
Abstract
Among numerous cardiovascular diseases, heart failure is a final and fatal stage, and its morbidity, mortality, and rehospitalization rate remain high, which reduces the exercise tolerance of patients and brings great medical burden and economic pressure to the society. Inflammation takes on a major influence in the occurrence, development, and prognosis of heart failure (HF). The NLRP3 inflammasome is a key node in a chronic inflammatory response, which can accelerate the production of pro-inflammatory cytokines IL-1β and IL-18, leading to the inflammatory response. Therefore, whether it is possible to suppress the downstream factors of NLRP3 inflammasome and its signaling path is expected to provide a new intervention mediator for the therapy of heart failure. This article synopsizes the research progress of NLRP3 inflammasome in heart failure, to provide a reference for clinical treatment.
Clinical Relevance
This study explored the downstream factors of NLRP3 inflammasome and its signal pathway. Targeted drug therapy for NLRP3 inflammasome is expected to provide a new intervention target for the treatment of heart failure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) is a complex clinical condition characterized by abnormal heart structure and/or function, and confirmed by elevated natriuretic peptide levels and/or objective evidence of pulmonary or systemic congestive [1]. The morbidity and mortality of HF will further increase with the aging of the population and the continuous rise of the number of cardiovascular diseases. The latest epidemiological survey data revealed that nearly 6.2 million adults suffer from HF in America and more than 37.7 million individuals worldwide [2, 3]. The prevalence of HF among Chinese patients is 1.3% (about 13.7 million patients), and the total prevalence has increased by 44% in the last 15 years [4]. HF is the fatal and terminal stage of numerous cardiovascular diseases. These patients have a high risk of rehospitalization, poor quality of life, and a heavy economic burden. It is one of the most important chronic cardiovascular diseases of the twenty-first century.
The basic pathological mechanism of the occurrence and development of HF is ventricular remodeling. Inhibiting the excessive activation of neuroendocrine has become the therapeutic basis for delaying and reversing ventricular remodeling. Therefore, the treatment of HF mainly starts by improving symptoms, enhancing the quality of life, improving prognosis, preventing and delaying ventricular remodeling, and reducing the rates of rehospitalization and mortality [5]. At present, the main therapeutic regimes for HF treatment include antagonizing sympathetic and neuroendocrine anti-heart failure drugs, cardiac resynchronization therapy (CRT), implantable cardioverter-defibrillators (ICDs), ventricular assist devices, and heart transplants. Among them, the drug treatment of HF has changed from the original “Golden Triangle”—angiotensin-converting enzyme inhibitor (ACEI)/angiotensin receptor neprilysin inhibitor (ARNI), beta-blockers, and mineralocorticoid receptor antagonists (MRA)—to “New Quadruple”—ACEI/ARNI, beta-blockers, MRA, and sodium-glucose cotransporter 2 inhibitor (SGLT2i). Currently, the treatment of chronic heart failure (CHF) has stepped into a new era of multi-mechanism and multi-target, bringing more benefits to the majority of HF patients [6]. Although remarkable achievements have been made in the pathological mechanism and prevention of HF in recent years, the overall prognosis of HF is poor. A study showed that the 1-year rehospitalization rates of inpatients and stable HF patients were 44% and 32%, respectively, and the mortality of HF patients during hospitalization was still as high as 4.1% [7, 8]. How to seek new therapeutic targets for HF, further reduce the mortality and rehospitalization rate, ameliorate symptoms, and improve the prognosis of patients has become the hotspot of modern medical research and a great challenge for global medicine. As we all know, in addition to the classic neurohumoral mechanism and sympathetic nervous system mechanism, inflammation takes on a significant influence on the development and prognosis of HF. As the key note of the chronic inflammatory response, nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) is the inflammasome with the most definite structure and function at present, which is expected to provide a new intervention mediator for the therapy of HF [9, 10]. This paper synopsizes the research progress of NLRP3 inflammasome in HF, to provide scientific reference for clinical prevention and treatment of HF.
The Importance of Inflammation in Heart Failure
Inflammation is the organism’s first immune defense against harmful stimuli, which occurs when the organism receives exogenous infection and endogenous cell tissue damage [11, 12]. Various inflammatory cytokines released by inflammatory cells induce cardiomyocyte apoptosis, decrease myocardial systolic and diastolic function, further reduce cardiac ejection and filling capacity, cause ventricular remodeling, and then exacerbate the deterioration of HF. Elevated inflammatory biomarkers are a hallmark feature of CHF and can better predict prognosis in patients with HF [13]. Many pro-inflammatory cytokines including tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), interleukin-6 (IL-6), and C-reactive protein (CRP) are elevated in HF patients, and elevated levels of them are strongly associated with severity and poor prognosis of HF [14]. These cytokines can regulate ventricular remodeling, apoptosis, myocardial fibrosis, and other structural changes. TNF-α is a pro-inflammatory cytokine that has been implicated in the pathogenesis of HF. TNF-α can promote myocardial cell protein synthesis, weaken myocardial contractility, and promote cell pyroptosis and myocardial hypertrophy, thereby accelerating the process of HF [15]. A CHF immunomodulation trial improved patient clinical endpoints by reducing TNF-α levels, suggesting that elevated TNF-α levels in HF patients were associated with worse outcomes [16]. Animal studies also showed that the TNF-α was significantly elevated in aortic tissue in the HFpEF pig model [17]. A substantial body of experimental evidence indicates that IL-1 is involved in the pathogenesis of HF and the occurrence of systolic dysfunction [18]. It has been reported that IL-1β can accumulate myocardial collagen, increase reactive oxygen species, and accelerate the process of HF in rats with HF [19]. However, the potential effectiveness of IL-1 targeting in patients with HF has not yet been determined. IL-6 is derived from monocytes, which can increase the production of intracellular superoxide anion (O2 -), reduce the expression of nitric oxide (NO), induce endoplasmic reticulum stress, and cause cardiomyocyte apoptosis. Clinical studies have confirmed that IL-6 promotes cardiomyocyte apoptosis, the expression of IL-6 mRNA in cardiomyocytes in patients with HF increases, and the level of plasma IL-6 increases [20]. CRP is a sensitive marker of the nonspecific inflammatory response, which can bind to receptors on monocytes, macrophages, and neutrophils, and promotes phagocytosis by activating the classical complement pathway [21]. The level of hs CRP in HF patients is higher than that of healthy people, and the level of hs CRP is positively correlated with the severity of inflammation [22].
Furthermore, other inflammatory markers such as nuclear factor κB (NF-κB) [23] and monocyte chemoattractant protein 1 (MCP-1) [24] have been reported. A large amount of evidence above suggests that immune and inflammatory responses are taking part in the pathological process of CHF via inflammatory cytokines.
NLRP3 Inflammasome
As the critical first line of defense against exogenous or endogenous stimuli, the innate immune system can recognize danger signals and initiate immune defense reactions in the hosts. The inflammasome is an intracellular pattern recognition receptor that discriminates various stimuli such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Signals that induce immune and inflammation-related gene expression play a major role in defending against infection and maintaining immune homeostasis [25]. Inflammasomes include nucleotide-binding oligomerization domain-like receptors (NLRs) family and pyrin and HIN domain-containing (PYHIN) family. Among the NLRs family, the NLRP3 inflammasome is the most widely and comprehensively characterized. NLRP3 inflammasome is a significant innate immune sensor that recognizes a variety of unsafe signals, induces inflammatory responses, and participates in the immune response of the body [26].
Structure and Function
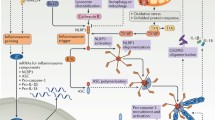
NLRP3 inflammasome is a complex composed of a variety of proteins, mainly composed of receptor protein NLRP3 (a sensor), apoptosis-associated speck-like protein containing a CARD (ASC, an adaptor), and pro-caspase-1 (an effector) [27]. The NLRP3 protein includes three regions; the C-terminal leucine-rich repeat domain (LRR) can recognize and bind PAMPs and DAMPs. The central terminal is the nucleotide-binding oligomerization domain (NOD), responsible for mediating nucleic acid ligation, and protein oligomerization. And an N-terminal effector domain consists of a caspase recruitment domain (CARD) or a pyrin domain (PYD), which is responsible for transmitting the signal downstream. The N-terminal PYD and the C-terminal CARD form the adaptor protein ASC, which connects NLRP3 and caspase-1. Pro-caspase-1 is the precursor molecule of the effector protein caspase-1, which generates active caspase-1 through self-cleavage, which promotes the maturation and secretion of IL-1β and IL-18 [28,29,30].
Activation Mechanism
Normally, NLRP3 inflammasome activation takes two steps: The first is to upregulate the expression of each component and downstream factors. DAMPs or PAMPs activate TLRs located on the cell membrane so that nuclear factor-κB (NF-κB) can upregulate the NLRP3 protein expression at the transcriptional level [31]. The second step is to activate NLRP3 to promote the assembly of multi-protein complexes. The receptor protein recognizes PAMPs or DAMPs through LRR, and through PYD and the PYD site of ASC is connected to form a complex next. Subsequently, ASC recruits pro-caspase-1 through CARD to activate the effector protein [32]. After the effector protein is activated, dependent on its proteolytic hydrolysis, active caspase-1 cleaves into pro-IL-1β and pro-IL-18, promotes the maturation and secretion of downstream IL-1β and IL-18, and initiates the downstream inflammatory response [33, 34]. Additionally, activated caspase-1 cleaves and decomposes GSDMD (gasdermin D), making it into pores in the cell membrane, inducing cell rupture, and triggering pyroptosis [35]. Currently, it is considered that the activation pathways of the NLRP3 inflammasome mainly include the following three: The first is the efflux of potassium ions. Studies have shown that potassium efflux is a trigger for the activation of the NLRP3 inflammasome. Under the stimulation of extracellular adenosine triphosphate (ATP), intracellular potassium ion is activated by the P2X7 receptor and pannexin-1 pore, causing the activation of NLRP3 inflammasome [36]. The second is the generation of reactive oxygen species (ROS). The NLRP3 ligand thioredoxin-interacting protein (TXNIP) is activated in a ROS-sensitive manner upon ROS accumulation. The binding of free TXNIP to NLRP3 protein makes the NLRP3 inflammasome active. However, some studies have shown that ROS is involved in the sensitization process of NLRP3, but not in the activation of NLRP3 [37]. The third is the lysosome damage. After macrophages phagocytose microbial toxins and other substances, the continuous accumulation causes lysosomal swelling, leakage, and release of contents such as cathepsin B, thereby activating the NLRP3 inflammasome. Furthermore, cathepsin B can also exert downstream effects by activating TXNIP [38, 39]. Thus, investigating the activation pathway of the NLRP3 inflammasome will help to further understand the pathogenesis of HF.
The Role of NLRP3 Inflammasome in the Pathogenesis of Heart Failure
Inflammasome has significant implications for chronic inflammation and affects the process of HF. Activation of NLRP3 inflammasome can stimulate the expression and secretion of IL-1β and IL-18, thereby aggravating the occurrence and development of HF [10].
Experimental Research Progress
Experimental studies suggest that compared to wild-type mice, the levels of NLRP3 and pro-caspase-1 in myocardial tissue, and IL-1β in serum are significantly increased in cardiac-specific transgenic heterozygous (CNTg) mice overexpressing phosphatase. Myocardial inflammation and myocardial contractility are significantly improved in NLRP3-knockout mice with HF and IL-1β antagonist intervention in CNTg mice. This experiment demonstrates that the NLRP3 inflammasome in CNTg mice leads to myocarditis and cardiac systolic dysfunction by activating IL-1β, thereby triggering HF [40]. The experiment carried out two mouse models of HF: pressure overload and LAD ligation; hematopoietic or myeloid Tet2 deficiency exacerbates cardiac remodeling and dysfunction in mice while increasing IL-1 expression. Simultaneously, the therapy with NLRP3 inflammasome inhibitor slowed the HF process, and there is no significant difference in cardiac parameters between Tet2-deficient and wild-type mice [41]. The NLRP3 inflammasome is significantly increased in a model of ventricular hypertrophy induced by transverse aortic constriction (TAC) and is associated with inflammatory mediators, pro-fibrotic factors, and cardiac dysfunction [42]. Expression of NLRP3, IL-1β, and IL-18 in myocardial infarction mice with left coronary artery ligation (CAL) is significantly decreased after MCC950 treatment, indicating that NLRP3 inhibitors can reduce myocardial fibrosis and delay the development of HF after myocardial infarction (MI) [43]. Similarly, it has been shown that coronary ligation and doxorubicin were used to establish ischemic and non-ischemic myocardial injury models in mice, and after intervention with NLRP3 inflammasome inhibitors, the results showed that the left ventricular systolic function of the two myocardial injury models was improved, and protected primary and cultured cardiomyocytes from the deleterious effects of inflammasome activation [44]. Cardiomyocyte calmodulin-dependent protein kinase IIδ (CaMKIIδ)-dependent activation of the NLRP3 inflammasome mediates fibrosis during HF, while NLRP3 knockout also reduces macrophage accumulation, which attenuated the inflammatory response and the development of fibrosis (Table 1) [45].
Clinical Research Progress
Clinical studies have found that serum inflammatory factors and chemokines are proportional to the deterioration of cardiac function. Stable coronary artery disease (SCAD) patients with elevated CRP, fibrinogen, and leukocytes are associated with the incidence of HF [46]. A study of 7 patients with HF who received 2-week anakinra (IL-1 antagonist) injections showed that the treatment group significantly increased oxygen consumption, decreased carbon dioxide retention, and improved exercise tolerance, and the serum IL-1β levels decreased by 89% [47]. Comparing the plasma IL-18 levels of 48 CHF patients and 10 healthy people who died unexpectedly, the results described that compared with the normal control group, the plasma IL-18 level in patients with CHF was upwards. After treatment, the IL-18 level with improved symptoms of HF decreased significantly, and the IL-18 level was found to be significantly different in CHF patients with different cardiac functions and increased with the severity of HF [48]. In addition, studies have found that IL-18 levels have no significant correlation with the underlying cause of CHF, but are positively correlated with the severity of CHF [49]. IL-18 may accelerate the progression of HF by promoting myocardial fibrosis [50]. A prospective study of 54 patients with HF showed that patients with HF had a downward trend in ASC methylation levels and IL-1β and ASC mRNA levels trended upward, while the exact opposite results were obtained after exercise. The reason for this result may be that the changes in ASC methylation and expression have a connection to the decline of IL-1β in exercisers [51]. Many studies have presented that targeting the NLRP3 inflammasome and downstream IL-1 and IL-18 therapy has promise as a new direction for the treatment of HF (Table 2).
Therapeutic Strategies for Heart Failure Associated with the NLRP3 Inflammasome
Study shows the NLRP3 inflammasome has been proposed as a potential intervention mediator to treat multiple inflammatory diseases [52]. With further research of its activation mechanism, the development of drugs acting on NLRP3 or related signaling pathways has become a research hotspot of NLRP3 inflammasome inhibitors, and targeting the NLRP3 inflammasome supplies a novel idea for the therapy of HF (Table 3).
NLRP3 Inhibitors
Direct Inhibitors
MCC950
MCC950, a diarylsulfonylurea-containing compound, can not only blockade the in-depth handling of IL-1β by caspase-1 but can also block classical and non-classical NLRP3 inflammasome activation and IL-1β secretion by eliminating ASC oligomerization [53]. MCC950 specifically acts on the NLRP3 inflammasome, but not activation of the NLRP1, NLRC4, or AIM [54]. Recent studies have found that MCC950 acts through direct binding to the Walker B motif of the NLRP3 NACHT domain preventing ASC oligomerization and NLRP3 inflammasome composition [54]. Studies have shown that in a porcine MI model, MCC950 can reduce the level of IL-1β in the myocardium, obviously decline the size of MI, and improve cardiac function [55]. MCC950 effectively improves diabetic kidney injury by inhibiting the NLRP3/caspase-1/IL-1β pathway [56]. MCC950 can delay the progression of HF caused by Tet2 mutation in hematopoietic cells by reducing IL-1β levels [41].
OLT1177
OLT1177 is an active β-sulfonyl nitrile molecule, and its two most defined roles are specific inhibition of the NLRP3 inflammasome and reversal of the metabolic cost of inflammation. OLT1177 blocks classical and non-classical of the NLRP3 inflammasome. It also acts directly with NLRP3 and blockades ATPase activity [57]. Meanwhile, OLT1177 cuts down IL-18 and IL-1β secretion, with no effect on NLRC4 or AIM2 inflammasomes. The study found that OLT1177 preserves β-adrenergic responsiveness and prevents left ventricular diastolic dysfunction in a mouse model of non-reperfused anterior MI [58]. At present, OLT1177 is undergoing clinical trials, and phase I trials of OLT1177 in healthy subjects showed good tolerability and a favorable safety profile. OLT1177 reduces joint pain in patients with acute gouty arthritis [59]. Furthermore, it has longer half-lives and the subjects did not show any toxicity (organ/blood) despite the different doses [60]. In a phase 1B double-blind trial, patients with stable HFrEF showed significant improvement in LVEF and exercise time after 14 days of treatment with OLT1177, which was well tolerated and safe [61]. Therefore, OLT1177 has the potential to be developed into NLRP3-targeting therapeutics against various NLRP3-related diseases.
CY90
CY90, as an analog of CFTR(inh)-172 (C172), inhibits the cystic fibrosis transmembrane conductance regulator (CFTR) channel [62]. Studies have shown that CY90 compounds can directly act on the NLRP3 protein and specifically prevent the composition of the NLRP3 inflammasome and the secretion of IL-1β. CY90 achieves the purpose of blocking ATPase activity by directly binding to the NLRP3 Walker A motif, and it does not affect NOD2/RIG, NLRP1, and NLRC4 [63]. After receiving CY90 treatment in diabetic stroke model mice, the cardiac dysfunction of the model mice was significantly improved [64]. Furthermore, CY90 also has good oral bioavailability, safety, and stability, and the drug development targeting CY90 has great potential. However, there is no direct evidence for the relationship between CY90 and HF, and further investigation of its anti-inflammatory effect as a potential NLRP3 inhibitor for the treatment of HF is warranted.
Other NLRP3 direct inhibitors include tranilast [65], 3,4-methylenedioxy-beta-nitrostyrene (MNS) [66], and oridonin [67]. By directly targeting the NLRP3 inflammasome, they can more accurately prevent and treat NLRP3-related diseases.
Indirect Inhibitors
Glyburide
Glyburide is a sulfonylurea drug mainly used to treat type II diabetes, which blockades ATP-sensitive K-channels (KATP) channels by stimulating insulin release [68]. The study found that glyburide can inhibit the NLRP3 inflammasome, thereby reducing the inflammatory response in fetal mice with acute lung injury induced by hyperoxia exposure [69]. Glyburide prevents the activation of the NLRP3 inflammasome and has a specific inhibitory effect on the NLRP3 inflammasome, and makes no difference to other inflammasomes (NLRC4, AIM2, or NLRP1) [68]. Glyburide can treat cardiovascular abnormalities caused by Cantu syndrome by inhibiting the K ATP channel. However, the relationship between glyburide and HF is not yet clear, and further research is needed [70].
16673-34-0
16673-34-0 is an intermediate formed in the synthesis of glyburide, but it lacks cyclohexylurea, which is associated with insulin release, and the lack of the cyclohexylurea moiety is inessential for the inhibition of NLRP3 inflammasome activity. Consequently, it has a specific inhibitory effect on the NLRP3 inflammasome but has no huge influence on glucose metabolism, and can also hinder the aggregation of the structure by interfering with NLRP3 activation or aggregation with ASC [71]. The pericardial effusion and pericardial thickening in experimental pericarditis model mice were markedly decreased by the effect of 16673-34-0 [72]. 16673-34-0 prevents cardiac insufficiency in obese mice induced by a high-sugar, high-fat diet (Western diet), and dietary treatment with 16673-34-0 prevents systolic and diastolic dysfunction [73].
Caspase-1, IL-1, and IL-18 Inhibitors
Direct blockade of caspase-1, IL-1β, and IL-18 can overlap with inhibition of NLRP3. Nevertheless, they are not exclusive to the NLRP3 inflammasome, so blocking caspase-1, IL-1β, and IL-18 may affect the normal physiological function of the organism.
VX-765 is a highly selective caspase-1 inhibitor combined with a P2Y 12 receptor antagonist for the ischemia-reperfusion rat model which can further reduce the size of myocardial infarction and improve cardiac function in rats [74]. VX-765 helps reduce cognitive impairment and AD severity in mice [75]. However, the exact inhibitory mechanism of HF effects is not fully understood, and further studies are required to fully define its inhibitory potential. The inflammatory response induced by cytokines of the IL-1 family further exacerbates the development of HF. Anakinra was previously approved for the treatment of rheumatoid arthritis [76]. Recent studies have shown that in the acute decompensated heart failure (ADHF) patients, acute inflammatory responses were significantly reduced with anakinra initiated within 24 h of admission compared with placebo, and LVEF improved after 14 days of treatment [77]. In another study hospitalized for ADHF, anakinin was taken at discharge. After 12 weeks, it was observed that the expression of N-terminal pro-B-type natriuretic peptide (NT proBNP) decreased, and cardiopulmonary function and quality of life improved [78]. Canakinumab has anti-inflammatory effects and is beneficial for IL-1β-mediated inflammatory diseases, including acute gouty arthritis [79] and type 2 diabetes [80]. A recently published analysis of the CANTOS trial found, when compared with placebo, participants who responded to canakinumab experienced a significant 38% reduction in HF hospitalization and a combined 32% reduction in HF hospitalization and all-cause mortality after IL-1β blockade [81]. IL-18 can promote myocardial fibrosis and may be involved in the occurrence and development of HF [50]. Compared with controls, the infarct size of the ischemia-reperfusion injury mice pretreated with IL-18 antibody was significantly reduced [82]. Recombinant IL-18 binding protein (IL-18BP) can improve right ventricular function in mice with chronic alveolar hypoxia [83]. IL-18BP prevents left ventricular systolic dysfunction in mice treated with plasma from decompensated HF patients [84]
Innovation and Limitations
In inhibiting inflammatory response, identifying and effectively intervening in new inflammatory pathways are one of the important means to delay the development of HF. Due to many redundant and compensatory reactions in this process, anti-inflammatory drug treatment may lead to the impairment of body defense function or the amplification of the inflammatory process. Therefore, targeting specific inflammatory pathways, such as the NLRP3 inflammasome, can more precisely reduce harmful inflammation in HF and protect host defense barriers. Furthermore, this novel therapy that inhibits the NLRP3 inflammasome for the treatment of HF may be a novel mechanism of investigation for many existing drugs, with high applicability in the clinical environment, and may broaden the therapeutic field of HF. Some limitations should be fully considered before recommending NLRP3 inhibitors for clinical practice. First of all, most data on inflammation are obtained in laboratory acute animal models; how to translate experimental animal data into human medicine is still in the research stage, and HF is accompanied by complex multi-system lesions; gender and age differences, early and late differences in inflammation, and so on should be considered, which is difficult to simulate in the laboratory animal model. Second, we tend to overlook whether the detected inflammatory parameters are the cause of cardiovascular pathology or simple markers, and the reliability of these biomarkers in clinical settings or human clinical trials is also worth considering. In addition, correlation does not reflect causality; and finally, how to avoid the side effects of these NLRP3 inhibitors. In conclusion, in-depth studies should be carried out to investigate the exact role of NLRP3 in animal models and HF patients.
Future Development Directions
In recent years, small molecules have been developed as specific NLRP3 inhibitors and have been confirmed in animal and cell experiments, but are less studied in clinical trials and need to be proven effective and safe in more patients. The future research direction should focus on clinical trials. In addition, the future can also focus on the development of compound derivatives with improved pharmacokinetic characteristics to enhance the specificity of targeting NLRP3 inhibitors. Finally, the complex disease of HF cannot adopt a unified treatment method. It is necessary to master individualized strategies, but also to go beyond the heart, focus on comorbidities, and provide new treatment opportunities for the prevention and management of HF.
Conclusions
After the NLRP3 inflammasome receives stimulatory signals, it induces inflammatory responses by upregulating the expression of inflammatory factors such as IL-1β and IL-18. NLRP3 inflammasome is inseparable from the formation and deterioration of HF. Therefore, by intervening in the downstream molecules of NLRP3 inflammasome and its signal transduction pathway, the activation of NLRP3 inflammasome can be effectively inhibited, providing a new idea for the prophylaxis and cure of HF (Fig. 1). Although the research on inflammation-modulating drugs is still in the preliminary stage, it is believed that with advancing science and research, targeted drug therapy for the NLRP3 inflammasome is expected to provide new ideas and new solutions for the diagnosis and treatment of HF.

The NLRP3 inflammasome plays an important role in the pathogenesis of HF, and inhibiting its downstream factors and signaling pathways is expected to provide a new therapeutic target for the treatment of HF. ASC, apoptosis-associated speck-like protein containing a CARD; CARD, caspase recruitment domain; DAMPs, danger-associated molecular patterns; GSDMD, Gasdermin D; GSDMD-NT, N-terminal fragment of Gasdermin D; IL-1β, interleukin-1β; IL-18, interleukin-18; IL-18BP, interleukin-18 binding protein; LRR, leucine-rich repeat domain; NF-kB, nuclear factor κB; NLRP3, nucleotide-binding oligomerization domain-like receptor protein 3; PAMPs, pathogen-associated molecular patterns; Pro-IL-1β, pro-interleukin 1β; Pro-IL-18, pro-interleukin 18; PYD, pyrin domain; TLR4, toll-like receptor4.
References
Bozkurt, B., Coats, A. J. S., Tsutsui, H., et al. (2021). Universal definition and classification of heart failure: A report of the Heart Failure Society of America, Heart Failure Association of the European Society of Cardiology, Japanese Heart Failure Society and Writing Committee of the Universal Definition of Heart Failure: Endorsed by the Canadian Heart Failure Society, Heart Failure Association of India, Cardiac Society of Australia and New Zealand, and Chinese Heart Failure Association. European Journal of Heart Failure, 23(3), 352–380. https://doi.org/10.1002/ejhf.2115
Virani, S. S., Alonso, A., Benjamin, E. J., et al. (2020). Heart Disease and Stroke Statistics-2020 Update: A report from the American Heart Association. Circulation, 141(9), e139–e596. https://doi.org/10.1161/CIR.0000000000000757
Ziaeian, B., & Fonarow, G. C. (2016). Epidemiology and aetiology of heart failure. Nature Reviews Cardiology, 13(6), 368–378. https://doi.org/10.1002/ehf2.13910
Hao, G., Wang, X., Chen, Z., et al. (2019). Prevalence of heart failure and left ventricular dysfunction in China: The China Hypertension Survey, 2012–2015. European Journal of Heart Failure, 21(11), 1329–1337. https://doi.org/10.1002/ejhf.1629
Wang, H., Liang, Y. C. (2018). 2018 Guidelines for diagnosis and treatment of heart failure in China. Zhong Hua Xin Xue Guan Bing Za Zhi, 46(10), 760-789. [In Chinese] http://rs.yiigle.com/CN112148201810/1071728.htm
McDonagh, T. A., Metra, M., Adamo, M., et al. (2021). 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. European Heart Journal, 42(36), 3599–3726. https://doi.org/10.1016/j.rec.2021.11.023
Maggioni, A. P., Dahlström, U., Filippatos, G., et al. (2013). EURObservational Research Programme: Regional differences and 1-year follow-up results of the Heart Failure Pilot Survey (ESC-HF Pilot). European Journal of Heart Failure, 15(7), 808–817. https://doi.org/10.1093/eurjhf/hft050
Zhang, Y., Zhang, J., Butler, J., et al. (2017). Contemporary epidemiology, management, and outcomes of patients hospitalized for heart failure in China: Results from the China Heart Failure (China-HF) Registry. Journal of Cardiac Failure, 23(12), 868–875. https://doi.org/10.1016/j.cardfail.2017.09.014
Shirazi, L. F., Bissett, J., Romeo, F., et al. (2017). Role of inflammation in heart failure. Current Atherosclerosis Reports, 19(6), 27. https://doi.org/10.1007/s11883-017-0660-3
Butts, B., Gary, R. A., Dunbar, S. B., et al. (2015). The importance of NLRP3 inflammasome in heart failure. Journal of Cardiac Failure, 21(7), 586–593. https://doi.org/10.1016/j.cardfail.2015.04.014
Feehan, K. T., & Gilroy, D. W. (2019). Is resolution the end of inflammation? Trends in Molecular Medicine, 25(3), 198–214. https://doi.org/10.1016/j.molmed.2019.01.006
Ip, W. K., & Medzhitov, R. (2015). Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nature Communications, 6, 6931. https://doi.org/10.1038/ncomms7931
Benz, A. P., Aeschbacher, S., Krisai, P., et al. (2021). Biomarkers of inflammation and risk of hospitalization for heart failure in patients with atrial fibrillation. Journal of the American Heart Association, 10(8), be019168. https://doi.org/10.1161/JAHA.120.019168
Segers, V. F. M., Brutsaert, D. L., & De Keulenaer, G. W. (2018). Cardiac remodeling: Endothelial cells have more to say than just NO. Frontiers in Physiology, 9, 382. https://doi.org/10.3389/fphys.2018.00382
Xu, Y., Chen, S., Cao, Y., et al. (2018). Discovery of novel small molecule TLR4 inhibitors as potent anti-inflammatory agents. European Journal of Medicinal Chemistry, 154, 253–266. https://doi.org/10.1016/j.ejmech.2018.05.033
Dick, S. A., & Epelman, S. (2016). Chronic heart failure and inflammation: What do we really know? Circulation Research, 119(1), 159–176. https://doi.org/10.1161/CIRCRESAHA.116.308030
Zhang, N., Feng, B., Ma, X., et al. (2019). Dapagliflozin improves left ventricular remodeling and aorta sympathetic tone in a pig model of heart failure with preserved ejection fraction. Cardiovascular Diabetology, 18(1), 107. https://doi.org/10.1186/s12933-019-0914-1
Abbate, A., Toldo, S., Marchetti, C., et al. (2020). Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circulation Research, 126(9), 1260–1280. https://doi.org/10.1161/CIRCRESAHA.120.315937
Harouki, N., Nicol, L., Remy-Jouet, I., et al. (2017). The IL-1β antibody gevokizumab limits cardiac remodeling and coronary dysfunction in rats with heart failure. JACC Basic Translational Science, 2(4), 418–430. https://doi.org/10.1016/j.jacbts.2017.06.005
Shao, T., Zhang, Y., Tang, R., et al. (2018). Effects of milrinone on serum IL-6, TNF-α, Cys-C and cardiac functions of patients with chronic heart failure. Experimental and Therapeutic Medicine, 16(5), 4162–4166. https://doi.org/10.3892/etm.2018.6672
Mega, J. L., Stitziel, N. O., Smith, J. G., et al. (2015). Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: An analysis of primary and secondary prevention trials. Lancet, 385(9984), 2264–2271. https://doi.org/10.1016/S0140-6736(14)61730-X
Antunovic, T., Stefanovic, A., Gligorovic Barhanovic, N., et al. (2017). Prooxidant-antioxidant balance, hsTnI and hsCRP: mortality prediction in haemodialysis patients, two-year follow-up. Renal Failure, 39(1), 491–499. https://doi.org/10.1080/0886022X.2017.1323645
Xue, G. L., Li, D. S., Wang, Z. Y., et al. (2021). Interleukin-17 upregulation participates in the pathogenesis of heart failure in mice via NF-κB-dependent suppression of SERCA2a and Cav1.2 expression. Acta Pharmacologica Sinica, 42(11), 1780–1789. https://doi.org/10.1038/s41401-020-00580-6
Suciu-Petrescu, M., Truta, A., Suciu, M. D., et al. (2021). Clinical impact of echocardiography parameters and molecular biomarkers in heart failure: Correlation of ACE2 and MCP-1 polymorphisms with echocardiography parameters: A comparative study. Experimental and Therapeutic Medicine, 22(1), 686. https://doi.org/10.3892/etm.2021.10118
Fenini, G., Contassot, E., & French, L. E. (2017). Potential of IL-1, IL-18 and inflammasome inhibition for the treatment of inflammatory skin diseases. Frontiers in Pharmacology, 8, 278. https://doi.org/10.3389/fphar.2017.00278
Chen, H., Mao, X., Meng, X., et al. (2019). Hydrogen alleviates mitochondrial dysfunction and organ damage via autophagy-mediated NLRP3 inflammasome inactivation in sepsis. International Journal of Molecular Medicine, 44(4), 1309–1324. https://doi.org/10.3892/ijmm.2019.4311
Mangan, M. S. J., Olhava, E. J., Roush, W. R., et al. (2018). Targeting the NLRP3 inflammasome in inflammatory diseases. Nature Reviews. Drug Discovery, 17(8), 588–606. https://doi.org/10.1038/nrd.2018.97
Yang, X., Lin, G., Han, Z., et al. (2019). Structural biology of NOD-like receptors. Advances in Experimental Medicine and Biology, 1172, 119–141. https://doi.org/10.1007/978-981-13-9367-9_6
Swanson, K. V., Deng, M., & Ting, J. P. (2019). The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nature Reviews Immunology, 19(8), 477–489. https://doi.org/10.1038/s41577-019-0165-0
Freeman, T. L., & Swartz, T. H. (2020). Targeting the NLRP3 inflammasome in severe COVID-19. Frontiers in Immunology, 11, 1518. https://doi.org/10.3389/fimmu.2020.01518
Liang, N., Yang, Y. P., Li, W., et al. (2018). Overexpression of NLRP3, NLRC4 and AIM2 inflammasomes and their priming-associated molecules (TLR2, TLR4, Dectin-1, Dectin-2 and NFκB) in Malassezia folliculitis. Mycoses, 61(2), 111–118. https://doi.org/10.1111/myc.12711
Zhou, W., Chen, C., Chen, Z., et al. (2018). NLRP3: A novel mediator in cardiovascular disease. Journal of Immunology Research, 2018, 5702103. https://doi.org/10.1155/2018/5702103
He, Y., Hara, H., & Núñez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends in Biochemical Sciences, 41(12), 1012–1021. https://doi.org/10.1016/j.tibs.2016.09.002
Jo, E. K., Kim, J. K., Shin, D. M., et al. (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cellular & Molecular Immunology, 13(2), 148–159. https://doi.org/10.1038/cmi.2015.95
Karmakar, M., Minns, M., Greenberg, E. N., et al. (2020). N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nature Communications, 11(1), 2212. https://doi.org/10.1038/s41467-020-16043-9
Muñoz-Planillo, R., Kuffa, P., Martínez-Colón, G., et al. (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity, 38(6), 1142–1153. https://doi.org/10.1016/j.immuni.2013.05.016
Tschopp, J., & Schroder, K. (2010). NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nature Reviews Immunology, 10(3), 210–215. https://doi.org/10.1038/nri2725
An, N., Gao, Y., Si, Z., et al. (2019). Regulatory mechanisms of the NLRP3 inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Frontiers in Immunology, 10, 1592. https://doi.org/10.3389/fimmu.2019.01592
Chen, W., Zhao, M., Zhao, S., et al. (2017). Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: A novel inhibitory effect of minocycline. Inflammation Research, 66(2), 157–166. https://doi.org/10.1007/s00011-016-1002-6
Bracey, N. A., Beck, P. L., Muruve, D. A., et al. (2013). The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Experimental Physiology, 98(2), 462–472. https://doi.org/10.1113/expphysiol.2012.068338
Sano, S., Oshima, K., Wang, Y., et al. (2018). Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. Journal of the American College of Cardiology, 71(8), 875–886. https://doi.org/10.1016/j.jacc.2017.12.037
Wang, Y., Wu, Y., Chen, J., et al. (2013). Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology, 126(1), 1–11. https://doi.org/10.1159/000351179
Gao, R., Shi, H., Chang, S., et al. (2019). The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. International Immunopharmacology, 74, 105575. https://doi.org/10.1016/j.intimp.2019.04.022
Marchetti, C., Toldo, S., Chojnacki, J., et al. (2015). Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. Journal of Cardiovascular Pharmacology, 66(1), 1–8. https://doi.org/10.1097/FJC.0000000000000247
Willeford, A., Suetomi, T., Nickle, A., et al. (2018). CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight, 3(12), e97054. https://doi.org/10.1172/jci.insight.97054
Eisen, A., Benderly, M., Behar, S., et al. (2014). Inflammation and future risk of symptomatic heart failure in patients with stable coronary artery disease. American Heart Journal, 167(5), 707–714. https://doi.org/10.1016/j.ahj.2014.01.008
Van Tassell, B. W., Arena, R. A., Toldo, S., et al. (2012). Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS ONE, 7(3), e33438. https://doi.org/10.1371/journal.pone.0033438
Mallat, Z., Heymes, C., Corbaz, A., et al. (2004). Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. The FASEB Journal, 18(14), 1752–1754. https://doi.org/10.1096/fj.04-2426fje
Mao, Y., Xia, C. Y., Chen, X., et al. (2008) Study of the serum levels and roles of cytokines IL-12, IL-18 in patients with chronic heart failure. Lin Chuang Xin Xue Guan Bing Za Zhi, (7), 494–496. [In Chinese]. https://doi.org/10.3969/j.issn.1001-1439.2008.07.005
Bracey, N. A., Gershkovich, B., Chun, J., et al. (2014). Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. Journal of Biological Chemistry, 289(28), 19571–19584. https://doi.org/10.1074/jbc.M114.550624
Butts, B., Butler, J., Dunbar, S. B., et al. (2018). Effects of exercise on ASC methylation and IL-1 cytokines in heart failure. Medicine and Science in Sports and Exercise, 50(9), 1757–1766. https://doi.org/10.1249/MSS.0000000000001641
Xu, C., Lu, Z., Luo, Y., et al. (2018). Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nature Communications, 9(1), 4092. https://doi.org/10.1038/s41467-018-06522-5
Coll, R. C., Robertson, A. A., Chae, J. J., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature Medicine, 21(3), 248–255. https://doi.org/10.1038/nm.3806
Coll, R. C., Hill, J. R., Day, C. J., et al. (2019). MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nature Chemical Biology, 15(6), 556–559. https://doi.org/10.1038/s41589-019-0277-7
van Hout, G. P. J., Bosch, L., Ellenbroek, G. H. J. M., et al. (2017). The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. European Heart Journal, 38(11), 828–836. https://doi.org/10.1093/eurheartj/ehw247
Zhang, C., Zhu, X., Li, L., et al. (2019). A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes Metab Syndr Obes, 12, 1297–1309. https://doi.org/10.2147/DMSO.S199802
Marchetti, C., Swartzwelter, B., Koenders, M. I., et al. (2018). NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Research & Therapy, 20(1), 169. https://doi.org/10.1186/s13075-018-1664-2
Aliaga, J., Bonaventura, A., Mezzaroma, E., et al. (2021). Preservation of contractile reserve and diastolic function by inhibiting the NLRP3 inflammasome with OLT1177® (Dapansutrile) in a mouse model of severe ischemic cardiomyopathy due to non-reperfused anterior wall myocardial infarction. Molecules, 26(12), 3534. https://doi.org/10.3390/molecules26123534
Klück, V., Jansen, T. L. T. A., Janssen, M., et al. (2020). Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol, 2(5), e270–e280. https://doi.org/10.1016/S2665-9913(20)30065-5
Marchetti, C., Swartzwelter, B., Gamboni, F., et al. (2018). OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A, 115(7), E1530–E1539. https://doi.org/10.1073/pnas.1716095115
Wohlford, G. F., Van Tassell, B. W., Billingsley, H. E., et al. (2020). Phase 1B, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor dapansutrile in subjects with NYHA II-III systolic heart failure. Journal of Cardiovascular Pharmacology, 77(1), 49–60. https://doi.org/10.1097/FJC.0000000000000931
Ma, T., Thiagarajah, J. R., Yang, H., et al. (2002). Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. The Journal of Clinical Investigation, 110(11), 1651–1658. https://doi.org/10.1172/JCI16112
Jiang, H., He, H., Chen, Y., et al. (2017). Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. Journal of Experimental Medicine, 214(11), 3219–3238. https://doi.org/10.1084/jem.20171419
Lin, H. B., Wei, G. S., Li, F. X., et al. (2020). Macrophage-NLRP3 inflammasome activation exacerbates cardiac dysfunction after ischemic stroke in a mouse model of diabetes. Neuroscience Bulletin, 36(9), 1035–1045. https://doi.org/10.1007/s12264-020-00544-0
Huang, Y., Jiang, H., Chen, Y., et al. (2018). Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med, 10(4), e8689. https://doi.org/10.15252/emmm.201708689
He, Y., Varadarajan, S., Muñoz-Planillo, R., et al. (2014). 3,4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. Journal of Biological Chemistry, 289(2), 1142–1150. https://doi.org/10.1074/jbc.M113.515080
Zhao, G., Zhang, T., Ma, X., et al. (2017). Oridonin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-induced RAW264.7 cells and acute lung injury. Oncotarget, 8(40), 68153–68164. https://doi.org/10.18632/oncotarget.19249
Lamkanfi, M., Mueller, J. L., Vitari, A. C., et al. (2009). Glyburide inhibits the cryopyrin/Nalp3 inflammasome. Journal of Cell Biology, 187(1), 61–70. https://doi.org/10.1083/jcb.200903124
Liao, J., Kapadia, V. S., Brown, L. S., et al. (2015). The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nature Communications, 6, 8977. https://doi.org/10.1038/ncomms9977
McClenaghan, C., Huang, Y., Yan, Z., et al. (2020). Glibenclamide reverses cardiovascular abnormalities of Cantu syndrome driven by KATP channel overactivity. The Journal of Clinical Investigation, 130(3), 1116–1121. https://doi.org/10.1172/JCI130571
Marchetti, C., Chojnacki, J., Toldo, S., et al. (2014). A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. Journal of Cardiovascular Pharmacology, 63(4), 316–322. https://doi.org/10.1097/FJC.0000000000000053
Mauro, A. G., Bonaventura, A., Vecchié, A., et al. (2021). The role of NLRP3 inflammasome in pericarditis: potential for therapeutic approaches. JACC Basic to Translational Science, 6(2), 137–150. https://doi.org/10.1016/j.jacbts.2020.11.016
Carbone, S., Mauro, A. G., Prestamburgo, A., et al. (2018). An orally available NLRP3 inflammasome inhibitor prevents western diet-induced cardiac dysfunction in mice. Journal of Cardiovascular Pharmacology, 72(6), 303–307. https://doi.org/10.1097/FJC.0000000000000628
Audia, J. P., Yang, X. M., Crockett, E. S., et al. (2018). Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Research in Cardiology, 113(5), 32. https://doi.org/10.1007/s00395-018-0692-z
Flores, J., Noël, A., Foveau, B., et al. (2018). Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nature Communications, 9(1), 3916. https://doi.org/10.1038/s41467-018-06449-x
Ramírez, J., & Cañete, J. D. (2018). Anakinra for the treatment of rheumatoid arthritis: A safety evaluation. Expert Opinion on Drug Safety, 17(7), 727–732. https://doi.org/10.1080/14740338.2018.1486819
Van Tassell, B. W., Abouzaki, N. A., Erdle, C. O., et al. (2016). Interleukin-1 blockade in acute decompensated heart failure: A randomized, double-blinded, placebo-controlled pilot study. Journal of Cardiovascular Pharmacology, 67(6), 544–551. https://doi.org/10.1097/FJC.0000000000000378
Van Tassell, B. W., Canada, J., Carbone, S., et al. (2017). Interleukin-1 blockade in recently decompensated systolic heart failure: results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circulation. Heart Failure, 10(11), e004373. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004373
Jena, M., Tripathy, A., Mishra, A., et al. (2021). Effect of canakinumab on clinical and biochemical parameters in acute gouty arthritis: A meta-analysis. Inflammopharmacology, 29(1), 35–47. https://doi.org/10.1007/s10787-020-00753-z
Everett, B. M., Donath, M. Y., Pradhan, A. D., et al. (2018). Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. Journal of the American College of Cardiology, 71(21), 2392–2401. https://doi.org/10.1016/j.jacc.2018.03.002
Everett, B. M., Cornel, J. H., Lainscak, M., et al. (2019). Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation, 139(10), 1289–1299. https://doi.org/10.1161/CIRCULATIONAHA.118.038010
Venkatachalam, K., Prabhu, S. D., Reddy, V. S., et al. (2009). Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. Journal of Biological Chemistry, 284(12), 7853–7865. https://doi.org/10.1074/jbc.M808824200
Hillestad, V., Espe, E. K., Cero, F., et al. (2015). IL-18 neutralization during alveolar hypoxia improves left ventricular diastolic function in mice. Acta Psychologica, 213(2), 492–504. https://doi.org/10.1111/apha.12376
Toldo, S., Mezzaroma, E., O’Brien, L., et al. (2014). Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. American Journal of Physiology. Heart and Circulatory Physiology, 306(7), H1025–H1031. https://doi.org/10.1152/ajpheart.00795.2013
Acknowledgements
We gratefully acknowledge the Tianjin Municipal Health and Family Planning Industry High-level Talent Selection and Training Project for their funding support for this paper, and thanks to all participants for their contributions to this paper.
Funding
This work was supported by the Tianjin Municipal Health and Family Planning Industry High-level Talent Selection and Training Project.
Author information
Authors and Affiliations
Contributions
Yunjiao Wang and Yanyang Li contributed to the conception of and writing of the manuscript. Wanqin Zhang and Zhuo Yuan contributed significantly to analysis and revision of the manuscript. Shichao Lv and Junping Zhang helped perform the analysis with constructive discussions.
Corresponding authors
Ethics declarations
Ethics Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Research Involving Human Participants and/or Animals
No human/animal studies were carried out by the authors for this article.
Informed Consent
Patient consent for publication is not required.
Consent to Participate
Patient and public involvement and patients and/or the public were not involved in the design, conduct, reporting, or dissemination plans of this research.
Competing Interests
The authors declare no competing interests.
Additional information
Associate Editor Junjie Xiao oversaw the review of this article.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, Y., Li, Y., Zhang, W. et al. NLRP3 Inflammasome: a Novel Insight into Heart Failure. J. of Cardiovasc. Trans. Res. 16, 166–176 (2023). https://doi.org/10.1007/s12265-022-10286-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-022-10286-1