC) on levels of plasma cytokines, cell surface characteristics of monocytes and in-vitro angiogenesis: a pilot study. Basic Res Cardiol. 2021;116:60.
Gao J, Min F, Wang S, et al. Effect of Rho-kinase and autophagy on remote ischemic conditioning-induced cardioprotection in rat myocardial ischemia/reperfusion injury model. Cardiovasc Ther. 2022;2022:6806427.
Yan Z, Du L, Liu Q, et al. Remote limb ischaemic conditioning produces cardioprotection in rats with testicular ischaemia-reperfusion injury. Exp Physiol. 2021;106:2223–34.
Zhao Y, Ding M, Yan F, et al. Inhibition of the JAK2/STAT3 pathway and cell cycle re-entry contribute to the protective effect of remote ischemic pre-conditioning of rat hindlimbs on cerebral ischemia/reperfusion injury. CNS Neurosci Ther. 2022. https://doi.org/10.1111/cns.14023.
Mi L, Zhang N, Wan J, et al. Remote ischemic post-conditioning alleviates ischemia/reperfusion-induced intestinal injury via the ERK signaling pathway-mediated RAGE/HMGB axis. Mol Med Rep. 2021:24.
Ajoolabady A, Aslkhodapasandhokmabad H, Aghanejad A, et al. Mitophagy receptors and mediators: therapeutic targets in the management of cardiovascular ageing. Ageing Res Rev. 2020;62:101129.
Santin Y, Fazal L, Sainte-Marie Y, et al. Mitochondrial 4-HNE derived from MAO-A promotes mitoCa(2+) overload in chronic postischemic cardiac remodeling. Cell Death Differ. 2020;27:1907–23.
Beretta M, Wölkart G, Schernthaner M, et al. Vascular bioactivation of nitroglycerin is catalyzed by cytosolic aldehyde dehydrogenase-2. Circ Res. 2012;110:385–93.
Ma H, Guo R, Yu L, et al. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J. 2011;32:1025–38.
Zhai M, Li B, Duan W, et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res. 2017;63.
Harris PS, Gomez JD, Backos DS, et al. Characterizing sirtuin 3 deacetylase affinity for aldehyde dehydrogenase 2. Chem Res Toxicol. 2017;30:785–93.
Koeppen M, Lee JW, Seo SW, et al. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat Commun. 2018;9:816.
Cai Z, Luo W, Zhan H, et al. Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc Natl Acad Sci U S A. 2013;110:17462–7.
Kalakech H, Tamareille S, Pons S, et al. Role of hypoxia inducible factor-1α in remote limb ischemic preconditioning. J Mol Cell Cardiol. 2013;65:98–104.
Zhao Y, Wang B, Zhang J, et al. ALDH2 (aldehyde dehydrogenase 2) protects against hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2019;39:2303–19.
Laitakari A, Ollonen T, Kietzmann T, et al. Systemic inactivation of hypoxia-inducible factor prolyl 4-hydroxylase 2 in mice protects from alcohol-induced fatty liver disease. Redox Biol. 2019;22:101145.
Ji W, Wei S, Hao P, et al. Aldehyde dehydrogenase 2 has cardioprotective effects on myocardial ischaemia/reperfusion injury via suppressing mitophagy. Front Pharmacol. 2016;7:101.
Kornfeld OS, Hwang S, Disatnik MH, et al. Mitochondrial reactive oxygen species at the heart of the matter: new therapeutic approaches for cardiovascular diseases. Circ Res. 2015;116:1783–99.
Guéraud F. 4-Hydroxynonenal metabolites and adducts in pre-carcinogenic conditions and cancer. Free Radic Biol Med. 2017;111:196–208.
Mak S, Lehotay DC, Yazdanpanah M, et al. Unsaturated aldehydes including 4-OH-nonenal are elevated in patients with congestive heart failure. J Card Fail. 2000;6:108–14.
Eng MY, Luczak SE, Wall TL. ALDH2, ADH1B, and ADH1C genotypes in Asians: a literature review. Alcohol Res Health. 2007;30:22–7.
Yukawa Y, Muto M, Hori K, et al. Combination of ADH1B*2/ALDH2*2 polymorphisms alters acetaldehyde-derived DNA damage in the blood of Japanese alcoholics. Cancer Sci. 2012;103:1651–5.
Li H, Borinskaya S, Yoshimura K, et al. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann Hum Genet. 2009;73:335–45.
Funding
This research was funded by the Jiangxi Provincial Natural Science Foundation Project (20071BBG70067, 20181074), and the National Natural Science Foundation of China (81160019 and 81360031).
Author information
Authors and Affiliations
Contributions
Rifeng Gao, Chunyu Lv, and Yanan Qu designed the research and wrote the paper. Rifeng Gao, Heng Yang, Xiaolei Sun, and Chuangze Hao performed the experiments. Rifeng Gao, Xiaosheng Hu, Yiqing Yang, and Yanhua Tang contributed new reagents/analytic tools and provide critical suggestions. Rifeng Gao, Chunyu Lv, and Yanan Qu analyzed the data. Xiaosheng Hu, Yiqing Yang, and Yanhua Tang edited the paper.
Corresponding authors
Ethics declarations
Consent for Publication
Not applicable.
Consent to Participate
Not applicable.
Animal Studies
All animal experiments were approved by the Animal Care Ethics Committee of Fudan University and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The mice were kept under a 12:12-h light/dark cycle at a consistent temperature and humidity and were also given ad libitum access to food and water. Additional dose of analgesics was given if the animals appeared to be experiencing pain (based on criteria such as immobility and failure to eat). At the indicated time points, mice were euthanized by CO2/cervical dislocation, and tissues were subsequently harvested for analyses.
Conflict of Interest
The authors declare no competing interests.
Additional information
Associate Editor Yihua Bei oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Supplementary Figure 1
RIC treatment preserves cardiac function after I/R. Representative images of echocardiography tracing in I/R mice treated with RIC in each group after 24 h reperfusion (a). Heart rate (BPM, n>6, b). Left ventricular ejection fraction (LVEF, n>6, c). Left ventricular fractional shortening (LVFS, n>6, D). Peak shortening (% cell lengthening, n>6, e). Maximal shortening velocity (-dl/dt, n>6, f). Data are depicted as the mean ± SEM. Statistical significance was determined by two-way ANOVA with a post-hoc Holm-Sidak test, ns, not significant; *P<0.05; ***P<0.001; compared with the control group. (PNG 1135 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Gao, R., Lv, C., Qu, Y. et al. Remote Ischemic Conditioning Mediates Cardio-protection After Myocardial Ischemia/Reperfusion Injury by Reducing 4-HNE Levels and Regulating Autophagy via the ALDH2/SIRT3/HIF1α Signaling Pathway. J. of Cardiovasc. Trans. Res. 17, 169–182 (2024). https://doi.org/10.1007/s12265-023-10355-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-023-10355-z