
Abstract
Neurological manifestations of coronavirus disease 2019 (COVID-19) are less noticeable than the respiratory symptoms, but they may be associated with disability and mortality in COVID-19. Even though Omicron caused less severe disease than Delta, the incidence of neurological manifestations is similar. More than 30% of patients experienced “brain fog”, delirium, stroke, and cognitive impairment, and over half of these patients presented abnormal neuroimaging outcomes. In this review, we summarize current advances in the clinical findings of neurological manifestations in COVID-19 patients and compare them with those in patients with influenza infection. We also illustrate the structure and cellular invasion mechanisms of SARS-CoV-2 and describe the pathway for central SARS-CoV-2 invasion. In addition, we discuss direct damage and other pathological conditions caused by SARS-CoV-2, such as an aberrant interferon response, cytokine storm, lymphopenia, and hypercoagulation, to provide treatment ideas. This review may offer new insights into preventing or treating brain damage in COVID-19.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As of Jan 5, 2023, the global pandemic coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has resulted in over 600 million confirmed cases and 6 million deaths (https://covid19.who.int/). The majority of COVID-19 patients initially suffer respiratory symptoms, which can progress to severe substantial pulmonary diseases, such as pneumonia or acute respiratory distress syndrome (ARDS) [1]. With increasing clinical evidence, researchers have discovered that SARS-CoV-2 causes neurological manifestations in ~30% of patients who recovered from COVID-19. The most common neurological manifestations include cognitive impairment, depression, and psychosis [2, 3]. Further investigations have revealed brain pathology in COVID-19 patients with neurological manifestations [4]. Although Omicron causes less severe disease than Delta, it still leads to brain pathology in many patients. However, the mechanisms of brain disease in COVID-19 continue to be a mystery because of the complexity of brain structure and function, the uncertain course of central SARS-CoV-2 invasion, and the diverse responses to the virus. In this review, we summarize the clinical manifestations and brain pathological features of COVID-19 and discuss the mechanisms of brain damage caused by SARS-CoV-2.
Clinical Neurological Manifestations in COVID-19 Patients
Neurological manifestations have been reported in COVID-19 patients of all ages. For instance, in a study conducted in Wuhan, China, more than a third of COVID-19 patients developed new neurological symptoms after being infected, as shown in Table 1 [5]. Another retrospective cohort study that included 236,379 patients also found a similar rate (33.62%) of patients with neurological manifestations [6]. During the acute phase of infection, patients most frequently experience headache, anosmia, and stroke [7], while "brain fog", headache, anxiety, anosmia, ageusia, and cognitive impairment are common post-acute neurological sequelae of SARS-CoV-2 infection [8]. Many patients had anosmia, and further investigations showed that their nasal components [9] and olfactory gyrus [10] had been damaged. A recent study suggested that sensory and neurological disorders are more evident in younger patients, whereas mental health, musculoskeletal, and episodic disorders are more noticeable in older patients [11]. While most of these symptoms gradually disappear within a few months after COVID-19 recovery, more research is required to confirm the duration of these symptoms and subsequent changes.
Compared to the patients infected by influenza, COVID-19 patients with neurological manifestations are older and exhibit a higher incidence of altered mental status, headache, anosmia, dysgeusia, and ischemic stroke [12, 13]. In addition, COVID-19 patients develop symptoms more quickly. As shown in Tables 3 and 5, flu patients may suffer from influenza-associated encephalitis/encephalopathy (IAE) with symptoms such as altered consciousness and seizures. Flu patients may also experience post-influenza encephalitis, Guillain-Barre syndrome (GBS), Reye's syndrome, and Parkinsonian symptoms (PD) [14].
Neuroimaging Findings of COVID-19 Patients
Neuroimaging applies quantitative techniques to visualize the brain's structure and function. As listed in Table 2, medical imaging examinations, particularly magnetic resonance imaging (MRI) and positron emission computed tomography (PET) reveal the brain structure and function of patients with neurological manifestations [15]. The most frequent imaging features (affecting >60% of patients) include ischemic infarcts, intracerebral hemorrhages, perfusion abnormalities, and leptomeningeal enhancement. Hypometabolism in the pons, cerebellum, bilateral insula, bilateral medial lobes, and prefrontal cortex indicates brain function dysregulation in COVID patients. The high incidence of cerebrovascular events such as ischemic stroke and intracerebral hemorrhage, indicates potential endothelial injury and coagulation dysfunction. Other investigations have also reported hypoxic alterations in the cerebellum and cerebrum, metabolic alterations of astrocytes, microglial activation [16], neuro-axonal damage, and neuronal loss [17], which provide additional evidence for brain damage in COVID patients.
As for flu patients, the most prevalent imaging features include brain lesions, edema, rapid and fulminant demyelination, and inflammation. These abnormalities are mainly in the cortex, white matter, or brainstem (Tables 4 and 5). In addition, glial activation, metabolic disorders, and genetic factors may be involved [18, 19].
Variant and Neurological Complications in Patients with COVID-19
After the appearance of the Alpha variant, there was no discernible difference in the prevalence of neurological manifestations in patients with COVID. However, the risk of anxiety disorders, insomnia, cognitive deficit, and ischemic stroke were significantly higher after the appearance of the Delta variant. Patients diagnosed with COVID-19 after the emergence of the Omicron variant have a lower death rate, but their neurological outcomes still carry a similar risk to those with the Alpha variant [20]. Therefore, despite the reduced rate of severe cases of Omicron, we cannot underestimate the high incidence of neurological sequelae. It is important to note that this aspect has not been adequately studied because of the difficulty of differentiating the variants of viruses that infect patients in those published clinical studies.
Patients’ Characteristics and Brain Pathology in COVID-19
Cohort studies on the neurological manifestations of COVID-19 have shown that older age and pre-existing neurological disorders are risk factors for neuropathy and mortality [21]. It is known that aging results in immunosenescence, which is a progressive weakening of the ability to mount efficient immune responses against infections [22]. In addition, older people are more likely to have pre-existing neurodegenerative diseases, including Alzheimer’s disease (AD) and PD [23]. Previous studies have suggested that patients with AD are vulnerable to experiencing severe conditions and passing away during COVID-19 [24]. It is worth noticing that the apolipoprotein E4 (APOE4) allele, which is strongly associated with AD, is also a risk factor for severe COVID-19 [25]. Neuropathy in COVID-19 may be exacerbated by APOE4 because it can increase fibrinogenesis, blood-brain barrier (BBB) permeability, and cerebral amyloid angiopathy in the brain [26]. In contrast to female patients, aged male patients have fewer activated and differentiated T cells, as well as poorer CD8+ T cell activation and IFNγ production [27]. However, the incidence of COVID-19 sequelae does not differ significantly by gender or exhibits a modest overrepresentation of female patients [28]. Patients with pre-existing diseases, such as hypertension, diabetes, and cardiac diseases, are at a higher risk of infection and brain pathology [29]. Pre-existing pathological conditions, such as arteriosclerosis, contribute to acute ischemia, stroke, dementia, and other neurological manifestations [30]. Moreover, drugs used to treat pre-existing diseases must also be considered for their potential impact on the incidence and severity of infection. One study has suggested that patients treated with drugs that increase angiotensin-converting enzyme 2 (ACE2) expression may be more vulnerable to severe COVID-19 infection [31]. Therefore, patient characteristics, including age, pre-existing conditions, and medication use, should be carefully evaluated when making clinical decisions.
Evidence for Central Invasion of SARS-CoV-2
In several post-mortem studies, small amounts of SARS-CoV-2 have been detected in the brains of patients, but in almost all cases [60], the patients had at least one site in the brain with low but positive amounts of SARS-CoV-2 RNA, with the cerebellum being the most frequently affected [61]. The astrocyte has been identified to be the preferred target for SARS-CoV-2 infection and replication. However, SARS-CoV-2 is difficult to detect in microglia, although the activation of microglia is prominent in the brain of severe COVID patients and is similar to the state of human neurodegenerative disease [62]. In addition, SARS-CoV-2 has also been detected in brain capillary endothelial cells [63]. Notably, SARS-CoV-2 has been found to infect and kill neurons in human brain organoids [64]. A recent study identified virus-specific protein expression in the hypothalamus and spinal ganglia neurons by immunofluorescence. Furthermore, this study demonstrated that SARS-CoV-2 RNA persists in the brain for months, even after the virus has been eliminated from the plasma [65]. However, additional research is required to confirm the virus's capacity for replication in the brain. It should be emphasized that the existing studies are insufficient to demonstrate direct viral invasion of neurons or detect low virus levels in the brain. Currently, no conclusive information about the brain invasion of SARS-CoV-2 is available in human post-mortem studies. Besides, unlike animal studies with a good perfusion process before sampling, the SARS-CoV-2 detected in the human brain might originate in the blood. Thus, it is more likely that the virus indirectly causes damage to the brain by triggering a series of responses. The importance (even the existence) of direct viral brain invasion of SARS-CoV-2 still needs further study.
Virus Structure and Cellular Invasion Mechanisms
SARS-CoV-2, known as the main culprit of this global pandemic, possesses single-stranded positive-sense RNA (+ssRNA), four structural proteins (N, E, M, and S), and sixteen non-structural proteins (nsp1−16) [66, 67]. The spike (S) protein, which consists of S1 and S2 subunits, plays a prominent role in viral invasion and is regarded as a crucial target for antiviral treatment [68, 69] (Fig. 1). In addition, the nucleocapsid (N), envelope (E), and membrane (M) proteins are all involved in viral production and combine with host cell organelles to cause various dysregulation of physiological processes [70, 71]. ACE2 is a significant receptor for SARS-CoV-2 entering host cells [72]. After binding to ACE2 via the S1 unit, SARS-CoV-2 can initiate membrane fusion depending on a furin-like cleavage site on the S1/S2 and S2 units [73]. The S2 unit is cleaved by the endosomal proteases cathepsin B and L (CatB/L) [74], or the transmembrane protease serine 2 (TMPRSS2) [75], to form a fusion pore, which allows the viral genome to enter cells [76]. The replication and transcription complex discontinuously processes the viral RNA, mainly formed by nsp12 cooperating with nsp7 and nsp8 [77]. Viral transcription, replication, and translation depend on host cell organelles, including the endoplasmic reticulum and Golgi [78]. It is worth noting that the S protein of Omicron has a higher ACE2 binding affinity than the S protein of other variants, which might explain the higher infection of Omicron [79]. In addition, since different receptors are highly expressed on different types of cells, it is important to consider the impact of additional receptors, such as neuropilin-1, in bringing about pathological changes.

The structure of SARS-CoV-2. SARS-CoV-2 consists of 4 structural proteins (S, M, E, and N) and 16 non-structural proteins (nsp1−16). After the S1 component binds to ACE2, the S1/S2 cleavage primes the fusion and creates extra cell surface receptors, like NRP1. Then, SARS-CoV-2 starts entry by CatB/L and TMPRSS2. The viral RNA is released after the entry and translated into corresponding viral structures. The open reading frame 1a (ORF1a) and ORF1b of RNA are translated into polyprotein 1a (pp1a) and pp1ab, further being translated into nsp1−11 and nsp12−16. Host cell organelles, such as the Golgi, are involved in virus replication. SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; ACE2: angiotensin-converting enzyme 2; NRP-1: neuropilin-1; TMPRSS2: transmembrane protease serine 2.
As SARS-CoV-2 has been shown to cause damage and dysregulation in other systems [80], its detection in the brain by polymerase chain reaction [81] and immunohistochemistry [82] suggests that it may also harm brain cells and tissues. Typically, the virus causes damage through two mechanisms: (1) replication in cells to produce daughter viruses and breaking the cell to release more viruses to invade other cells [83]; (2) the body’s response against the virus inevitably results in cell destruction. Thus, SARS-CoV-2 might also enter the brain and directly or indirectly damage brain cells. In direct damage, while the daughter virus is released by secretory vesicles and causes light damage to cell structure, interference by viral protein in the cell cycle can result in severe cell damage and death [84]. The nonstructural proteins (nsps) of SARS-CoV-2 disturb host DNA replication, protein synthesis, and transport [85], leading to some pathological brain changes. Besides, the virus can directly kill cells by inducing apoptosis, necroptosis, and autophagy in infected cells [86].
Possible Brain Entry Mechanisms of SARS-CoV-2
Via the BBB
The BBB typically serves as a barrier and a transporter. It transports ions, macromolecules, nutrients, and toxins into or out of the brain [87]. The BBB is mostly composed of vascular endothelial cells, pericytes, and astrocytes. The tight junctions (Tjs) and adhesion junctions between endothelial cells act as significant parts of the barrier [88]. Pericytes, embedded in the basement membrane of microvessels, regulate BBB formation and permeability and serve in CNS immune surveillance, such as the leukocyte migration across endothelial cells [89]. Astrocytes exhibit a strong link with BBB formation and dominantly enhance the frequency, length, and complexity of Tjs [90, 91].
As remarked above, ACE2 is the main receptor of SARS-CoV-2 and has been detected in several tissues, especially in arterial and venous endothelial cells [92]. In the human brain, the substantial nigra, paraventricular nuclei of the thalamus, raphe nuclei, tuberomammillary nucleus, central glial substance (an area of gray matter surrounding the central canal), and choroid plexus (ChP) of the lateral ventricle may have high ACE2 expression [93]. SARS-CoV-2 might infect brain microvascular endothelial cells through ACE2 and replicate in them [94]. However, entry through endothelial cells may be limited, because numerous studies have not yet demonstrated that infection and replication can occur there effectively. Besides, the basement membrane disruption of the BBB induced by SARS-CoV-2 through matrix metalloproteinase-9 is considered a significant factor for virus entry [95]. Indeed, BBB disruption and leakage have also been reported in COVID-19 patients with neurological manifestations [96].
The SRAS-CoV-2 may also use host cells to convey traffic across the BBB [97]. The virus is believed to preferentially invade lipid-secreting cells and use lipid metabolism for replication and spread [98]. A study has also shown replication of SRAS-CoV-2 in infected monocytes and macrophages [99]. Since the immune cells can enter the brain through the BBB and the ChP, SARS-CoV-2 may get a ride and get out after brain entry [100, 101]. However, it is not yet known whether viral replication in monocytes produces infectious viruses [102]. Further research is needed to investigate this mechanism
Via the CSF
As some cases have reported the presence of SARS-CoV-2 in cerebrospinal fluid (CSF), it is worth discussing the possibility of the CSF route [103]. The blood-cerebrospinal fluid barrier (BCSFB) is one of the selectively permeable barriers which regulates the transportation of substances between the blood and CSF. The BCSFB is formed by epithelial cells of the ChP, which separate the fenestrated capillaries from the CSF [104] (Fig. 2A and B). Similar to the BBB, The Tjs between ChP endothelial cells function as the barrier [105]. The ChP also secretes pro-inflammatory cytokines and provides space for transporting immune cells into the stroma [106]. ACE2 and its co-receptors TMPRSS2 and TMPRSS4 are highly expressed in the ChP, through which SARS-CoV-2 destroys the structure and function of the BCSFB in organoids [107, 108]. The circumventricular organs (CVOs) are located in the third and fourth ventricles and have continually fenestrated and highly permeable vessels. ACE2 has been detected at a high level in CVOs, which provide a facility for virus entry [109, 110]. However, due to the limited detection of SARS-CoV-2 in CSF, the efficiency of virus entry through this route must be further studied.

The CSF and retrograde nerve transmission pathways for virus entry. A, B The CSF pathway. SARS-CoV-2 traverses the permeable vessels of the choroid plexus (ChP) to reach the stroma. In the stroma, it binds to ACE2 and TMPRSS2/4 to enter endothelial cells and then be released into the CSF. C The retrograde nerve transmission pathway. The virus invades the olfactory sensory nerves (OSNs), moves retrogradely along nerves, and finally enters the neurons of the CNS. Besides, the virus can invade nervous terminal neurons that contact Bowman’s gland through cathepsin B/L (CatB/L). Tj, tight junction; ACE2, angiotensin-converting enzyme 2; TMPRSS2, transmembrane protease serine 2; OB, olfactory bulb.
Via Retrograde Nerve Transmission
Olfactory dysfunction has been frequently reported in COVID-19 patients [111] and may be correlated with the invasion of olfactory cells by viruses (leading to the downregulation of odor detection pathways) [112]. Olfactory mucosa is in contact with droplets by ciliated cells and perceives them through the olfactory sensory nerves (OSNs) in the basal cells, which are supported and nourished by sustentacular cells. Signals are then transferred to the olfactory bulb (OB) where they reach the mitral cells. The virus might invade the peripheral nerves, move retrogradely along them, and finally enter the CNS [113]. Given that many viruses enter the brain in this way, it might also be a possible route for SARS-CoV-2 to invade the brain [114] (Fig. 2C). Meinhardt et al. analyzed regional mapping of the consecutive olfactory nervous tracts and defined CNS regions in 33 individuals, suggesting that neuroinvasion can occur by transmucosal entry via regional nervous structures [115]. Besides, the invasion of OSNs by SARS-CoV and SARS-CoV-2 has also been revealed in hamsters [116]. However, many other studies conversely showed a low possibility of the OB route in humans. Based on 88 post-mortem cases of COVID-19 patients, Khan et al. found that SARS-CoV-2 infects ciliated cells, sustentacular cells, and even leptomeningeal layers surrounding the OB, while the OSNs and OB are uninfected [117]. Another post-mortem research also obtained the same result and suggested that inflammation might cause axon and vascular injury [118]. Butowt et al. pointed out that a lack of quantitative analysis and false positives might explain the contradictory reports on OSN infection [119]. In addition to the OSNs, Bowman's gland, a branching tubular gland in the olfactory mucosa, has been found to show both CatB/L and furin expression [120]. Thus, the nerve terminals of neurons might receive SARS-CoV-2 from the infected Bowman's gland. Overall, retrograde nerve transmission at least is a theoretically possible means of SARS-CoV-2 invasion but needs further determination. Besides, retrograde nerve transmission is also being considered in the infection of the eye, intestine, and lung through the vagus nerve [121] or optic nerve [122].
Other Mechanisms for Brain Pathology in COVID-19
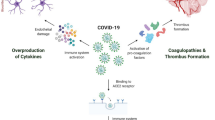
Apart from the direct damage caused by the cellular invasion, several pathological conditions, such as an aberrant IFN response, cytokine storm, lymphopenia, and hypercoagulation in the brain, may result in more severe and widespread damage to the brain. In this section, we elucidate the periphery-related potential mechanisms of central pathological conditions (Fig. 3).

Pathological conditions in the brain. SARS-CoV-2 may cause several pathological conditions, including an aberrant IFN response, cytokine storm, lymphopenia, and hypercoagulation, resulting in pathological brain changes. NETs, neutrophil extracellular traps.
Aberrant IFN Response
The innate immune system plays a significant role in defending against infection and determining the disease severity [123]. Dysregulation of type I and II IFN is the main cause of the insufficient innate immune response. In the early stages of infection, a large amount of type I IFN may be present in the patient’s body to fight the virus, and the ChP is the most vulnerable area to infection in the brain. A study by Suzzi et al. has demonstrated that the increased IFN response may disrupt the ChP and be involved in the process of brain damage [124]. After a peak of type I IFN in the early stage of infection, a delayed aberrant IFN response has been reported in most severe cases [125]. For example, in the later period of COVID-19, a decrease of IFN occurs in the CSF of severe patients with neurological manifestations, and this may be related to the deficient innate immune response [126]. SARS-CoV-2 interferes the IFN production and activation in two ways: (1) the nsp1 of SARS-CoV-2 inhibits the gene expression and translation of IFN, and (2) pattern-recognition receptor (PRR)-mediated signaling pathways are suppressed to downregulate the IFN signaling. PRRs, including membrane-bound C-type lectin receptors, NOD-like receptors (NLRs), retinoic acid-inducible gene I (RIG-I)-like receptors, and Toll-like receptors (TLRs), are commonly expressed on sensory neurons, astrocytes, and microglia [127, 128]. SARS-CoV-2 RNA and M proteins can bind to RIG-I and melanoma differentiation-associated gene 5, blocking the activation of interferon regulatory factor 3 and the type I IFN signaling [129]. In addition, it has been demonstrated that several SARS-CoV-2 components, including nsp6, nsp13, and open reading frame 3a, inhibit the type I IFN response in vitro [130]. As the balance of the innate immune response is closely related to the disease severity of COVID-19 patients with neurological manifestations, the potential strategy to rectify the IFN response is worth discussing.
Cytokine Storm
The cytokine storm is a dangerous sharp increase of cytokines in body fluid that is also a concern in COVID-19 patients, especially in severe cases. As inflammation has been considered to be involved in several CNS diseases such as AD and PD, the elevated cytokines induced by SARS-CoV-2, including IL-6, TNF-α, IL-1, and IL-10 [131], might also be related to the pathological brain changes in COVID-19. It is worth mentioning that the increase of cytokines not only occurs in the acute phase but also may persist in patients for 3 months after recovery from COVID-19. Low-grade chronic inflammation may be associated with brain injury and vascular injury-related stroke in patients [132].
The generation of inflammation in the brain may be closely relevant to the brain barriers. SRAS-CoV-2 can infect components of barriers such as endothelial cells and astrocytes to process pro-inflammatory cytokines [133] and influence BBB permeability [134]. The neurofilament light chain protein and glial fibrillary acidic protein are at high levels in both the plasma serum and CSF of COVID-19 patients, indicating that astrocytic and neuronal injury indeed occurs in these patients [135]. Notably, C-C motif chemokine ligand 11, which can specifically cause hippocampal microglial reactivity and impaired neurogenesis, has been found to be elevated in CSF only in COVID-19 patients with cognitive syndrome [136]. In addition to elevated CSF cytokines [137], the choroid-to-cortex network across inflammatory pathways also increases in COVID-19 patients. This indicates that the inflammatory signals are sent from the choroid plexus into the brain more effectively to activate the glia. Microglia, the innate immune cells in the CNS, respond to the virus and form clusters with infiltrating T cells, which are associated with the brain and perivascular inflammation [138]. Elevated complements like C1q and C3, combined with microglia, are linked to the synaptic loss and toxicity of soluble β-amyloid (Aβ) oligomers [139]. Moreover, the mast cells, as innate immune cells participating in the adaptive immune response, can be activated by SARS-CoV-2 and release pro-inflammatory cytokines, including IL-1β, C-C motif chemokine ligand 2, IL-6, granulocyte-macrophage colony-stimulating factor, and TNF-α, which makes them significant components in neuroinflammation [140, 141].
SARS-CoV-2 may bind to receptors on host cells, including ACE2 and PRRs, to promote inflammation. ACE2 plays a key part in the renin-angiotensin system (RAS) [142]. As SARS-CoV-2 interacts with ACE2 and influences its catalytic activity, it can further regulate the RAS and contribute to the cytokine storm [143]. Besides, the reduction of ACE2 leads to an increase of serum Ang II, which activates the production of reactive oxygen species (ROS) and nuclear factor-kappa B pathways. These activations accordingly promote the expression of pro-inflammatory cytokines and inactive NOD-like receptor family pyrin domain containing 3 (NLRP3) [144, 145]. Excessive ROS can trigger apoptosis through the mitochondria, which causes morphological and functional changes in the host cell [146]. ROS also leads to the overactivation of the immune response and other pathological reactions [147]. NLRP3 inflammasomes, activated by ROS production and mitochondrial dysfunction, show a high correlation with apoptosis, dysregulation of tau protein phosphorylation, and neurofibrillary tangles [148]. Uncontrolled activation of NLRP3 has also been found in endothelial progenitor cells of COVID-19 patients [149, 150].
As discussed above, PRRs, including TLRs, NLRs, and RIG-I, participate in the signaling pathways of IFN. PRRs in the CNS, especially TLRs, are also involved in the production and release of several cytokines including IL-1, IL-6, and TNF-α [151]. The S protein of SARS-CoV-2 can bind to TLR2 and TLR4 and activate the myeloid differentiation factor 88 signaling pathway to promote the cytokine storm [152]. Besides, TLR4-mediated inflammatory cytokines are reported to be upregulated in peripheral blood mononuclear cells and CSF [153].
Lymphopenia
Lymphopenia has been found in severe COVID-19 patients, and several factors may account for this phenomenon [154, 155]. Firstly, ACE2 has remarkable expression in lymphocyte cells, through which the SARS-CoV-2 can directly destroy them. SRAS-CoV-2 RNA has also been detected in macrophages, neutrophils, B cells, T cells, and natural killer cells [156]. Secondly, the elevated cytokines, especially TNF and IFNγ, can contribute to apoptosis and pyroptosis, which leads to the death of lymphocytes and atrophy of the spleen [157, 158]. The systemic reduction of lymphocytes also results in lymphopenia in the brain [60], which directly decreases the body’s defense against viruses and leads to pathological changes. Several studies have suggested that changes in immune cells might induce neural and neuroendocrine dysregulation, such as anxiety [159], depression [160], and neurodegenerative diseases [161]. Meanwhile, these psychiatric sequelae have been reported in COVID-19 patients with an aberrant immune system and include significant post-traumatic stress disorder [162]. In addition, lymphopenia is also considered a hallmark of poor prognosis in COVID-19 patients, further highlighting the importance of lymphopenia [163, 164].
Hypercoagulation
Hypercoagulation-related neurological manifestations, such as ischemic stroke, have been reported in COVID-19 patients [165]. In addition, risk factors of coagulation are also considerably elevated in COVID-19 patients, including D-dimer, von Willebrand Factor (vWF), vWF antigen, and FVIII [1, 166]. Both white and red thrombi have been observed in microvascular, macrovascular, and venous systems [167]. Affected individuals may experience neuropsychiatric diseases, neurocognitive (dementia-like) syndrome, and altered mental status as a result of the thrombi in the cerebrovascular, system; they obstruct the bloodstream and kill endothelial cells [21]. The term "coagulation cascade" refers to the sequence of events that leads to the formation of clots during the coagulation process.
The coagulation process includes the extrinsic, intrinsic, and complement pathways. (1) The extrinsic pathway starts with tissue factor, which is expressed on neutrophils and monocytes and can be activated by cytokines and neutrophil extracellular traps (NETs) [168]. NETs consist of cytosolic and granule proteins and mitochondrial DNA. Triggered by ROS against infection, NETs regulate the activation of the extrinsic pathway [169]. An elevated number of neutrophils and myeloperoxidase /DNA complexes, a well-defined marker of NETosis, indicates a high incidence of NET formation in COVID-19 patients [170]. (2) The intrinsic pathway begins with factor 12 and ultimately participates in fibrin formation. The intrinsic pathway is triggered by an endothelial injury, which can be caused by infection with SRAS-CoV-2. Factor 8 and vWF, a carrier of factor 8, and fibrinogen are highly expressed in COVID-19 patients [171]. In addition, patients with COVID-19 have remarkably elevated levels of the plasminogen activator inhibitor-1 (PAI-1), which downregulates the plasminogen activation to plasmin by inhibiting tissue-type plasminogen activator (tPA). Contradictory opinions have been generated by the fact that some other COVID-19 patients have elevated tPA. That is, the PAI-1 could counteract the local tPA effect to create a net prothrombotic hypofibrinolytic state, while the fibrinolysis led by tPA is predominant [172, 173]. Furthermore, despite the decrease or maintenance of platelet numbers in COVID-19 patients, the plasma thrombopoietin levels, platelet surface P-selectin expression (a marker of platelet activation), and circulating platelet-leukocyte aggregations are prominently high. These results may partly apply to the hypercoagulation state in COVID-19 patients and also suggest that the JAK3-MAPK pathway is involved in the process [174, 175]. The platelet-to-lymphocyte ratio level is significantly higher in severe patients than in non-severe patients with COVID-19 and shows a correlation with the cytokine storm [176]. The D-dimer, a protein produced when clots dissolve, is elevated in COVID-19 patients, which also suggests a serious hypercoagulation syndrome [177]. (3) The complement pathway is a cascade of events that leads to hemostasis. It has been found that the N protein of SARS-CoV-2 can cause abnormal complement activation [178]. Indeed, complements such as C5, C6, C5a, and C8 also increase in severe COVID-19 patients [179]. The complement dysregulation might impel thrombosis and endothelial injury, ultimately leading to brain damage. In summary, the “coagulation cascade” might be started by SARS-CoV-2 and result in a pathological hypercoagulation state, which induces endothelial injury and subsequently causes pathological brain changes.
Other Potential Mechanisms
Several studies have detected anti-neuronal and anti-glial autoantibodies in the serum or CSF of COVID-19 patients with neurological symptoms, suggesting that autoantibodies may be involved in the brain injury of COVID-19 [180, 181]. Autoantibodies may directly damage neurons and can also lead to immune abnormalities. In addition, Franke et al. found that autoantibodies have undetermined antigenic epitopes, which may be related to the molecular mimicry of the SARS-CoV-2 virus [182].
Hypoxia due to pulmonary fibrosis can cause multi-organ damage, which is more prominent in patients with severe disease [183]. Hypoxia in COVID-19 may be correlated with the expression of ACE2 and TMPRSS2 as well as Aβ deposits in the neocortex [184, 185]. Zilberman-Itskovich et al. found a lower incidence of neurological manifestations in COVID-19 patients subjected to hyperbaric oxygen therapy, suggesting that improving hypoxia may reduce neurological pathology by improving brain perfusion and neuroplasticity [186].
Conclusion
COVID-19 is still a problem that affects the entire world and interferes with people's daily lives. Approximately 30% of COVID-19 patients experience new neurological manifestations or exhibit pathological brain changes. However, the current treatments have hitherto been insufficient. In this review, we emphasize the need for clinical practice to consider patient features, such as pre-existing diseases, when treating COVID-19 patients with neurological symptoms. As for the mechanisms, in this review, we indicate that SARS-CoV-2 can infect host cells through ACE2 (accompanied by TMPRSS2 or CatB/L) and may further enter the brain through the BBB, CSF, and retrograde nerve transmission. SARS-CoV-2 may cause cell and tissue damage once it has entered the brain by preventing replication and promoting apoptosis. Apart from these direct effects, several systemic or peripheral pathological conditions, including an aberrant IFN response, cytokine storm, and hypercoagulation, may also contribute to brain pathology. The aberrant IFN response can result in a delayed innate immune response, rendering the viral defense ineffective. The cytokine storm produces excessive tissue damage and cell death, even in the brain, despite its original function as a defense mechanism against the virus. Besides, severe COVID-19 patients exhibit remarkable lymphopenia, indicating a high risk of neurological pathology. Hypercoagulation may be related to endothelial injuries, infarcts, and hemorrhages in the brain. Thus, in addition to host receptors (such as ACE2 and TMPRSS2) and virus structures (such as the S protein and nsps) as potential targets, we suggest that protecting the brain barrier, regulating immune responses, and ameliorating the hypercoagulation state via specific targets also potentially prevent or suggest treatment strategies for pathological brain changes in patients with COVID-19.
References
Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395: 1054–1062.
Gupta A, Madhavan MV, Sehgal K, Nair N, Mahajan S, Sehrawat TS, et al. Extrapulmonary manifestations of COVID-19. Nat Med 2020, 26: 1017–1032.
Spudich S, Nath A. Nervous system consequences of COVID-19. Science 2022, 375: 267–269.
Chougar L, Shor N, Weiss N, Galanaud D, Leclercq D, Mathon B, et al. Retrospective observational study of brain MRI findings in patients with acute SARS-CoV-2 infection and neurologic manifestations. Radiology 2020, 297: E313–E323.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan. China. JAMA Neurol 2020, 77: 683–690.
Taquet M, Geddes JR, Husain M, Luciano S, Harrison PJ. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: A retrospective cohort study using electronic health records. Lancet Psychiatry 2021, 8: 416–427.
Balcom EF, Nath A, Power C. Acute and chronic neurological disorders in COVID-19: Potential mechanisms of disease. Brain 2021, 144: 3576–3588.
Moghimi N, Di Napoli M, Biller J, Siegler JE, Shekhar R, McCullough LD, et al. The neurological manifestations of post-acute sequelae of SARS-CoV-2 infection. Curr Neurol Neurosci Rep 2021, 21: 44.
Niesen M, Trotta N, Noel A, Coolen T, Fayad G, Leurkin-Sterk G, et al. Structural and metabolic brain abnormalities in COVID-19 patients with sudden loss of smell. Eur J Nucl Med Mol Imaging 2021, 48: 1890–1901.
Morand A, Campion JY, Lepine A, Bosdure E, Luciani L, Cammilleri S, et al. Similar patterns of[18F]-FDG brain PET hypometabolism in paediatric and adult patients with long COVID: A paediatric case series. Eur J Nucl Med Mol Imaging 2022, 49: 913–920.
Xu E, Xie Y, Al-Aly Z. Long-term neurologic outcomes of COVID-19. Nat Med 2022, 28: 2406–2415.
Zayet S, Gendrin V, Royer PY, Toko L, Kadiane-Oussou NJ, Klopfenstein T. Comparison of COVID-19 and influenza in adult patients: Neurological symptoms input. J Med Virol 2021, 93: 1893–1895.
Zarifkar P, Peinkhofer C, Benros ME, Kondziella D. Frequency of neurological diseases after COVID-19, influenza A/B and bacterial pneumonia. Front Neurol 2022, 13: 904796.
Iosifescu AL, Hoogenboom WS, Buczek AJ, Fleysher R, Duong TQ. New-onset and persistent neurological and psychiatric sequelae of COVID-19 compared to influenza: A retrospective cohort study in a large New York City healthcare network. Int J Methods Psychiatr Res 2022, 31: e1914.
Liu X, Yan W, Lu T, Han Y, Lu L. Longitudinal abnormalities in brain structure in COVID-19 patients. Neurosci Bull 2022, 38: 1608–1612.
Crunfli F, Carregari VC, Veras FP, Silva LS, Nogueira MH, Antunes ASLM, et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc Natl Acad Sci U S A 2022, 119: e2200960119.
Zingaropoli MA, Iannetta M, Piermatteo L, Pasculli P, Latronico T, Mazzuti L, et al. Neuro-axonal damage and alteration of blood-brain barrier integrity in COVID-19 patients. Cells 2022, 11: 2480.
Li SG, Liang H, Chen YW, Pang YS. Death in children with influenza A (H3N2) virus infection-associated encephalopathy: Two case reports. J Int Med Res 2023, 51: 3000605221149879.
Sellers SA, Hagan RS, Hayden FG, Fischer WA 2nd. The hidden burden of influenza: A review of the extra-pulmonary complications of influenza infection. Influenza Other Respir Viruses 2017, 11: 372–393.
Taquet M, Sillett R, Zhu L, Mendel J, Camplisson I, Dercon Q, et al. Neurological and psychiatric risk trajectories after SARS-CoV-2 infection: An analysis of 2-year retrospective cohort studies including 1 284 437 patients. Lancet Psychiatry 2022, 9: 815–827.
Varatharaj A, Thomas N, Ellul MA, Davies NWS, Pollak TA, Tenorio EL, et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: A UK-wide surveillance study. Lancet Psychiatry 2020, 7: 875–882.
Santoro A, Bientinesi E, Monti D. Immunosenescence and inflammaging in the aging process: Age-related diseases or longevity? Ageing Res Rev 2021, 71: 101422.
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet 2011, 377: 1019–1031.
Xiong N, Schiller MR, Li J, Chen X, Lin Z. Severe COVID-19 in alzheimer’s disease: APOE4’s fault again? Alzheimers Res Ther 2021, 13: 111.
Kuo CL, Pilling LC, Atkins JL, Masoli JAH, Delgado J, Kuchel GA, et al. APOE e4 genotype predicts severe COVID-19 in the UK biobank community cohort. J Gerontol A Biol Sci Med Sci 2020, 75: 2231–2232.
Hultman K, Strickland S, Norris EH. The APOE ɛ4/ɛ4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J Cereb Blood Flow Metab 2013, 33: 1251–1258.
Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, Klein J, et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature 2020, 588: 315–320.
Ortona E, Malorni W. Long COVID: To investigate immunological mechanisms and sex/gender related aspects as fundamental steps for tailored therapy. Eur Respir J 2022, 59: 2102245.
Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, et al. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int J Infect Dis 2020, 94: 91–95.
Qureshi AI, Baskett WI, Huang W, Shyu D, Myers D, Raju M, et al. Acute ischemic stroke and COVID-19: An analysis of 27 676 patients. Stroke 2021, 52: 905–912.
Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med 2020, 8: e21.
Ruz-Caracuel I, Pian-Arias H, Corral Í, Carretero-Barrio I, Bueno-Sacristán D, Pérez-Mies B, et al. Neuropathological findings in fatal COVID-19 and their associated neurological clinical manifestations. Pathology 2022, 54: 738–745.
Cho SM, White N, Premraj L, Battaglini D, Fanning J, Suen J, et al. Neurological manifestations of COVID-19 in adults and children. Brain 2023, 146: 1648–1661.
Pun BT, Badenes R, La Calle GH, Orun OM, Chen W, Raman R, et al. Prevalence and risk factors for delirium in critically ill patients with COVID-19 (COVID-D): A multicentre cohort study. Lancet Respir Med 2021, 9: 239–250.
Frontera JA, Sabadia S, Lalchan R, Fang T, Flusty B, Millar-Vernetti P, et al. A prospective study of neurologic disorders in hospitalized patients with COVID-19 in New York city. Neurology 2021, 96: e575–e586.
Vanichkachorn G, Newcomb R, Cowl CT, Murad MH, Breeher L, Miller S, et al. Post-COVID-19 syndrome (long haul syndrome): Description of a multidisciplinary clinic at mayo clinic and characteristics of the initial patient cohort. Mayo Clin Proc 2021, 96: 1782–1791.
Meppiel E, Peiffer-Smadja N, Maury A, Bekri I, Delorme C, Desestret V, et al. Neurologic manifestations associated with COVID-19: A multicentre registry. Clin Microbiol Infect 2021, 27: 458–466.
Méndez R, Balanzá-Martínez V, Luperdi SC, Estrada I, Latorre A, González-Jiménez P, et al. Short-term neuropsychiatric outcomes and quality of life in COVID-19 survivors. J Intern Med 2021, 290: 621–631.
Garcez FB, Aliberti MJR, Poco PCE, Hiratsuka M, de F Takahashi S, Coelho VA, et al. Delirium and adverse outcomes in hospitalized patients withCOVID-19. J Am Geriatr Soc 2020, 68: 2440–2446.
Forget MF, Del Degan S, Leblanc J, Tannous R, Desjardins M, Durand M, et al. Delirium and inflammation in older adults hospitalized for COVID-19: A cohort study. Clin Interv Aging 2021, 16: 1223–1230.
Ray STJ, Abdel-Mannan O, Sa M, Fuller C, Wood GK, Pysden K, et al. Neurological manifestations of SARS-CoV-2 infection in hospitalised children and adolescents in the UK: A prospective national cohort study. Lancet Child Adolesc Health 2021, 5: 631–641.
Kremer S, Lersy F, de Sèze J, Ferré JC, Maamar A, Carsin-Nicol B, et al. Brain MRI findings in severe COVID-19: A retrospective observational study. Radiology 2020, 297: E242–E251.
Lambrecq V, Hanin A, Munoz-Musat E, Chougar L, Gassama S, Delorme C, et al. Association of clinical, biological, and brain magnetic resonance imaging findings with electroencephalographic findings for patients with COVID-19. JAMA Netw Open 2021, 4: e211489.
Helms J, Kremer S, Merdji H, Schenck M, Severac F, Clere-Jehl R, et al. Delirium and encephalopathy in severe COVID-19: A cohort analysis of ICU patients. Crit Care 2020, 24: 1–11.
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 2020, 382: 2268–2270.
Lu Y, Li X, Geng D, Mei N, Wu PY, Huang CC, et al. Cerebral micro-structural changes in COVID-19 patients-an MRI-based 3-month follow-up study. EClinicalMedicine 2020, 25: 100484.
Kas A, Soret M, Pyatigoskaya N, Habert MO, Hesters A, Le Guennec L, et al. The cerebral network of COVID-19-related encephalopathy: A longitudinal voxel-based 18F-FDG-PET study. Eur J Nucl Med Mol Imaging 2021, 48: 2543–2557.
Okuno H, Yahata Y, Tanaka-Taya K, Arai S, Satoh H, Morino S, et al. Characteristics and outcomes of influenza-associated encephalopathy cases among children and adults in Japan, 2010–2015. Clin Infect Dis 2018, 66: 1831–1837.
Mastrolia MV, Rubino C, Resti M, Trapani S, Galli L. Characteristics and outcome of influenza-associated encephalopathy/encephalitis among children in a tertiary pediatric hospital in Italy, 2017–2019. BMC Infect Dis 2019, 19: 1012.
Muhammad IH, Teh CM, Lee YL. Neurologic manifestations and complications of pandemic influenza A H1N1 in Malaysian children: What have we learnt from the ordeal? Brain Dev 2015, 37: 120–129.
Jantarabenjakul W, Paprad T, Paprad T, Anugulruengkitt S, Pancharoen C, Puthanakit T, et al. Neurological complications associated with influenza in hospitalized children. Influenza Other Respir Viruses 2023, 17: e13075.
Zeng H, Quinet S, Huang W, Gan Y, Han C, He Y, et al. Clinical and MRI features of neurological complications after influenza A (H1N1) infection in critically ill children. Pediatr Radiol 2013, 43: 1182–1189.
Thabet FI, Khalil S, Naz F, Dyme IZ. Cerebellar mutism and reversible cytotoxic edema in influenza B-associated encephalopathy. Pediatr Neurol 2013, 49: 489–492.
Britton PN, Dale RC, Blyth CC, MacArtney K, Crawford NW, Marshall H, et al. Influenza-associated encephalitis/encephalopathy identified by the Australian childhood encephalitis study 2013–2015. Pediatr Infect Dis J 2017, 36: 1021–1026.
Dadak M, Pul R, Lanfermann H, Hartmann H, Hehr U, Donnerstag F, et al. Varying patterns of CNS imaging in influenza A encephalopathy in childhood. Clin Neuroradiol 2020, 30: 243–249.
Li XF, Ai B, Ye JW, Tan LM, Yang HM, Fang CX, et al. Clinical characteristics of H1N1 influenza A-associated mild encephalopathy with reversible splenial lesion: 4 pediatric cases. Curr Med Sci 2021, 41: 815–820.
Meijer WJ, Linn FHH, Wensing AMJ, Leavis HL, van Riel D, GeurtsvanKessel CH, et al. Acute influenza virus-associated encephalitis and encephalopathy in adults: A challenging diagnosis. JMM Case Rep 2016, 3: e005076.
Shinya K, Makino A, Hatta M, Watanabe S, Kim JH, Hatta Y, et al. Subclinical brain injury caused by H5N1 influenza virus infection. J Virol 2011, 85: 5202–5207.
Wang GF, Li W, Li K. Acute encephalopathy and encephalitis caused by influenza virus infection. Curr Opin Neurol 2010, 23: 305–311.
Poloni TE, Medici V, Moretti M, Visonà SD, Cirrincione A, Carlos AF, et al. COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol 2021, 31: e12997.
Thakur KT, Miller EH, Glendinning MD, Al-Dalahmah O, Banu MA, Boehme AK, et al. COVID-19 neuropathology at Columbia university Irving medical center/new york Presbyterian hospital. Brain 2021, 144: 2696–2708.
Yang AC, Kern F, Losada PM, Agam MR, Maat CA, Schmartz GP, et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595: 565–571.
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, et al. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol 2020, 92: 699–702.
Song E, Zhang C, Israelow B, Lu-Culligan A, Prado AV, Skriabine S, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med 2021, 218: e20202135.
Stein SR, Ramelli SC, Grazioli A, Chung JY, Singh M, Yinda CK, et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612: 758–763.
Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep 2020, 19: 100682.
Watanabe C, Okiyama Y, Tanaka S, Fukuzawa K, Honma T. Molecular recognition of SARS-CoV-2 spike glycoprotein: Quantum chemical hot spot and epitope analyses. Chem Sci 2021, 12: 4722–4739.
Caruso IP, Dos Santos AV, do Amaral MJ, de Andrade GC, de Araújo GR, de Stelling AT, et al. Insights into the specificity for the interaction of the promiscuous SARS-CoV-2 nucleocapsid protein N-terminal domain with deoxyribonucleic acids. Int J Biol Macromol 2022; 203: 466–480.
Barnes CO, Jette CA, Abernathy ME, Dam KMA, Esswein SR, Gristick HB, et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588: 682–687.
Venkatagopalan P, Daskalova SM, Lopez LA, Dolezal KA, Hogue BG. Coronavirus envelope (E) protein remains at the site of assembly. Virology 2015, 478: 75–85.
Neuman BW, Kiss G, Kunding AH, Bhella D, Baksh MF, Connelly S, et al. A structural analysis of M protein in coronavirus assembly and morphology. J Struct Biol 2011, 174: 11–22.
Chen C, Boorla VS, Banerjee D, Chowdhury R, Cavener VS, Nissly RH, et al. Computational prediction of the effect of amino acid changes on the binding affinity between SARS-CoV-2 spike RBD and human ACE2. Proc Natl Acad Sci U S A 2021, 118.
Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol Cell 2020, 78: 779-784.e5.
Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci U S A 2005, 102: 11876–11881.
Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol 2019, 93: e01815–e01818.
Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol 2022, 23: 3–20.
Yan L, Ge J, Zheng L, Zhang Y, Gao Y, Wang T, et al. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell 2021, 184: 184-193.e10.
Wang Y, Gandy S. The Golgi apparatus: Site for convergence of COVID-19 brain fog and Alzheimer’s disease? Mol Neurodegener 2022, 17: 67.
Meng B, Abdullahi A, Ferreira IATM, Goonawardane N, Saito A, Kimura I, et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603: 706–714.
Berlin DA, Gulick RM, Martinez FJ. Severe covid-19. N Engl J Med 2020, 383: 2451–2460.
Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med 2020, 383: 590–592.
Matschke J, Lütgehetmann M, Hagel C, Sperhake JP, Schröder AS, Edler C, et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol 2020, 19: 919–929.
Danthi P. Viruses and the diversity of cell death. Annu Rev Virol 2016, 3: 533–553.
Lapointe CP, Grosely R, Johnson AG, Wang J, Fernández IS, Puglisi JD. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc Natl Acad Sci U S A 2021, 118: e2017715118.
Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS-CoV-2 disrupts splicing, translation, and protein trafficking to suppress host defenses. Cell 2020, 183: 1325-1339.e21.
Yapasert R, Khaw-On P, Banjerdpongchai R. Coronavirus infection-associated cell death signaling and potential therapeutic targets. Molecules 2021, 26: 7459.
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis 2010, 37: 13–25.
Berndt P, Winkler L, Cording J, Breitkreuz-Korff O, Rex A, Dithmer S, et al. Tight junction proteins at the blood-brain barrier: Far more than claudin-5. Cell Mol Life Sci 2019, 76: 1987–2002.
Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468: 557–561.
Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325: 253–257.
Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat Neurosci 2016, 19: 771–783.
Hamming I, Timens W, Bulthuis MC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004, 203: 631–637.
Chen R, Wang K, Yu J, Howard D, French L, Chen Z, et al. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front Neurol 2021, 11: 573095.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395: 1417–1418.
Zhang L, Zhou L, Bao L, Liu J, Zhu H, Lv Q, et al. SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther 2021, 6: 337.
Bellon M, Schweblin C, Lambeng N, Cherpillod P, Vazquez J, Lalive PH, et al. Cerebrospinal fluid features in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) reverse transcription polymerase chain reaction (RT-PCR) positive patients. Clin Infect Dis 2021, 73: e3102–e3105.
Hallmann R, Zhang X, Di Russo J, Li L, Song J, Hannocks MJ, et al. The regulation of immune cell trafficking by the extracellular matrix. Curr Opin Cell Biol 2015, 36: 54–61.
Lam SM, Huang X, Shui G. Neurological aspects of SARS-CoV-2 infection: Lipoproteins and exosomes as Trojan horses. Trends Endocrinol Metab 2022, 33: 554–568.
Percivalle E, Sammartino JC, Cassaniti I, Arbustini E, Urtis M, Smirnova A, et al. Macrophages and monocytes: Trojan horses in COVID-19. Viruses 2021, 13: 2178.
Wolburg H, Wolburg-Buchholz K, Engelhardt B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol 2005, 109: 181–190.
Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol 2017, 18: 123–131.
Junqueira C, Crespo Â, Ranjbar S, de Lacerda LB, Lewandrowski M, Ingber J, et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 2022, 606: 576–584.
Huang YH, Jiang D, Huang JT. SARS-CoV-2 detected in cerebrospinal fluid by PCR in a case of COVID-19 encephalitis. Brain Behav Immun 2020, 87: 149.
Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol 2018, 135: 337–361.
Lun MP, Monuki ES, Lehtinen MK. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat Rev Neurosci 2015, 16: 445–457.
Schwerk C, Tenenbaum T, Kim KS, Schroten H. The choroid plexus-a multi-role player during infectious diseases of the CNS. Front Cell Neurosci 2015, 9: 80.
Piras M, Cau F, Manchia M, Paribello P, Saba L, Suri JS, et al. Strong ACE-2 expression in the choroidal vessels: Do high choroid plexuses serve as a gateway for SARS-CoV-2 infection on the human brain? Eur Rev Med Pharmacol Sci 2022, 26: 3025–3029.
Pellegrini L, Albecka A, Mallery DL, Kellner MJ, Paul D, Carter AP, et al. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell 2020, 27: 951-961.e5.
Ong WY, Satish RL, Herr DR. ACE2, circumventricular organs and the hypothalamus, and COVID-19. Neuromolecular Med 2022, 24: 363–373.
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 2007, 292: R373–R381.
Subramanian A, Nirantharakumar K, Hughes S, Myles P, Williams T, Gokhale KM, et al. Symptoms and risk factors for long COVID in non-hospitalized adults. Nat Med 2022, 28: 1706–1714.
Zazhytska M, Kodra A, Hoagland DA, Frere J, Fullard JF, Shayya H, et al. Non-cell-autonomous disruption of nuclear architecture as a potential cause of COVID-19-induced anosmia. Cell 2022, 185: 1052-1064.e12.
Dubé M, Le Coupanec A, Wong AHM, Rini JM, Desforges M, Talbot PJ. Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J Virol 2018, 92: e00404–e00418.
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J Virol 2008, 82: 7264–7275.
Meinhardt J, Radke J, Dittmayer C, Franz J, Thomas C, Mothes R, et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci 2021, 24: 168–175.
Sia SF, Yan LM, Chin AWH, Fung K, Choy KT, Wong AYL, et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020, 583: 834–838.
Khan M, Yoo SJ, Clijsters M, Backaert W, Vanstapel A, Speleman K, et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell 2021, 184: 5932-5949.e15.
Ho CY, Salimian M, Hegert J, O’Brien J, Choi SG, Ames H, et al. Postmortem assessment of olfactory tissue degeneration and microvasculopathy in patients with COVID-19. JAMA Neurol 2022, 79: 544–553.
Butowt R, Meunier N, Bryche B, Bartheld CS. The olfactory nerve is not a likely route to brain infection in COVID-19: A critical review of data from humans and animal models. Acta Neuropathol 2021, 141: 809–822.
Bilinska K, von Bartheld CS, Butowt R. Expression of the ACE2 virus entry protein in the nervus Terminalis reveals the potential for an alternative route to brain infection in COVID-19. Front Cell Neurosci 2021, 15: 674123.
Rangon CM, Krantic S, Moyse E, Fougère B. The vagal autonomic pathway of COVID-19 at the crossroad of alzheimer’s disease and aging: A review of knowledge. J Alzheimers Dis Rep 2020, 4: 537–551.
Chen Y, Yang W, Chen F, Cui L. COVID-19 and cognitive impairment: Neuroinvasive and blood-brain barrier dysfunction. J Neuroinflammation 2022, 19: 222.
van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of genetic variants among young men with severe COVID-19. JAMA 2020, 324: 663–673.
Suzzi S, Tsitsou-Kampeli A, Schwartz M. The type I interferon antiviral response in the choroid plexus and the cognitive risk in COVID-19. Nat Immunol 2023, 24: 220–224.
Ramasamy S, Subbian S. Critical determinants of cytokine storm and type I interferon response in COVID-19 pathogenesis. Clin Microbiol Rev 2021, 34: e00299–e00220.
Heming M, Li X, Räuber S, Mausberg AK, Börsch AL, Hartlehnert M, et al. Neurological manifestations of COVID-19 feature T cell exhaustion and dedifferentiated monocytes in cerebrospinal fluid. Immunity 2021, 54: 164-175.e6.
Kumar V. Toll-like receptors in the pathogenesis of neuroinflammation. J Neuroimmunol 2019, 332: 16–30.
Loo YM, Gale MJ. Immune signaling by RIG-I-like receptors. Immunity 2011, 34: 680–692.
Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther 2020, 5: 299.
Xia H, Cao Z, Xie X, Zhang X, Chen JYC, Wang H, et al. Evasion of type I interferon by SARS-CoV-2. Cell Rep 2020, 33: 108234.
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395: 1033–1034.
Zhou M, Yin Z, Xu J, Wang S, Liao T, Wang K, et al. Inflammatory profiles and clinical features of coronavirus 2019 survivors 3 months after discharge in Wuhan, China. J Infect Dis 2021, 224: 1473–1488.
Chen Z, Li G. Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun 2021, 27: 109–117.
Daniels BP, Jujjavarapu H, Durrant DM, Williams JL, Green RR, White JP, et al. Regional astrocyte IFN signaling restricts pathogenesis during neurotropic viral infection. J Clin Invest 2017, 127: 843–856.
Paterson RW, Benjamin LA, Mehta PR, Brown RL, Athauda D, Ashton NJ, et al. Serum and cerebrospinal fluid biomarker profiles in acute SARS-CoV-2-associated neurological syndromes. Brain Commun 2021, 3: fcab099.
Fernández-Castañeda A, Lu P, Geraghty AC, Song E, Lee MH, Wood J, et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell 2022, 185: 2452-2468.e16.
Guasp M, Muñoz-Sánchez G, Martínez-Hernández E, Santana D, Carbayo Á, Naranjo L, et al. CSF biomarkers in COVID-19 associated encephalopathy and encephalitis predict long-term outcome. Front Immunol 2022, 13: 866153.
Schwabenland M, Salié H, Tanevski J, Killmer S, Lago MS, Schlaak AE, et al. Deep spatial profiling of human COVID-19 brains reveals neuroinflammation with distinct microanatomical microglia-T-cell interactions. Immunity 2021, 54: 1594-1610.e11.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352: 712–716.
Elieh Ali Komi D, Wöhrl S, Bielory L. Mast cell biology at molecular level: A comprehensive review. Clin Rev Allergy Immunol 2020, 58: 342–365.
Kempuraj D, Selvakumar GP, Ahmed ME, Raikwar SP, Thangavel R, Khan A, et al. COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist 2020, 26: 402–414.
Horio N, Clermont AC, Abiko A, Abiko T, Shoelson BD, -E Bursell S, et al. Angiotensin AT1 receptor antagonism normalizes retinal blood flow and acetylcholine-induced vasodiliation in normotensive diabetic rats. Diabetologia 2004, 47: 113–123.
Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med 2020, 76: 14–20.
Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: Celebrating the 20th anniversary of the discovery of ACE2. Circ Res 2020, 126: 1456–1474.
Dai Q, Xu M, Yao M, Sun B. Angiotensin AT1 receptor antagonists exert anti-inflammatory effects in spontaneously hypertensive rats. Br J Pharmacol 2007, 152: 1042–1048.
Cornillez-Ty CT, Liao L, Yates JR 3rd, Kuhn P, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J Virol 2009, 83: 10314–10318.
Saleh J, Peyssonnaux C, Singh KK, Edeas M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54: 1–7.
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575: 669–673.
Ratajczak MZ, Bujko K, Ciechanowicz A, Sielatycka K, Cymer M, Marlicz W, et al. SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45- precursors of hematopoietic and endothelial cells and in response to virus spike protein activates the Nlrp3 inflammasome. Stem Cell Rev Rep 2021, 17: 266–277.
Potere N, Del Buono MG, Caricchio R, Cremer PC, Vecchié A, Porreca E, et al. Interleukin-1 and the NLRP3 inflammasome in COVID-19: Pathogenetic and therapeutic implications. EBioMedicine 2022, 85: 104299.
Bsibsi M, Persoon-Deen C, Verwer RWH, Meeuwsen S, Ravid R, Van Noort JM. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006, 53: 688–695.
Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10: e68563.
Sohn KM, Lee SG, Kim HJ, Cheon S, Jeong H, Lee J, et al. COVID-19 patients upregulate toll-like receptor 4-mediated inflammatory signaling that mimics bacterial Sepsis. J Korean Med Sci 2020, 35: e343.
Wiersinga WJ, Rhodes A, Cheng AC, Peacock SJ, Prescott HC. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): A review. JAMA 2020, 324: 782–793.
Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C. Cytokine storm in COVID-19: The Current evidence and treatment strategies. Front Immunol 2020, 11: 1708.
Ren X, Wen W, Fan X, Hou W, Su B, Cai P, et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell 2021, 184: 1895-1913.e19.
Chan JFW, Zhang AJ, Yuan S, Poon VKM, Chan CCS, Lee ACY, et al. Simulation of the clinical and pathological manifestations of coronavirus disease 2019 (COVID-19) in a golden Syrian Hamster model: Implications for disease pathogenesis and transmissibility. Clin Infect Dis 2020, 71: 2428–2446.
Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological findings and complications of COVID-19. Am J Hematol 2020, 95: 834–847.
Alves de Lima K, Rustenhoven J, Da Mesquita S, Wall M, Salvador AF, Smirnov I, et al. Meningeal γδ T cells regulate anxiety-like behavior via IL-17a signaling in neurons. Nat Immunol 2020, 21: 1421–1429.
Bekhbat M, Treadway MT, Goldsmith DR, Woolwine BJ, Haroon E, Miller AH, et al. Gene signatures in peripheral blood immune cells related to insulin resistance and low tyrosine metabolism define a sub-type of depression with high CRP and anhedonia. Brain Behav Immun 2020, 88: 161–165.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 2018, 19: 610–621.
Mazza MG, De Lorenzo R, Conte C, Poletti S, Vai B, Bollettini I, et al. Anxiety and depression in COVID-19 survivors: Role of inflammatory and clinical predictors. Brain Behav Immun 2020, 89: 594–600.
Goubet AG, Dubuisson A, Geraud A, Danlos FX, Terrisse S, Silva CAC, et al. Prolonged SARS-CoV-2 RNA virus shedding and lymphopenia are hallmarks of COVID-19 in cancer patients with poor prognosis. Cell Death Differ 2021, 28: 3297–3315.
Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang YQ, et al. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal Transduct Target Ther 2020, 5: 33.
Nasrallah MP, Mourelatos Z, Lee EB. Neuropathology associated with SARS-CoV-2 infection. Lancet 2021, 397: 277.
Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med 2020, 46: 1089–1098.
Wu C, Chen X, Cai Y, Xia JA, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med 2020, 180: 934–943.
Iba T, Miki T, Hashiguchi N, Tabe Y, Nagaoka I. Is the neutrophil a ‘prima donna’ in the procoagulant process during sepsis? Crit Care 2014, 18: 230.
Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 2018, 18: 134–147.
Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest 2020, 130: 6151–6157.
Chan NC, Weitz JI. COVID-19 coagulopathy, thrombosis, and bleeding. Blood 2020, 136: 381–383.
Nougier C, Benoit R, Simon M, Desmurs-Clavel H, Marcotte G, Argaud L, et al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J Thromb Haemost 2020, 18: 2215–2219.
Zuo Y, Warnock M, Harbaugh A, Yalavarthi S, Gockman K, Zuo M, et al. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci Rep 2021, 11: 1580.
Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136: 1317–1329.
Bonaventura A, Vecchié A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol 2021, 21: 319–329.
Qu R, Ling Y, Zhang YHZ, Wei LY, Chen X, Li XM, et al. Platelet-to-lymphocyte ratio is associated with prognosis in patients with coronavirus disease-19. J Med Virol 2020, 92: 1533–1541.
Elberts SJ, Bateman R, Koutsoubis A, London KS, White JL, Fields JM. The impact of COVID-19 on the sensitivity of D-dimer for pulmonary embolism. Acad Emerg Med 2021, 28: 1142–1149.
Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol 2021, 17: 46–64.
Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell 2020, 182: 59-72.e15.
Lavi Y, Vojdani A, Halpert G, Sharif K, Ostrinski Y, Zyskind I, et al. Dysregulated levels of circulating autoantibodies against neuronal and nervous system autoantigens in COVID-19 patients. Diagnostics (Basel) 2023, 13: 687.
Bartley CM, Johns C, Ngo TT, Dandekar R, Loudermilk RL, Alvarenga BD, et al. Anti-SARS-CoV-2 and autoantibody profiles in the cerebrospinal fluid of 3 teenaged patients with COVID-19 and subacute neuropsychiatric symptoms. JAMA Neurol 2021, 78: 1503–1509.
Franke C, Ferse C, Kreye J, Reincke SM, Sanchez-Sendin E, Rocco A, et al. High frequency of cerebrospinal fluid autoantibodies in COVID-19 patients with neurological symptoms. Brain Behav Immun 2021, 93: 415–419.
Ordookhanian C, Amidon RF, Kaloostian SW, Vartanian T, Kaloostian P. COVID-19-induced hypoxia with accompanying Syncope event and traumatic injury. Cureus 2021, 13: e14602.
Imperio GE, Lye P, Mughis H, Hamada H, Bloise E, Lye SJ, et al. Hypoxia alters the expression of ACE2 and TMPRSS2 SARS-CoV-2 cell entry mediators in hCMEC/D3 brain endothelial cells. Microvasc Res 2021, 138: 104232.
Priemer DS, Rhodes CH, Karlovich E, Perl DP, Goldman JE. Aβ deposits in the neocortex of adult and infant hypoxic brains, including in cases of COVID-19. J Neuropathol Exp Neurol 2022, 81: 988–995.
Zilberman-Itskovich S, Catalogna M, Sasson E, Elman-Shina K, Hadanny A, Lang E, et al. Hyperbaric oxygen therapy improves neurocognitive functions and symptoms of post-COVID condition: randomized controlled trial. Sci Rep 2022, 12: 11252. https://doi.org/10.1038/s41598-022-15565-0.
Acknowledgements
We disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This review was supported by the National Natural Science Foundation of China (82174005) and The Natural Science Foundation of Zhejiang Province (LY22H280007 and LEZ20H190001).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Zhong Chen is an Editorial Board member of Neuroscience Bulletin and a co-author of this article. To minimize bias, he was excluded from all editorial decision-making related to the acceptance of this article for publication.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, Z., Wang, H., Jiang, S. et al. Brain Pathology in COVID-19: Clinical Manifestations and Potential Mechanisms. Neurosci. Bull. 40, 383–400 (2024). https://doi.org/10.1007/s12264-023-01110-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-023-01110-0