
Abstract
Hv1 is the only voltage-gated proton-selective channel in mammalian cells. It contains a conserved voltage-sensor domain, shared by a large class of voltage-gated ion channels, but lacks a pore domain. Its primary role is to extrude protons from the cytoplasm upon pH reduction and membrane depolarization. The best-known function of Hv1 is the regulation of cytosolic pH and the nicotinamide adenine dinucleotide phosphate oxidase-dependent production of reactive oxygen species. Accumulating evidence indicates that Hv1 is expressed in nervous systems, in addition to immune cells and others. Here, we summarize the molecular properties, distribution, and physiological functions of Hv1 in the peripheral and central nervous systems. We describe the recently discovered functions of Hv1 in various neurological diseases, including brain or spinal cord injury, ischemic stroke, demyelinating diseases, and pain. We also summarize the current advances in the discovery and application of Hv1-targeted small molecules in neurological diseases. Finally, we discuss the current limitations of our understanding of Hv1 and suggest future research directions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ion channels play critical roles in controlling and shaping the physiological functions of neurons and glial cells in the nervous system [1, 2]. Dysfunctional ion channels have been implicated in several neurological diseases [3,4,5,6,7,8]. The voltage-gated proton channel, Hv1, is an ion channel with high selectivity for H+. Findings in the early 1980s in snail neurons described a voltage-gated proton current for the first time [9]. However, the identity of Hv1, encoded by the gene HVCN1, was not determined until 2006; it was identified via a bioinformatics approach, based on known voltage-sensing domains and cation channels from the human and mouse genomes [10, 11]. Regarding the nomenclature “Hv1”: H represents transported H+, V represents voltage gating, and isoform number 1 indicates a unique isoform. In addition, the prefix denotes the species, such as “mHv1” stands for the Hv1 channel of the mouse, and “hHv1” indicates the channel in humans. The gene encoding Hv1 is called HVCN1 (hydrogen voltage-gated channel 1) [12]. Soon after the identification of the Hv1 channel gene, HVCN1 knockout mice (Hvcn1–/–) were generated [13, 14]. Since then, research on Hv1 channel function in vivo has accelerated.
The production of reactive oxygen species (ROS) and tissue acidosis can damage glia and neurons, contributing to the pathophysiology of neuroinflammatory disorders [15, 16], while Hv1 has been reported to play a critical role in regulating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) activity and pH in phagocytes [14]. Therefore, in this regard, there is a mounting focus on the potential role of Hv1 in nervous system-related disorders. Early research showed that Hv1 is selectively expressed and functions in CNS-resident microglia in the mammalian nervous system [17], and that microglial Hv1 is critical for NOX-dependent ROS production and enhances the CNS damage caused by ischemic stroke [17,18,19,20], traumatic brain injury [21], spinal cord injury [22,23,24], and demyelinating lesions [25,26,27]. Moreover, Hv1 has been identified as contributing to the pathogenesis of neuropathic pain by regulating ROS production of microglia and microglia-astrocyte communication [28]. Recently, Hv1 was determined to be expressed not only in the microglia but also in the sensory neurons of the dorsal root ganglion (DRG) of the mammalian nervous system including mice and humans [29, 30]. In parallel, a selective Hv1 inhibitor, YHV98-4, which is suitable for in vivo use, has been identified using a structure-based approach, exhibiting an analgesic effect in several animals models of pain, including neuropathic pain, inflammatory pain, and opioid-induced hyperalgesia [30].
In this review, we summarize the molecular properties of the voltage-gated proton channel Hv1, and its distribution and functional expression in the central and peripheral nervous systems, particularly the latest research progress on Hv1 in various diseases of the nervous system. We also summarize the current progress in small molecule discovery targeting Hv1 and their applications in various diseases. Moreover, we discuss the current limitations of Hv1 research and suggest directions for future research on Hv1 in the nervous system.
Properties of the Voltage-Gated Proton Channel Hv1
Structure and Physiological Function of Hv1
Hv1 belongs to a family of voltage-gated ion channels, its structure is highly conserved among mammalian cells [31, 32], and it has some exceptional characteristics that distinguish it from the classic voltage-gated ion channels (Fig. 1).

Schematic structure of the classical voltage-gated ion channel and voltage-gated H+ channel. A Classical voltage-gated ion channels are composed of a tetrameric structure including four subunits (such as Kv, Na+, and Ca2+), where the ion pathway is located at the center axis. Each subunit consists of six transmembrane domains (S1–S6). The S1-S4 segments harbor the voltage sensor domain (VSD), where the gating charges are located. The S5–S6 segments form the pore domain (PD) and the ion-selectivity filter structure. B The Hv1 channel is a homodimer involving two subunits. Each subunit corresponds to an isolated VSD with an intrinsic conduction pathway serving as the PD, lacking the S5–S6 domain. T29: phosphorylation site, enhancement of gating, T29A is the mutation of T29 with the loss of phosphorylation function [45]; M91T: mutation site, reduced pH sensitivity [44]; D112: the functional site of the selectivity filter for H+ [37]; R205, R208, R211: the functional sites of voltage sensors [38]. On the left are the schematic topologies of the channels, and on the right are the schematic quaternary structures of the channels.
Being structurally unique, the Hv1 channel lacks the typical pore domains (PDs) of ordinary ion channels [10, 11]. The classic voltage-gated ion channels typically comprise six transmembrane (TM) segments, where the TM segments S1–S4 comprise the voltage-sensing domain (VSD) that is critical for sensing changes in the cell membrane potential. Within the S4 domain there are a series of positively-charged residues that are responsible for voltage sensing, while the TM segments S5–S6 form the ionically conductive PD responsible for selective ion permeation, and the electrical energy generated by the displacement of these S4 residues during voltage activation is then transduced to the S5–S6 PD, leading to channel opening [33] (Fig. 1A). Remarkably, the Hv1 channel only has S1–S4, which serve as both VSD and PD, and the channel is assembled as a homodimer, with each subunit containing its conduction pathway [34, 35]. The channel is therefore able to function without the typical center-of-the-tetramer S5–S6 PD structure as found in other classic voltage-gated ion channels. (Fig. 1B).
From a functional standpoint, Hv1 channels are exclusively selective for H+, a feature that is almost absent in other ion channels [36]. This remarkably high selectivity is determined by Asp112 (labeled D112 in Fig. 1B), where neutralization of this acidic residue converts Hv1 into an anion channel [37]. This configuration enables the Hv1 channel to efficiently transport H+. In addition, the gating function of Hv1 is exceptional, as it senses both the pH gradient and the TM voltage. Thus, a depolarizing voltage, an acidic pH in the intracellular environment, and an alkaline pH outside the cell favor the activation of Hv1. The Hv1 S4 TM segment is voltage-dependent due to three charged Arg residues (labeled R205, R208, and R211 in Fig. 1B), which are analogous to the VSD of K+ channels [38]. The relative H+ concentrations on each side of the membrane result in a pH gradient, which establishes the voltage range over which Hv1 channels open. This regulation by the pH gradient under normal conditions results in Hv1 channels opening only when doing so will produce an outward current. “Countercharge” and “electrostatic” mechanisms have been proposed to explain the production of ΔpH-dependent gating [39], while pH sensing does not yet have a well-defined structural basis. Hv1 has also been found to be sensitive to temperature [40, 41], showing temperature-dependent activation and its temperature-dependence is far greater than that of almost any other channel [42, 43], which indicates the requirement for temperature control in electrophysiology-related experiments in Hv1 channel research.
A methionine-to-threonine mutation of Hv1 at site 91 (labeled M91T in Fig. 1B) produces a channel that requires an additional 0.5 pH units of pH difference between the intracellular and extracellular domains than wild-type (WT) Hv1 in epithelial cells. This reduced pH sensitivity provides both functional and molecular evidence for the mediation of pH-regulated acid secretion in the airway epithelium by Hv1 [44]. Furthermore, it has been found that the activation of phagocytes by phorbol esters strikingly affects the gating properties of the Hv1 channel, by shifting the current-voltage relationship in a positive direction. This is mediated by phosphorylation of the Hv1 protein at the Thr29 site, as Thr29Ala or Thr29Asp substitutions (labeled T29A in Fig. 1B) completely abolish this activation promoting effect [45]. This provides insights that the regulation of Hv1 activation might be achieved through regulating the activity of protein kinase C (PKC), which can phosphorylate the Thr29 site of the Hv1 channel. Exploration of the potential regulation of Hv1 by other kinases, such as PKA, MAPK, and CaMKII, is an important research direction for the field.
Cellular Mechanisms and Regulation of Hv1 Activation
NOX activity and the pH difference between the intracellular and extracellular regions are crucial stimuli for regulating the activation of the Hv1 channel.
The Hv1 Channel Mediates the NOX-dependent ROS Signaling Pathway
NOX enzymes consist of seven members, NOX1–NOX5 and DUOX1-2, that produce ROS, including superoxide O2- and hydrogen peroxide H2O2, using O2 and NADPH as substrates [46]. These enzymes are widely expressed in a variety of species and tissues, with different functions depending on location and expression. In addition to contributing to the progress of many oxidative stress-induced diseases, ROS produced by NOX enzymes is involved in host defense and redox-dependent signaling [46]. NOX2 is so far the most well-characterized member of the NOX family, and a high level of expression has been detected in neutrophils, which are involved in the respiratory bursts that are required for phagocytosis [47].
To date, the most studied function of Hv1 is to regulate NOX2 activity in phagocytes [48]. During the respiratory burst catalyzed by NOX2, NADPH provides two electrons that are transferred across the plasma membrane, binding the O2 molecule outside of the cell, leading to the production of ROS and membrane depolarization; at the same time, it generates two H+ in the cytoplasm, resulting in acidification of the cytosol (Fig. 2). Depending on the amount of ROS produced by phagocytes, NOX activity can cause a large enhancement of cytoplasmic acid production [49, 50]. However, the intracellular pH is not reduced and can even be up-regulated during NOX activation [50,51,52], reminding that this cytoplasmic acidification can be remedied by proton extrusion mechanisms, and Hv1 channels may be involved in NOX2 activation. Furthermore, Morgan et al. demonstrated that NOX activity in neutrophils can cause cytoplasmic pH to drop by 1 unit or more when Hv1 is blocked or genetically knocked out [53], providing evidence for the involvement of Hv1 channels in NOX2 activation. In contrast to electroneutral exchangers such as Na+-H+ exchangers, Hv1 has the advantage that its electrogenic H+ extrusion prevents the massive depolarization that would otherwise result from the electrogenic electron transport mediated by NOX activity. In neutrophils, Henderson et al. reported that voltage-gated H+ channels have been implicated in pH homeostasis by balancing the effects of acid production and depolarization caused by NOX production in the phagocyte [54].

Scheme of the cooperation between the NADPH oxidase complex (NOX) and the Hv1 channel. The NOX produces the radical superoxide anion O2- by using NADPH as an electron donor and molecular oxygen as an electron acceptor. The activity of the NOX results in an accumulation of protons, H+, in the cytosol and depolarizes the membrane. Hv1 extrudes these H+ to maintain intracellular physiological pH and repolarize the membrane. H+ extruded by Hv1 is used in the reactions of converting the highly unstable reactive oxygen species (ROS, O2-) to more stable ROS such as hydrogen peroxide (H2O2). NADPH oxidases, especially NOX2, are well-recognized sources contributing to cytosolic acidification. Nevertheless, other sources, such as the non-oxidative breakdown of glucose in cancer cells, might also contribute to the production of intracellular protons.
It has been noted that either membrane depolarization or cytosolic acidification can inhibit the activity of NOX2, while Hv1-mediated H+ extrusion from the cytoplasm is able to reverse it, which can maintain physiological membrane potential and rebuild normal pH [55]. Blunted Hv1 activity would lead to the suppression of H+ extrusion capacity, which could lead to intracellular acidosis. For instance, a study has found that phorbol myristate acetate (PMA) treatment of Hv1-deficient neutrophils results in a more acidic cytoplasm than in comparable WT neutrophils [48]. It has also been reported that Hv1-deficient eosinophils produce significantly less ROS while undergoing significantly greater cell death following PMA stimulation than WT eosinophils, which suggests that intracellular acidosis during respiratory bursts might be much more severe in this kind of cell [56]. In B cells, the key to adaptive immunity, Hv1 modulates the signal strength of the B cell antigen receptor (BCR) [57]. ROS production is reduced in Hv1-deficient B cells, leading to impaired BCR signaling through attenuated oxidation of the protein Src homology region 2 domain-containing phosphatase-1 (SHP-1). In Hv1-deficient mice, activation of the spleen tyrosine kinase and the serine/threonine kinase Akt signaling pathways is reduced, mitochondrial respiration and glycolysis are impaired, and antibody responses are diminished [57]. In T cells, it has been reported that the deletion of Hv1 in Jurkat T cells induces acidification, which promotes the death of apoptotic cells [58]. In addition, the indirect function of Hv1 in myeloid-derived suppressor cells (MDSCs) on T cells has also been revealed recently: Alvear et al. showed that the activation of Hv1 channel in MDSCs increase NOX2-dependent ROS generation, which would react with the T cell receptor complex, resulting in the abolishment of antigen presentation, and blocking the immune system from eliminating tumor cells [59]. This study also suggested that Hv1 inhibition facilitates the attack of tumor cells by the immune system [59]. In microglia, the function of the Hv1 channel has mostly been reported to transfer H+ to the extracellular space as charge compensation and to regulate the generation of NOX2-dependent ROS [17]. Nevertheless, it has also been reported that a deficiency of Hv1 in primary cultured microglia enhances the extracellular ROS signal [20, 60], as well as in Kupffer cells [61]. This is seemingly paradoxical, considering the numerous pieces of evidence indicating that Hv1 boosts ROS generation. On the basis that the results are reliable, these results emphasize that the complex regulations of Hv1 may depend on the diverse physiological states of microglia. Further, the functional link between NOX2 expression and Hv1 channel activity may apply to all NOX isoforms. In sperm cells, the NOX5 isoform has been found to be a major source of ROS in human spermatozoa, and a role for the Hv1 channel of mediating NOX5-dependent ROS generation in the motility of human spermatozoa has also been suggested [62, 63]. Taken together, a large body of evidence shows that the NOX-dependent ROS signaling pathway plays a critical modulatory role in the activity of Hv1 channels.
pH Regulation
As a proton channel, Hv1 also functions independently of NOX in many instances. A function of Hv1 is to regulate H+ homeostasis. In the spermatozoa of humans, Hv1 is reported to be highly expressed in the flagellum. The activation of Hv1 leads to intracellular alkalization and the activation of spermatozoa [64, 65]. The important functions of sperm, such as motility, capacitation, the interaction between sperm and zona pellucida, the acrosome reaction, and sperm-oocyte fusion, are likely regulated by Hv1 [63,64,65,66,67].
In the airway and pulmonary epithelial cells, Hv1 plays a role in maintaining the physiological pH levels of the environment in the airways and lungs [44, 68,69,70,71,72]. In osteoclast cells, Hv1 acidifies the extracellular environment and breaks down mineralized bone by conducting outward H+ ions, which promotes bone resorption [73]. In basophils, Hv1 activation by PMA or anti-IgE incites histamine release, associated with the ability of Hv1 to stabilize the intracellular pH but is not linked to NOX2, since basophils do not express the enzyme [74]. In mast cells, the same mechanism is exhibited to control histamine release [75].
Apart from regulating cytosolic pH, Hv1 is also responsible for the regulation of phagocytic vacuole pH in neutrophils. In Hv1-deficient neutrophils, the phagocytic vacuolar pH is approximately two pH units greater than that pH in WT neutrophils, which means the vacuolar cytoplasm is much more alkalinized while the cell cytosol is excessively acidified, showing that Hv1 also plays an important role in H+ conduction on the phagocytic vacuolar membrane within neutrophils [76]. Furthermore, in plasmacytoid dendritic cells (pDCs), Hv1 deficiency delays the acidification of pDC endosomes, decreasing the secretion of type I interferon\s (IFN-I), consequently limiting protease activity and toll-like receptor 9 (TLR9) signaling [77], which reveals an important function of Hv1 for innate immune responses. In cells of the renal medullary thick ascending limb, Hv1 modulates the ROS production of mitochondria through complex I rather than through NOX. This provides a novel mechanism by which Hv1 regulates ROS production [78]. Taken together, the involvement of Hv1 in the regulation of ROS and H+ in mammalian cells is multifaceted, and the NOX-independent role of Hv1 is fascinating and requires further investigation.
Expression and Function of Hv1 in the Nervous System
Over the last decades, the Hv1 channel has been found with high expression in immune cells including basophils [74], eosinophils [56, 79], neutrophils [76, 80,81,82], B cells [57, 83], T cells [58, 59, 83], monocytes [84], macrophages [61, 85], mast cells [42], and dendritic cells [77, 86], demonstrating that the functions of Hv1 are involved in various immunologic processes. In the nervous system, studies on Hv1 have mostly focused on mammalian microglia [87,88,89]. Until recently, a large amount of Hv1 expression in neurons was identified in the DRG of the peripheral nervous system (PNS) [30]. The nervous system primarily comprises two cell types: neurons and glial cells. The CNS primarily comprises three main types of glial cells: microglia, astrocytes, and oligodendrocyte lineage cells, whereas they are mainly Schwann cells in the PNS [90, 91]. The expression and function of Hv1 in these cells of the nervous system are illustrated in detail below. The expression and function of the Hv1 channel in other non-nervous system cell types across multiple species have been systematically reviewed in previous studies [55, 87].
Hv1 in Glia
Microglia are the resident macrophage-like immune cells of the CNS, where they play a critical role in host defense and tissue repair [92]. In the past two decades, a large number of Hv1 studies in the CNS have predominantly focused on microglia [24, 87,88,89]. The expressions of Hv1 in microglia in rodents, and microglia collected from human brain tissue have been identified by electrophysiology, qPCR, western blot, and immunohistochemistry [17, 22, 24, 93]. Also, it has been reported that there are species differences in the level of Hv1 expression in microglia [17, 87]. Wu et al. found that Hv1 currents are much larger in mice than in rats by whole-cell recording in microglia from the hippocampus, and showed that the H+ current in the rat is only 8% of that in mice [17]. They also recorded appreciable Hv1 protein and Hv1 currents from cultured human microglia, while it was pointed out that whether human microglia in situ have large Hv1 currents is unknown. Furthermore, Wu et al. found that activating Hv1 leads to NOX-dependent ROS production in brain microglia. ROS production is significantly attenuated in Hv1-deficient mice, and mice lacking Hv1 are protected from NOX–mediated neuronal death and brain damage 24 h after stroke, as a result of reducing the amount of ROS from the activated microglia [17]. Paradoxically, Kawai et al. demonstrated that ROS production is drastically enhanced in isolated Hv1-deficient microglia compared with WT microglia in primary culture, and postulated that this is a result of the alteration of actin dynamics and the change of intracellular distribution of cytosolic NADPH oxidase subunit p67 in Hvcn1-/- microglia, although the underlying mechanisms are still not fully understood [20]. Therefore, it seems that the regulation of ROS by microglial Hv1 is complex, and may be determined by the physiological states of microglia since microglia are widely known to undergo complicated and multifaceted physiological states dependent on the CNS environment [94]. Kawai et al. also showed that the regulation of microglial ROS production by Hv1 is age-dependent, and that oxidative stress-responsive gene expression is dramatically altered with aging in Hv1- deficient mouse brains [60]. The age-dependent Hv1 function in microglia has also been reported by Zhang et al., who found that aged (18 months old) mice have upregulated Hv1 and NOX expression compared with that in adult (2-3 months old) mice, and that the percentage of pro-inflammatory M1 microglia co-labeling with Hv1 is higher in aged mice after tibial fracture surgery, indicating that aging alters the Hv1-mediated microglial polarization. This means that Hv1 upregulation in the aged brain might shift the dynamic equilibrium of microglial activation toward pro-inflammatory M1 polarization [95]. Considering the important role of microglia in nervous system development and maintenance [96, 97], Wu et al. reported a close interaction between microglia and neurons in vivo or in situ [98], and they suggested that microglial Hv1 function may be mediated by the activation of neuronal TRP channels [99] and acid-sensing ion channels (ASICs) [100] in the mouse brain. The microglia-derived protons can target neuronal ASICs and the microglia-derived ROS can target neuronal TRP channels, revealing that protons and ROS produced by Hv1 may play a pivotal role in microglia-neuron communication [98]. As for the acid-regulatory capacity of microglia, Ritzel et al. reported that microglia lacking Hv1 show a reduced ability to extrude protons and that Hv1-deficient mice exhibit reduced pathological acidosis after traumatic brain injury (TBI), suggesting that microglial Hv1 channels have an important role of exacerbating pathological acidosis within the injury microenvironment following head injury [21]. Hv1 also plays a role in regulating microglial migratory ability. Wang et al. found that blockade of Hv1 with an anti-Hv1 antibody in vivo promotes microglial migration in the brain, while increased migration is also detected in microglia with Hv1 genetic deletion in vitro [101]. However, previous studies have shown that microglial Hv1 is not involved in ATP-induced process chemotaxis [17, 102]. This discrepancy in Hv1 in regulating cell migration remains unclear and requires further clarification [101]. A large proportion of experimental data in the literature was collected by using Hv1 staining, while it should be kept in mind that the Hv1 staining using certain antibodies appears to be non-specific or due to channel bleed-through for the lack of specific a Hv1 antibody for immunostaining. Therefore, some negative controls should be applied, such as using Hv1-deficient mice or cell types with no Hv1 expression; or using multiple methods such as electrophysiology or qRT-PCR to verify expression and function.
In the CNS, astrocytes play important immune-inflammatory roles and support the function and survival of neurons [103, 104]. No Hv1 current has been detected in hippocampal astrocytes isolated from WT mice using electrophysiology [17], indicating that there may be no functional expression of Hv1 in astrocytes. However, it remains unclear whether astrocytes in other brain regions express Hv1. Peng et al. showed that the activation of astrocytes is significantly reduced in Hv1-deficient mice after spinal nerve transection (SNT) [28], and this is explained by the regulation of Hv1 in spinal microglia and that ROS production modulated by Hv1 in microglia promotes spinal astrocyte activation after SNT [28].
Oligodendrocyte lineage cells include oligodendrocytes (OLs) and oligodendrocyte progenitor cells (OPCs). So far, no solid evidence supports the existence of Hv1 expression in OPCs or OLs, whereas some studies have shown that Hv1-deficient mice show some OPC and OL dysfunctions. Liu et al. reported that Hv1-deficient mice are partially protected from demyelination and motor deficits compared with those in WT mice in a model of cuprizone-induced multiple sclerosis (MS) because the inhibition of microglial Hv1 leads to a reduction of ROS production, the impairment of OPC proliferation is reduced, and the number of mature OLs and remyelination is increased [25]. Later, Yu et al. showed that Hv1 deficiency in microglia attenuates OPC apoptosis and promotes OPC proliferation and differentiation following oxygen-glucose deprivation-induced injury in vitro, which is mediated by reducing ROS generation and pro-inflammatory cytokine production in Hv1-deficient microglia [105]. Chen et al. also found that the loss of Hv1 in microglia alleviates the damage caused by lysophosphatidylcholine (LPC) to the myelin sheath by decreasing ROS production, leading to an increase in mature OLs and a reduction in demyelination [26]. Recently, Wang et al. identified a low level of Hv1 expression in OPCs and OLs by the detection of Hvcn1 mRNA in primary OPCs cultured from rat brains through RT-qPCR, and this was confirmed by detection of the Hv1 protein in primary cultured OPCs and OLs by Western blotting [101]. Besides, Hv1 is also expressed in some OLs labeled by SOX10+ in the cerebral cortex of the mouse through immunofluorescence staining [101]. How Hv1 in OPCs and OLs, if any, contributes to cell-type-specific function in vivo requires further study.
Schwann cells (SCs) are a critical component of the PNS and are responsible for maintaining peripheral nerve structure and function by myelinating and ensheathing nerve fibers [106]. In addition, SCs play an important role in facilitating peripheral nerve regeneration by secreting neurotrophic factors and producing extracellular matrix molecules [107]. To our knowledge, no study has reported Hv1 expression in SCs, and this deserves further exploration.
The role of macrophages in the PNS is to some extent similar to that of microglia in the CNS. In the PNS, Yu et al. demonstrated that DRG macrophages contribute to both the initiation and persistence of neuropathic pain, with molecular cross-talk between axotomized sensory neurons and macrophages, revealing potential peripheral DRG targets for neuropathic pain management [108]. The Hv1 expression in macrophages of other systems has been well-defined in previous studies [61, 85]. Kawai et al. showed, using immunocytochemistry and electrophysiology, that Hv1 is specifically expressed in Kupffer cells (liver-resident macrophages), but not in hepatocytes, and regulates hepatic oxidative stress in vivo [61]. We detected low expression levels of Hv1 mRNA by RNAscope technology in a small percentage of macrophages (<16%) in the DRG of Cx3cr1GFP mice (the microglia/macrophage labeled with green fluorescence), which contribute to <8% of the total Hvcn1 expression in the DRG. This expression is not significantly increased in the chronic inflammation pain model constructed by the paw injection of complete Freund's adjuvant (CFA).
Hv1 in Neurons
Voltage-gated H+ currents were first recorded in snail neurons [9]. In later studies, Diarra et al. detected an H+ current by measuring the changes in intracellular and extracellular pH of neurons in the CA1 region of the rat hippocampus. They believed that there may be a voltage-gated H+ channel in neurons with an unknown molecular identity [109]. Cheng et al. [110] reported a proton current in rat hippocampal neurons, which was insensitive to Zn2 + and not temperature-dependent [110], indicating that it was probably not an Hv1 current. A similar H+ current was later presented in neurons from Hv1 KO mice [17], further suggesting that non-Hv1-mediated proton efflux may be present in neurons. Later, Li et al. found that Hv1 mRNA is almost undetectable by qPCR in primary cultured mouse cortical neurons [24]. Taken together, these previous studies failed to identify Hv1-mediated proton currents in mammalian neurons in the brain. The Hv1-mediated proton currents in mammalian neurons were unexpectedly identified in the PNS. Our latest findings demonstrated wide expression of Hv1 in sensory neurons in the DRG [30], with almost 60% of these neurons expressing Hv1 mRNA, as detected by RNAscope in situ hybridization; these included small and medium-to-large neurons [30]. In normal human abdominal skin and scar tissue, Hv1 has also been identified as co-localized with PGP9.5, a marker for nerve terminals. Hv1 mRNA expression in DRG neurons is significantly upregulated by inflammation, NOX activators, or nerve injury [30]. An Hv1-mediated proton current has been isolated in DRG neurons from naïve mice using the newly-discovered inhibitor YHV98-4 or Zn2+; this current is also significantly upregulated in inflammation and is largely reduced in Hv1 KO mice or mice with Hv1-specific knockdown in the DRG using a viral strategy [30]. Notably, YHV98-4 and Zn2+-sensitive currents display very fast activation kinetics, which is similar to the proton currents previously reported in snail neurons, in contrast to the slow activation rate of the Hv1 current in mammalian microglia. The mechanism underlying the differences in the kinetics of Hv1 currents in different cells remains incompletely understood. It has been found that it can be regulated by intracellular/extracellular H+ and the phosphorylation state of the channel [88]. Nevertheless, the fast kinetics of Hv1 currents in neurons seems to serve as an ideal mechanism for the rapid restoration of intracellular H+, as the action potential usually produces fast cytoplasmic acidification via Ca2+/H+ exchange.
Pathological Roles of Hv1 in the Nervous System
Traumatic Brain Injury
In patients with TBI, a common characteristic of brain damage is tissue acidosis, with an acute decrease of pH in the brain, contributing to prolonged coma, neurologic deficits, and poor long-term outcomes [111,112,113]. A recent study by Ritzel et al. demonstrated that Hv1 in the microglia plays a key role in tissue acidosis, oxidative stress, and neuroinflammation in TBI [21]. Hv1-deficient mice present with decreased levels of pathological acidosis and inflammation after TBI, leading to long-term neuroprotection and functional recovery [21]. In a controlled cortical impact model (CCI) of TBI, it has been shown that CCI caused acidosis both intracellularly and extracellularly, persisting for weeks after injury, and Hv1 expression is chronically upregulated in the brain after TBI. In Hv1 knockout mice however, extracellular acidosis is decreased, ROS production is attenuated, pro-inflammatory cytokines (IL-1β, IL6, and TNFα) are reduced, and anti-inflammatory cytokines (IL10, TGFβ, IL4Rα, arginase 1, and SOCS3) are increased in brain microglia during the acute period after TBI [21]. Moreover, the attenuation of brain acidosis in the acute period in Hv1-deficient mice also limits subsequent neurodegeneration and long-term neurological impairment 6 months after injury, indicating that the deficiency of Hv1 alleviates acidosis and could be beneficial even in the chronic stages of TBI [21]. These data suggest that targeting the Hv1 channel in microglia may be a novel therapeutic strategy for TBI.
However, the brain with traumatic injuries is often accompanied by blood-brain-barrier (BBB) disruption, concomitant with the infiltration of various blood-derived immune cells expressing a high level of Hv1 [21, 89]. Hv1 in blood-derived immune cells may contribute to the neuroprotective effect of TBI. Cell-specific Hv1 conditional knockout animals should be useful in addressing this issue.
Spinal Cord Injury
The pathophysiology of spinal cord injury (SCI) includes two phases, primarily spinal cord tissue is disrupted by an initial mechanical injury, followed by a secondary injury phase that involves vascular dysfunction, excitotoxicity, inflammation, ROS production, and cell death [114]. Upon SCI, the release of ROS and pro-inflammatory molecules by excessively activated microglia aggravates the secondary damage [115]. Recent studies have demonstrated that in contusion models of SCI in mice, Hv1 deficiency ameliorates the activation of microglia, leading to reduced production of ROS and pro-inflammatory cytokines, and attenuates tissue acidosis, resulting in alleviated secondary damage, which subsequently decreases neuronal loss and correlated with improved locomotor recovery [22,23,24, 116, 117]. Li et al. revealed that Hv1 deficiency attenuates NLRP3-inflammasome-mediated neuronal pyroptosis and apoptosis, facilitates myelin-axon regeneration, and improves motor function in mice after SCI [22]. It has been postulated that this effect is mediated by reduced ROS generation in microglia with Hv1 deletion. SCI induces elevated levels of ROS, whereas Hv1 deficiency downregulates microglial ROS generation. Furthermore, in vitro experiments using the PC12 cell line have shown that ROS activates the NLRP3 inflammasome pathway and induces neuronal pyroptosis, while the addition of a ROS scavenger reverses this effect [22]. These data indicate that microglial Hv1 mediates ROS production after SCI to regulate neuronal apoptosis and NLRP3-induced neuronal pyroptosis. However, it should be noted that the PC12 cells used in this study are a cell line from rat pheochromocytoma cells used to mimic mature neurons [118], but they cannot represent the realistic state of neurons in vivo. Li et al. have also shown that the absence of Hv1 reduces the OL apoptosis, demyelinated areas, and cavity formation induced by SCI [116]. Murugan et al. reported similar results, showing that mice lacking Hv1 have improved motor recovery following contusion SCI compared to WT mice [23]. It has been shown that deficiency of Hv1 results in a decrease in the number and soma size of spinal microglia, as well as their reduced outward rectifier K+ current density in Hv1 KO mice compared to WT mice, 7 days following SCI, suggesting that microglia are both morphologically and electrophysiologically less activated in Hv1 KO mice than they are in WT mice after SCI. How Hv1 mediates these changes remains elusive. A recent study has shown that Hv1 depletion significantly diminishes NOX2-mediated ROS production, pro-inflammatory cytokine production, microglial proliferation, tissue acidosis, phagocytic oxidative bursts, and leukocyte infiltration at 3 days post-SCI in adult female mice [24].
Overall, these data reveal an important role for Hv1 in regulating microglial activation, NOX2-mediated ROS production, extracellular acidosis, and functional outcomes following SCI, implying that Hv1 may be a promising potential therapeutic target to alleviate both primary and secondary damage following SCI.
Ischemic Stroke
During ischemic stroke, a large amount of ROS is produced in the brain that rapidly reacts with lipids, proteins, cofactors, and DNA, in turn activating apoptotic pathways [119, 120], and excessive oxidative stress produced by neurons and glia in ischemic area of the brain is one of the major mechanisms leading to the severe damage of brain cells [121, 122]. In 2012, Wu et al. first reported a detrimental role of Hv1 in ischemic stroke, in which Hv1-deficient mice were protected in the middle cerebral artery occlusion model of ischemic stroke, via the ROS production mechanism mediated by microglial Hv1 [17]. When microglial Hv1 is absent, NOX2 causes excessive membrane depolarization and inhibits electron transfer by the oxidase, thus eliminating ROS production. As the brain is also invaded by blood-borne leukocytes expressing high levels of NOX2 after stroke [123], this study also excluded the potential effect of peripheral Hv1 on ischemic brain damage through bone marrow transplantation experiments, by creating bone marrow chimeric mice devoid of either microglial- or bone marrow-derived Hv1 [17]. The synaptic transmission, plasticity, and NMDA receptor function are normal in Hv1−/− mice and Hv1-deficiency does not rescue the neuronal and microglial death induced by NMDA in primary cultures [17]. This denotes that the contribution of Hv1 to brain damage in the stroke model is dependent on its function in the regulation of microglial activity rather than its contribution to glutamate neurotoxicity.
Tian et al. also revealed the protective effect of inhibiting Hv1 by shifting microglia from pro-inflammatory-M1 toward an anti-inflammatory-M2 state in a photothrombotic stroke model of ischemic cerebral injury [19]. Kawai et al. reported that the regulation of microglial ROS production by Hv1 is age-dependent [20]. Compared to age-matched WT mice, the expression levels of ROS in the cerebral cortex are slightly decreased in Hv1 KO mice at 1 day, 5 days, and 3 weeks of age, but drastically increased at 6 months of age. Besides, the aged Hv1 KO mice (6 months old) show a smaller infarct volume in brain ischemia, while the young Hv1 KO mice (9–10 weeks old) show little difference [20]. Taken together, these findings indicate that the depletion of Hv1 may be a promising strategy to treat ischemic stroke, and age dependence in neuroprotection should be considered as a factor for therapies that target Hv1 in ischemic stroke in the future.
Demyelinating Diseases
Several CNS diseases are characterized by a pathological endpoint of demyelination, such as MS, subacute sclerosing panencephalomyelitis, metachromatic leukodystrophy, and Pelizaeus-Merzbacher disease [124]. In a cuprizone-induced demyelination mouse model of MS, mice with Hv1-deficiency are partially protected from demyelination and motor deficits compared to WT mice through ameliorated microglial activation, reduced ROS production, increased OPC proliferation, and an increased number of mature OLs [25]. In a model of demyelination using the two-point injection of LPC into the corpus callosum, a study by Chen et al. found that the myelin damage mediated by LPC is reduced in mice lacking Hv1, with decreased ROS production and autophagy of microglia in the demyelinated region, leading to an increase in mature OLs and a reduction in demyelination [26]. The autophagic pathway inhibition after Hv1 deficiency in microglia warrants further investigation. Later, using a mouse brain hypoperfusion model of white matter injury, Yu et al. found that ablation of Hv1 attenuates the bilateral common carotid artery stenosis-induced disruption of white matter myelin integrity and cognitive impairment in mice [27]. Furthermore, Wang et al. showed that the neutralization of Hv1 with antibody in vivo promotes myelin debris clearance by microglia/macrophages in a model of LPC-induced focal demyelinating lesions, by enhancing the migration of microglia/macrophages [101].
Therefore, microglial Hv1 contributes to pathological processes in CNS demyelinating diseases via unidentified non-cell-autonomous mechanisms. The interaction between microglia and OPCs/OLs is intriguing and worthy of further investigation. Another possibility is that Hv1 is functionally present in OPCs/OLs and contributes to demyelination via a cell-autonomous mechanism.
Pain
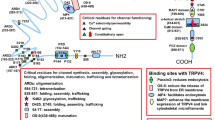
Using an SNT model of peripheral nerve injury, Peng et al. reported that the function of Hv1 in spinal cord microglia is crucial for the development of neuropathic pain [28]. In this study, microglial Hv1 contributed to ROS production, astrocyte activation, IFN‑γ upregulation, and subsequent hyperalgesia and allodynia following peripheral nerve injury [28]. This study highlights a microglial Hv1-astrocytic IFN-γ axis of communication in pain hypersensitivity (Fig. 3A). Interestingly, spinal microglial activation is not altered in Hv1 KO mice after SNT [28], given that microglial activation is one of the hallmarks in chronic pain.

Schematic for Hv1 in chronic pain. A The microglial Hv1 channel regulates neuropathic pain by promoting microglia‑astrocyte communication in the spinal cord. After peripheral nerve injury, spinal microglia are activated and this is accompanied by Hv1-dependent ROS production. ROS acts on both neurons and astrocytes, triggering IFN‑γ release. IFN‑γ in turn contributes to further microglial activation and microglia‑astrocyte interaction to maintain pain hypersensitivity after SNT [28]. B Schematic of neuronal Hv1 channel regulation of inflammation and pain via modulation of ROS production and the SHP-1/AKT pathway in the DRG. Complete Freund's adjuvant and chronic morphine exposure increased the production of Hv1-mediated ROS, inducing the downregulation of SHP-1 phosphorylation in DRG neurons, leading to activation of the PI3K/pAkt pathway with the up-regulation of pro-nociceptive inflammatory cytokines, contributing to the inflammatory pain and the morphine-induced tolerance and hyperalgesia, which can be reversed by pharmacological and genetic inhibition of Hv1.
The conventional view that Hv1 is not present in neurons was changed by a recent study, in which Hv1 was identified in peripheral primary sensory neurons and regulated the production of neuronal ROS and pHi homeostasis in the DRG [29, 30]. Another significant advance presented in this study is the discovery of a series of Hv1 inhibitors for in vivo application that preferentially bind to an intermediate conformation of the Hv1 channel, which results in improved specificity [30]. The selective inhibitor YHV98-4 reduces inflammation and has analgesic effects in mice for both inflammatory and neuropathic pain [30]. Neuronal Hv1 inhibition reduces intracellular alkalization and ROS production, recovers the imbalance in downstream SHP-1-PI3K/pAKT signaling, and decreases the release of several pro-inflammatory chemokines (Fig. 3B) [29, 30]. Notably, Hv1 inhibition in neurons prevents the development of morphine-induced hyperalgesia and tolerance, two common adverse side-effects of the repeated administration of opioids. Mechanistically, a strong production of ROS in the DRG is correlated with repeated chronic morphine exposure. This study suggests that inhibiting Hv1 is a promising strategy for reducing the occurrence of opioid-induced hyperalgesia and tolerance (Table 1).
Inhibitors Targeting Hv1
Most currently available Hv1 inhibitors have served as useful tools to identify the function of Hv1 at the cellular level. However, selective inhibitors for Hv1 with both good selectivity and in vivo pharmacokinetic profiles are still lacking. Thus, despite the expectation that Hv1 inhibition may be beneficial for treating many ROS-related diseases, including immune diseases, various cancers, respiratory diseases, and neurological disorders, the therapeutic potential of inhibiting Hv1 remains largely underexplored. Here, we briefly summarize the currently available Hv1 inhibitors and their applications are summarized in Table 2.
Zn2+
Zn2+ has been applied for many years as a proton current inhibitor for in vitro experiments, even serving as the gold standard for identifying putative H+ currents. The mechanism of Hv1 inhibition by Zn2+ is to act as a competitor of H+ for binding to the Hv1 channel's external surface, which has been comprehensively reviewed [12, 125]. Of note, though, several species have been found to lack the histidines at key locations that bind Zn2+, probably contributing to their insensitivity to Zn2+ [31, 126]. In addition, Zn2+ is involved in various other physiological processes, therefore, the utilization of Zn2+ as a specific blocker of the H+ channel is restricted.
Hanatoxin
The toxin hanatoxin, extracted from the venom of the tarantula Grammostola spatulata, is a nonspecific Hv1 inhibitor. A paddle motif is a modulator unit that is common to voltage sensors and shows high conservation among different voltage-gated ion channels such as voltage-activated K+ channels and Hv1 [126]. Hanatoxin interacts with the paddle region of Hv1 and inhibits proton currents in transfected HEK293 cells, while the D185A mutant efficiently eliminates the inhibitory effects of this molecule [126].
Guanidine Derivatives
As open-channel blockers, 2-guanidinobenzimidazole and 5‐chloro‐2‐guanidinobenzimidazole inhibit Hv1. The inhibition by guanidine derivatives is reversible. However, the selectivity of these compounds remains imprecisely determined. Moreover, guanidine derivatives are unable to permeate cytoplasmic membranes because they are too polar, and the inhibitory effect of guanidine derivatives has only been reported for intracellular application [127, 128]. These features thus prevent them from being considered Hv1 inhibitors suitable for most in vivo experiments.
Corza6 (C6)
Corza6 (C6) is a peptide inhibitor highly specific to hHv1. The polypeptide fragment of C6 is composed of 41 amino-acids, derived from three natural parental spider toxins [66]. The high specificity of C6 to hHv1 has been supported by the findings that there is no inhibition of CiHv1 (the proton channel from the sea squirt Ciona intestinalis), Kv2.1, and Nav1.4 with 1 μmol/L C6 [66]. C6 was identified by a phage-display strategy [66, 131]. The phage-display library was constructed by 1 million novel peptides with inhibitor cysteine knots that were synthesized by combining natural toxin elements, and C6-expressing phagemids were selected on the basis of their ability to bind to the purified hHv1 protein. Inhibition of channel function is contributed by two C6 peptides binding to the two VSDs in hHv1. The inhibitory effect of C6 decreases when cell membranes depolarize, resulting in partial inhibition [66]. Later, guided by a C6–hHv1 complex structural model, Zhao et al. designed a bivalent C6 peptide (named C62), which inhibits hHv1 current with picomolar affinity by binding simultaneously to both subunits [129]. C62 can fully inhibit hHv1 at depolarized voltages, and its affinity is ∼50-fold better than C6 at +40 mV [129]. However, the inhibitory effects of C6 and C62 have only been evaluated in cultured cells including spermatozoa and neutrophils [66, 129]; so far, there is no evidence to support their effectiveness in vivo.
YHV98-4
YHV98-4 was identified using a structure-based approach. Zhang et al. showed that YHV98-4 binds to an intermediate conformational state of Hv1, a transitional state between the resting and activated states. This may explain the high selectivity of YHV98-4 against other VSD-containing channels, including hERG, KCNQ2, BK, Nav1.7, TRP channels, and the Volume-Regulated Anion Channel, in which this particular intermediate conformation state may be absent. YHV98-4 inhibits the Hv1 channel with an IC50 of <1 μmol/L and a maximal inhibition rate of ~50%. Importantly, YHV98-4 is able to cross the BBB and achieves a relatively high concentration in the plasma with a half-life of 1.75 h, making it suitable for in vivo applications. YHV98-4 has been shown to be effective in alleviating chronic pain and opioid-induced hyperalgesia and tolerance in mice [29, 30].
Other Pharmacological Modulators of Hv1
Several proton current inhibitors indirectly affect Hv1 function, presumably by increasing intracellular pH. These include 4-aminopyridine, amantadine, imipramine, rimantadine, nicardipine, chlorpromazine, clozapine, amiloride, and haloperidol [55, 130,131,132]. In addition, the Hv1 antibody has been used as a blocker to neutralize Hv1 by injecting it into the mouse cerebral cortex [101]. This blocking effect might be further validated using Hv1-deficient mice. Zhao et al. reported that human albumin (Alb) can activate hHv1 in sperm [65]. In hHv1, one Alb binds both VSDs, shifting the activation of hHv1 to more negative potentials and speeding channel activation, resulting in an increase of proton current. Using a computational model of the Alb-hHv1 complex, they identified two VSD-interacting sites in Alb domain II, proposing a modification mechanism based on electrostatic gating that favors the active "up" sensor configuration [65]. The activators targeting Hv1 may hold promise for the treatment of male infertility.
Conclusions and Outlook
In the nervous system, the evidence thus far supports the conclusion that Hv1 is mostly expressed in the microglia in the CNS and the sensory neurons in the PNS. The expression and function of Hv1 in OLs/OPCs require further clarification. Hv1 has been shown to perform important functions in neurological disorders including TBI, SCI, ischemic stroke, and pain. Because of the evidence that microglia are highly involved in neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis [133, 134], it would not be surprising if Hv1 played a role in these diseases. This is an important direction for future research. The newly-discovered inhibitor YHV98-4 is BBB-penetrable and should be useful for exploring the therapeutic potential of inhibiting Hv1 in CNS-related diseases. As far as chronic pain is concerned, an important direction would be exploring the function of Hv1 in cancer pain, as YHV98-4 shows remarkable effects in inhibiting morphine-induced tolerance and hyperalgesia, two prevailing adverse side effects of pain management in cancer. Given its strong expression in immune cells [135], cancer cells [58, 59, 136,137,138,139,140,141,142,143,144,145], and sensory neurons, inhibiting Hv1 may have dual beneficial effects on pain and tumors [29], which is an exciting avenue for future research.
References
Smith RS, Walsh CA. Ion channel functions in early brain development. Trends Neurosci 2020, 43: 103–114.
Mir FA, Jha SK. Locus coeruleus acid-sensing ion channels modulate sleep-wakefulness and state transition from NREM to REM sleep in the rat. Neurosci Bull 2021, 37: 684–700.
Steinlein OK. Ion channel mutations in neuronal diseases: A genetics perspective. Chem Rev 2012, 112: 6334–6352.
Shibata M, Tang C. Implications of transient receptor potential cation channels in migraine pathophysiology. Neurosci Bull 2021, 37: 103–116.
Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA. Ion channels in genetic epilepsy: From genes and mechanisms to disease-targeted therapies. Pharmacol Rev 2018, 70: 142–173.
Wang R, Tu S, Zhang J, Shao A. Roles of TRP channels in neurological diseases. Oxid Med Cell Longev 2020, 2020: 7289194.
Wang HR, Hu SW, Zhang S, Song Y, Wang XY, Wang L. KCNQ channels in the mesolimbic reward circuit regulate nociception in chronic pain in mice. Neurosci Bull 2021, 37: 597–610.
Santoro B, Shah MM. Hyperpolarization-activated cyclic nucleotide-gated channels as drug targets for neurological disorders. Annu Rev Pharmacol Toxicol 2020, 60: 109–131.
Thomas RC, Meech RW. Hydrogen ion currents and intracellular pH in depolarized voltage-clamped snail neurones. Nature 1982, 299: 826–828.
Ramsey IS, Moran MM, Chong JA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature 2006, 440: 1213–1216.
Sasaki M, Takagi M, Okamura Y. A voltage sensor-domain protein is a voltage-gated proton channel. Science 2006, 312: 589–592.
DeCoursey TE. Voltage-gated proton channels: Molecular biology, physiology, and pathophysiology of the H(V) family. Physiol Rev 2013, 93: 599–652.
Okochi Y, Sasaki M, Iwasaki H, Okamura Y. Voltage-gated proton channel is expressed on phagosomes. Biochem Biophys Res Commun 2009, 382: 274–279.
Ramsey IS, Ruchti E, Kaczmarek JS, Clapham DE. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc Natl Acad Sci U S A 2009, 106: 7642–7647.
Qin L, Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation 2012, 9: 5.
von Leden RE, Yauger YJ, Khayrullina G, Byrnes KR. Central nervous system injury and nicotinamide adenine dinucleotide phosphate oxidase: Oxidative stress and therapeutic targets. J Neurotrauma 2017, 34: 755–764.
Wu LJ, Wu G, Akhavan Sharif MR, Baker A, Jia Y, Fahey FH, et al. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat Neurosci 2012, 15: 565–573.
Wu LJ. Microglial voltage-gated proton channel Hv1 in ischemic stroke. Transl Stroke Res 2014, 5: 99–108.
Tian DS, Li CY, Qin C, Murugan M, Wu LJ, Liu JL. Deficiency in the voltage-gated proton channel Hv1 increases M2 polarization of microglia and attenuates brain damage from photothrombotic ischemic stroke. J Neurochem 2016, 139: 96–105.
Kawai T, Okochi Y, Ozaki T, Imura Y, Koizumi S, Yamazaki M, et al. Unconventional role of voltage-gated proton channels (VSOP/Hv1) in regulation of microglial ROS production. J Neurochem 2017, 142: 686–699.
Ritzel RM, He J, Li Y, Cao T, Khan N, Shim B, et al. Proton extrusion during oxidative burst in microglia exacerbates pathological acidosis following traumatic brain injury. Glia 2021, 69: 746–764.
Li X, Yu Z, Zong W, Chen P, Li J, Wang M, et al. Deficiency of the microglial Hv1 proton channel attenuates neuronal pyroptosis and inhibits inflammatory reaction after spinal cord injury. J Neuroinflammation 2020, 17: 263.
Murugan M, Zheng J, Wu G, Mogilevsky R, Zheng X, Hu P, et al. The voltage-gated proton channel Hv1 contributes to neuronal injury and motor deficits in a mouse model of spinal cord injury. Mol Brain 2020, 13: 143.
Li Y, Ritzel RM, He J, Cao T, Sabirzhanov B, Li H, et al. The voltage-gated proton channel Hv1 plays a detrimental role in contusion spinal cord injury via extracellular acidosis-mediated neuroinflammation. Brain Behav Immun 2021, 91: 267–283.
Liu J, Tian D, Murugan M, Eyo UB, Dreyfus CF, Wang W, et al. Microglial Hv1 proton channel promotes cuprizone-induced demyelination through oxidative damage. J Neurochem 2015, 135: 347–356.
Chen M, Yang LL, Hu ZW, Qin C, Zhou LQ, Duan YL, et al. Deficiency of microglial Hv1 channel is associated with activation of autophagic pathway and ROS production in LPC-induced demyelination mouse model. J Neuroinflamm 2020, 17: 333.
Yu Y, Luo X, Li C, Ding F, Wang M, Xie M, et al. Microglial Hv1 proton channels promote white matter injuries after chronic hypoperfusion in mice. J Neurochem 2020, 152: 350–367.
Peng J, Yi MH, Jeong H, McEwan PP, Zheng J, Wu G, et al. The voltage-gated proton channel Hv1 promotes microglia-astrocyte communication and neuropathic pain after peripheral nerve injury. Mol Brain 2021, 14: 99.
Furutani K, Ji RR. Targeting Hv1 proton channel for pain control. Cell Res 2022, 32: 419–420.
Zhang Q, Ren Y, Mo Y, Guo P, Liao P, Luo Y, et al. Inhibiting Hv1 channel in peripheral sensory neurons attenuates chronic inflammatory pain and opioid side effects. Cell Res 2022, 32: 461–476.
Smith SME, Morgan D, Musset B, Cherny VV, Place AR, Hastings JW, et al. Voltage-gated proton channel in a dinoflagellate. Proc Natl Acad Sci U S A 2011, 108: 18162–18167.
Decoursey TE. Voltage-gated proton channels. Compr Physiol 2012, 2: 1355–1385.
Gouaux E, MacKinnon R. Principles of selective ion transport in channels and pumps. Science 2005, 310: 1461–1465.
Koch HP, Kurokawa T, Okochi Y, Sasaki M, Okamura Y, Larsson HP. Multimeric nature of voltage-gated proton channels. Proc Natl Acad Sci U S A 2008, 105: 9111–9116.
Tombola F, Ulbrich MH, Isacoff EY. The voltage-gated proton channel Hv1 has two pores, each controlled by one voltage sensor. Neuron 2008, 58: 546–556.
Decoursey TE. Voltage-gated proton channels and other proton transfer pathways. Physiol Rev 2003, 83: 475–579.
Musset B, Smith SME, Rajan S, Morgan D, Cherny VV, Decoursey TE. Aspartate 112 is the selectivity filter of the human voltage-gated proton channel. Nature 2011, 480: 273–277.
Gonzalez C, Rebolledo S, Perez ME, Larsson HP. Molecular mechanism of voltage sensing in voltage-gated proton channels. J Gen Physiol 2013, 141: 275–285.
Sokolov VS, Cherny VV, Ayuyan AG, DeCoursey TE. Analysis of an electrostatic mechanism for ΔpH dependent gating of the voltage-gated proton channel, HV1, supports a contribution of protons to gating charge. Biochim Biophys Acta Bioenerg 2021, 1862: 148480.
Kuno M, Ando H, Morihata H, Sakai H, Mori H, Sawada M, et al. Temperature dependence of proton permeation through a voltage-gated proton channel. J Gen Physiol 2009, 134: 191–205.
Fujiwara Y, Kurokawa T, Takeshita K, Kobayashi M, Okochi Y, Nakagawa A, et al. The cytoplasmic coiled-coil mediates cooperative gating temperature sensitivity in the voltage-gated H(+) channel Hv1. Nat Commun 2012, 3: 816.
Kuno M, Kawawaki J, Nakamura F. A highly temperature-sensitive proton current in mouse bone marrow-derived mast cells. J Gen Physiol 1997, 109: 731–740.
DeCoursey TE, Cherny VV. Temperature dependence of voltage-gated H+ currents in human neutrophils, rat alveolar epithelial cells, and mammalian phagocytes. J Gen Physiol 1998, 112: 503–522.
Iovannisci D, Illek B, Fischer H. Function of the HVCN1 proton channel in airway epithelia and a naturally occurring mutation, M91T. J Gen Physiol 2010, 136: 35–46.
Musset B, Capasso M, Cherny VV, Morgan D, Bhamrah M, Dyer MJS, et al. Identification of Thr29 as a critical phosphorylation site that activates the human proton channel Hvcn1 in leukocytes. J Biol Chem 2010, 285: 5117–5121.
Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev 2007, 87: 245–313.
Okochi Y, Okamura Y. Regulation of neutrophil functions by Hv1/VSOP voltage-gated proton channels. Int J Mol Sci 2021, 22: 2620.
El Chemaly A, Okochi Y, Sasaki M, Arnaudeau S, Okamura Y, Demaurex N. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J Exp Med 2010, 207: 129–139.
Demaurex N, Grinstein S, Jaconi M, Schlegel W, Lew DP, Krause KH. Proton Currents in human granulocytes: Regulation by membrane potential and intracellular pH. J Physiol 1993, 466: 329–344.
Grinstein S, Furuya W. Characterization of the amiloride-sensitive Na+-H+ antiport of human neutrophils. Am J Physiol 1986, 250: C283–C291.
Simchowitz L. Chemotactic factor-induced activation of Na+/H+ exchange in human neutrophils. II. Intracellular pH changes. J Biol Chem 1985, 260: 13248–13255.
Grinstein S, Furuya W, Biggar WD. Cytoplasmic pH regulation in normal and abnormal neutrophils. Role of superoxide generation and Na+/H+ exchange. J Biol Chem 1986, 261: 512–514.
Morgan D, Capasso M, Musset B, Cherny VV, Ríos E, Dyer MJS, et al. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc Natl Acad Sci U S A 2009, 106: 18022–18027.
Henderson LM, Chappell JB, Jones OT. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem J 1987, 246: 325–329.
Seredenina T, Demaurex N, Krause KH. Voltage-gated proton channels as novel drug targets: From NADPH oxidase regulation to sperm biology. Antioxid Redox Signal 2015, 23: 490–513.
Zhu X, Mose E, Zimmermann N. Proton channel HVCN1 is required for effector functions of mouse eosinophils. BMC Immunol 2013, 14: 24.
Capasso M, Bhamrah MK, Henley T, Boyd RS, Langlais C, Cain K, et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat Immunol 2010, 11: 265–272.
Asuaje A, Smaldini P, Martín P, Enrique N, Orlowski A, Aiello EA, et al. The inhibition of voltage-gated H+ channel (HVCN1) induces acidification of leukemic Jurkat T cells promoting cell death by apoptosis. Pflugers Arch - Eur J Physiol 2017, 469: 251–261.
Alvear-Arias JJ, Carrillo C, Villar JP, Garcia-Betancourt R, Peña-Pichicoi A, Fernandez A, et al. Expression of Hv1 proton channels in myeloid-derived suppressor cells (MDSC) and its potential role in T cell regulation. Proc Natl Acad Sci U S A 2022, 119: e2104453119.
Kawai T, Tatsumi S, Kihara S, Sakimura K, Okamura Y. Mechanistic insight into the suppression of microglial ROS production by voltage-gated proton channels (VSOP/Hv1). Channels 2018, 12: 1–8.
Kawai T, Kayama K, Tatsumi S, Akter S, Miyawaki N, Okochi Y, et al. Regulation of hepatic oxidative stress by voltage-gated proton channels (Hv1/VSOP) in Kupffer cells and its potential relationship with glucose metabolism. FASEB J 2020, 34: 15805–15821.
Lishko PV, Kirichok Y, Ren D, Navarro B, Chung JJ, Clapham DE. The control of male fertility by spermatozoan ion channels. Annu Rev Physiol 2012, 74: 453–475.
Musset B, Clark RA, DeCoursey TE, Petheo GL, Geiszt M, Chen Y, et al. NOX5 in human spermatozoa: Expression, function, and regulation. J Biol Chem 2012, 287: 9376–9388.
Lishko PV, Botchkina IL, Fedorenko A, Kirichok Y. Acid extrusion from human spermatozoa is mediated by flagellar voltage-gated proton channel. Cell 2010, 140: 327–337.
Zhao R, Dai H, Arias RJ, De Blas GA, Orta G, Pavarotti MA, et al. Direct activation of the proton channel by albumin leads to human sperm capacitation and sustained release of inflammatory mediators by neutrophils. Nat Commun 2021, 12: 3855.
Zhao R, Kennedy K, De Blas GA, Orta G, Pavarotti MA, Arias RJ, et al. Role of human Hv1 channels in sperm capacitation and white blood cell respiratory burst established by a designed peptide inhibitor. Proc Natl Acad Sci U S A 2018, 115: E11847–E11856.
Yeste M, Llavanera M, Mateo-Otero Y, Catalán J, Bonet S, Pinart E. HVCN1 channels are relevant for the maintenance of sperm motility during in vitro capacitation of pig spermatozoa. Int J Mol Sci 2020, 21: 3255.
Fischer H, Widdicombe JH, Illek B. Acid secretion and proton conductance in human airway epithelium. Am J Physiol Cell Physiol 2002, 282: C736–C743.
Cho DY, Hajighasemi M, Hwang PH, Illek B, Fischer H. Proton secretion in freshly excised sinonasal mucosa from asthma and sinusitis patients. Am J Rhinol Allergy 2009, 23: e10–e13.
Fischer H. Function of proton channels in lung epithelia. Wiley Interdiscip Rev Membr Transp Signal 2012, 1: 247–258.
Du H, Pang H, Gao Y, Zhou Y, Li SJ. Deficiency of voltage-gated proton channel Hv1 aggravates ovalbumin-induced allergic lung asthma in mice. Int Immunopharmacol 2021, 96: 107640.
Fischer H, Widdicombe JH. Mechanisms of acid and base secretion by the airway epithelium. J Membrane Biol 2006, 211: 139–150.
Nordström T, Rotstein OD, Romanek R, Asotra S, Heersche JN, Manolson MF, et al. Regulation of cytoplasmic pH in osteoclasts. contribution of proton pumps and a proton-selective conductance. J Biol Chem 1995, 270: 2203–2212.
Musset B, Morgan D, Cherny VV, MacGlashan DW Jr, Thomas LL, Ríos E, et al. A pH-stabilizing role of voltage-gated proton channels in IgE-mediated activation of human basophils. Proc Natl Acad Sci U S A 2008, 105: 11020–11025.
Marone G, Columbo M, de Paulis A, Cirillo R, Giugliano R, Condorelli M. Physiological concentrations of zinc inhibit the release of histamine from human basophils and lung mast cells. Agents Actions 1986, 18: 103–106.
Levine AP, Duchen MR, de Villiers S, Rich PR, Segal AW. Alkalinity of neutrophil phagocytic vacuoles is modulated by HVCN1 and has consequences for myeloperoxidase activity. PLoS One 2015, 10: e0125906.
Montes-Cobos E, Huscher B, Engler JB, Woo MS, Binkle L, Bauer S, et al. Voltage-gated proton channel Hv1 controls TLR9 activation in plasmacytoid dendritic cells. J Immunol 2020, 205: 3001–3010.
Patel B, Zheleznova NN, Ray SC, Sun J, Cowley AW Jr, O’Connor PM. Voltage gated proton channels modulate mitochondrial reactive oxygen species production by complex I in renal medullary thick ascending limb. Redox Biol 2019, 27: 101191.
DeCoursey TE, Cherny VV, DeCoursey AG, Xu W, Thomas LL. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J Physiol 2001, 535: 767–781.
DeCoursey TE, Cherny VV, Zhou W, Thomas LL. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc Natl Acad Sci U S A 2000, 97: 6885–6889.
Okochi Y, Aratani Y, Adissu HA, Miyawaki N, Sasaki M, Suzuki K, et al. The voltage-gated proton channel Hv1/VSOP inhibits neutrophil granule release. J Leukoc Biol 2016, 99: 7–19.
Okochi Y, Umemoto E, Okamura Y. Hv1/VSOP regulates neutrophil directional migration and ERK activity by tuning ROS production. J Leukoc Biol 2020, 107: 819–831.
Schilling T, Gratopp A, DeCoursey TE, Eder C. Voltage-activated proton currents in human lymphocytes. J Physiol 2002, 545: 93–105.
Musset B, Cherny VV, DeCoursey TE. Strong glucose dependence of electron current in human monocytes. Am J Physiol Cell Physiol 2012, 302: C286–C295.
Kapus A, Romanek R, Qu AY, Rotstein OD, Grinstein S. A pH-sensitive and voltage-dependent proton conductance in the plasma membrane of macrophages. J Gen Physiol 1993, 102: 729–760.
Szteyn K, Yang W, Schmid E, Lang F, Shumilina E. Lipopolysaccharide-sensitive H+ current in dendritic cells. Am J Physiol Cell Physiol 2012, 303: C204–C212.
Eder C, DeCoursey TE. Voltage-gated proton channels in microglia. Prog Neurobiol 2001, 64: 277–305.
DeCoursey TE. The voltage-gated proton channel: A riddle, wrapped in a mystery, inside an Enigma. Biochemistry 2015, 54: 3250–3268.
He J, Ritzel RM, Wu J. Functions and mechanisms of the voltage-gated proton channel Hv1 in brain and spinal cord injury. Front Cell Neurosci 2021, 15: 662971.
Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature 2009, 457: 675–677.
Schmidt CE, Leach JB. Neural tissue engineering: Strategies for repair and regeneration. Annu Rev Biomed Eng 2003, 5: 293–347.
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization—New prospects for brain repair. Nat Rev Neurol 2015, 11: 56–64.
Morihata H, Nakamura F, Tsutada T, Kuno M. Potentiation of a voltage-gated proton current in acidosis-induced swelling of rat microglia. J Neurosci 2000, 20: 7220–7227.
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev 2011, 91: 461–553.
Zhang ZJ, Zheng XX, Zhang XY, Zhang Y, Huang BY, Luo T. Aging alters Hv1-mediated microglial polarization and enhances neuroinflammation after peripheral surgery. CNS Neurosci Ther 2020, 26: 374–384.
Eyo UB, Wu LJ. Bidirectional microglia-neuron communication in the healthy brain. Neural Plast 2013, 2013: 456857.
Schafer DP, Lehrman EK, Stevens B. The, “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 2013, 61: 24–36.
Wu LJ. Voltage-gated proton channel HV1 in microglia. Neuroscientist 2014, 20: 599–609.
Wu LJ, Sweet TB, Clapham DE. International Union of Basic and Clinical Pharmacology. LXXVI. Current progress in the mammalian TRP ion channel family. Pharmacol Rev 2010, 62: 381–404.
Zeng WZ, Liu DS, Liu L, She L, Wu LJ, Xu TL. Activation of acid-sensing ion channels by localized proton transient reveals their role in proton signaling. Sci Rep 2015, 5: 14125.
Wang F, Ma XR, Wu Y, Xu YC, Gu HM, Wang DX, et al. Neutralization of Hv1/HVCN1 with antibody enhances microglia/macrophages myelin clearance by promoting their migration in the brain. Front Cell Neurosci 2021, 15: 768059.
Wu LJ, Vadakkan KI, Zhuo M. ATP-induced chemotaxis of microglial processes requires P2Y receptor-activated initiation of outward potassium currents. Glia 2007, 55: 810–821.
Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat Rev Neurosci 2005, 6: 626–640.
Barreto GE, Gonzalez J, Torres Y, Morales L. Astrocytic-neuronal crosstalk: Implications for neuroprotection from brain injury. Neurosci Res 2011, 71: 107–113.
Yu Y, Yu Z, Xie M, Wang W, Luo X. Hv1 proton channel facilitates production of ROS and pro-inflammatory cytokines in microglia and enhances oligodendrocyte progenitor cells damage from oxygen-glucose deprivation in vitro. Biochem Biophys Res Commun 2018, 498: 1–8.
Tian L, Prabhakaran MP, Ramakrishna S. Strategies for regeneration of components of nervous system: Scaffolds, cells and biomolecules. Regen Biomater 2015, 2: 31–45.
Jones LL, Oudega M, Bunge MB, Tuszynski MH. Neurotrophic factors, cellular bridges and gene therapy for spinal cord injury. J Physiol 2001, 533: 83–89.
Yu X, Liu H, Hamel KA, Morvan MG, Yu S, Leff J, et al. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat Commun 2020, 11: 264.
Diarra A, Sheldon C, Brett CL, Baimbridge KG, Church J. Anoxia-evoked intracellular pH and Ca2+ concentration changes in cultured postnatal rat hippocampal neurons. Neuroscience 1999, 93: 1003–1016.
Cheng YM, Kelly T, Church J. Potential contribution of a voltage-activated proton conductance to acid extrusion from rat hippocampal neurons. Neuroscience 2008, 151: 1084–1098.
Marmarou A. Intracellular acidosis in human and experimental brain injury. J Neurotrauma 1992, 9: S551–S562.
Marmarou A, Holdaway R, Ward JD, Yoshida K, Choi SC, Muizelaar JP, et al. Traumatic brain tissue acidosis: Experimental and clinical studies. Acta Neurochir Suppl (Wien) 1993, 57: 160–164.
Clausen T, Khaldi A, Zauner A, Reinert M, Doppenberg E, Menzel M, et al. Cerebral acid-base homeostasis after severe traumatic brain injury. J Neurosurg 2005, 103: 597–607.
Venkatesh K, Ghosh SK, Mullick M, Manivasagam G, Sen D. Spinal cord injury: Pathophysiology, treatment strategies, associated challenges, and future implications. Cell Tissue Res 2019, 377: 125–151.
Kroner A, Rosas Almanza J. Role of microglia in spinal cord injury. Neurosci Lett 2019, 709: 134370.
Li X, Liu R, Yu Z, He D, Zong W, Wang M, et al. Microglial Hv1 exacerbates secondary damage after spinal cord injury in mice. Biochem Biophys Res Commun 2020: S0006–S291X(20)30272–2.
Zheng J, Murugan M, Wang L, Wu LJ. Microglial voltage-gated proton channel Hv1 in spinal cord injury. Neural Regen Res 2022, 17: 1183–1189.
Wiatrak B, Kubis-Kubiak A, Piwowar A, Barg E. PC12 cell line: Cell types, coating of culture vessels, differentiation and other culture conditions. Cells 2020, 9: 958.
Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 2003, 4: 399–415.
Nathan C, Ding A. SnapShot: Reactive oxygen intermediates (ROI). Cell 2010, 140: 951-951.e2.
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci 2007, 8: 57–69.
Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron 2010, 67: 181–198.
Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J Leukoc Biol 2010, 87: 779–789.
Bradl M, Linington C. Animal models of demyelination. Brain Pathol 1996, 6: 303–311.
DeCoursey TE, Cherny VV. Pharmacology of voltage-gated proton channels. Curr Pharm Des 2007, 13: 2400–2420.
Alabi AA, Bahamonde MI, Jung HJ, Kim JI, Swartz KJ. Portability of paddle motif function and pharmacology in voltage sensors. Nature 2007, 450: 370–375.
Hong L, Kim IH, Tombola F. Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc Natl Acad Sci U S A 2014, 111: 9971–9976.
Hong L, Pathak MM, Kim IH, Ta D, Tombola F. Voltage-sensing domain of voltage-gated proton channel Hv1 shares mechanism of block with pore domains. Neuron 2013, 77: 274–287.
Zhao R, Shen R, Dai H, Perozo E, Goldstein SAN. Molecular determinants of inhibition of the human proton channel hHv1 by the designer peptide C6 and a bivalent derivative. Proc Natl Acad Sci U S A 2022, 119: e2120750119.
Shin H, Song JH. Antipsychotics, chlorpromazine and haloperidol inhibit voltage-gated proton currents in BV2 microglial cells. Eur J Pharmacol 2014, 738: 256–262.
Shin H, Kim J, Song JH. Clozapine and olanzapine inhibit proton currents in BV2 microglial cells. Eur J Pharmacol 2015, 755: 74–79.
Kim J, Song JH. Thioxanthenes, chlorprothixene and flupentixol inhibit proton currents in BV2 microglial cells. Eur J Pharmacol 2016, 779: 31–37.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl Neurodegener 2020, 9: 42.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science 2016, 353: 777–783.
Capasso M. Regulation of immune responses by proton channels. Immunology 2014, 143: 131–137.
Wang Y, Li SJ, Pan J, Che Y, Yin J, Zhao Q. Specific expression of the human voltage-gated proton channel Hv1 in highly metastatic breast cancer cells, promotes tumor progression and metastasis. Biochem Biophys Res Commun 2011, 412: 353–359.
Wang Y, Li SJ, Wu X, Che Y, Li Q. Clinicopathological and biological significance of human voltage-gated proton channel Hv1 protein overexpression in breast cancer. J Biol Chem 2012, 287: 13877–13888.
Ventura C, Leon IE, Asuaje A, Martín P, Enrique N, Núñez M, et al. Differential expression of the long and truncated Hv1 isoforms in breast-cancer cells. J Cell Physiol 2020, 235: 8757–8767.
Bare DJ, Cherny VV, DeCoursey TE, Abukhdeir AM, Morgan D. Expression and function of voltage gated proton channels (Hv1) in MDA-MB-231 cells. PLoS One 2020, 15: e0227522.
Wang Y, Wu X, Li Q, Zhang S, Li SJ. Human voltage-gated proton channel hv1: A new potential biomarker for diagnosis and prognosis of colorectal cancer. PLoS One 2013, 8: e70550.
Wang Y, Zhang S, Li SJ. Zn2+ induces apoptosis in human highly metastatic SHG-44 glioma cells, through inhibiting activity of the voltage-gated proton channel Hv1. Biochem Biophys Res Commun 2013, 438: 312–317.
Ribeiro-Silva L, Queiroz FO, da Silva AMB, Hirata AE, Arcisio-Miranda M. Voltage-gated proton channel in human glioblastoma multiforme cells. ACS Chem Neurosci 2016, 7: 864–869.
Asuaje A, Martín P, Enrique N, Zegarra LAD, Smaldini P, Docena G, et al. Diphenhydramine inhibits voltage-gated proton channels (Hv1) and induces acidification in leukemic Jurkat T cells- New insights into the pro-apoptotic effects of antihistaminic drugs. Channels 2018, 12: 58–64.
Krysiak K, Gomez F. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood 2017, 129: 473–483.
Fernández A, Pupo A, Mena-Ulecia K, Gonzalez C. Pharmacological modulation of proton channel Hv1 in cancer therapy: Future perspectives. Mol Pharmacol 2016, 90: 385–402.
Cherny VV, DeCoursey TE. pH-dependent inhibition of voltage-gated H+ currents in rat alveolar epithelial cells by Zn2+ and other divalent cations. J Gen Physiol 1999, 114: 819–838.
Droste A, Chaves G, Stein S, Trzmiel A, Schweizer M, Karl H, et al. Zinc accelerates respiratory burst termination in human PMN. Redox Biol 2021, 47: 102133.
Acknowledgements
This review was supported by the National Natural Science Foundation of China (32071003, 82271249, and 81873808), and the 1-3-5 Project for Disciplines of Excellence of West China Hospital of Sichuan University (ZYJC21034).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shen, Y., Luo, Y., Liao, P. et al. Role of the Voltage-Gated Proton Channel Hv1 in Nervous Systems. Neurosci. Bull. 39, 1157–1172 (2023). https://doi.org/10.1007/s12264-023-01053-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-023-01053-6