
Abstract
Neuroinflammation is a key contributor to the pathogenic cascades induced by hypoxic-ischemic (HI) insult in the neonatal brain. AD-16 is a novel anti-inflammatory compound, recently found to exert potent inhibition of the lipopolysaccharide-induced production of pro-inflammatory and neurotoxic mediators. In this study, we evaluated the effect of AD-16 on primary astrocytes and neurons under oxygen-glucose deprivation (OGD) in vitro and in mice with neonatal HI brain injury in vivo. We demonstrated that AD-16 protected against OGD-induced astrocytic and neuronal cell injury. Single dose post-treatment with AD-16 (1 mg/kg) improved the neurobehavioral outcome and reduced the infarct volume with a therapeutic window of up to 6 h. Chronic administration reduced the mortality rate and preserved whole-brain morphology following neonatal HI. The in vitro and in vivo effects suggest that AD-16 offers promising therapeutic efficacy in attenuating the progression of HI brain injury and protecting against the associated mortality and morbidity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neonatal brain injury resulting from hypoxemia and reduced cerebral blood perfusion is a leading cause of mortality and neurological morbidity in children. Incidence estimates of hypoxic-ischemic (HI) brain injury range between ~3/1000 in term infants (>36 weeks gestational age), to ~7/1000 in preterm infants (33–35 weeks gestational age), with higher rates in developing countries [1,2,3]. The severity and duration of the HI insult determine the subsequent evolution of clinical HI encephalopathy (HIE). Accordingly, the pathophysiology of HI is not characterized by a single event but rather an evolving cascade of molecular mechanisms that culminate in cell injury and death [4, 5]. Specifically, depletion of high-energy substrates in the primary phase of injury leads to increased intracellular lactate, rapid depolarization, and the accumulation of synaptic glutamate. These molecular cascades cause excessive Ca2+ influx, cytotoxic edema, and necrosis. Reperfusion and a quick recovery of cerebral metabolism promote secondary energy failure characterized by excitotoxicity, inflammation, and oxidative stress that induce delayed neuronal death hours to weeks after the initial HI insult [1, 4, 5]. This secondary deterioration is thought to be the predominant contributor to brain injury and the main predictor of neurodevelopmental outcome at 4 years of age [6].
Therapeutic hypothermia, the current standard of care for treating HIE, reduces the risk of death and disability at 18–22 months [7]. Head or whole-body cooling below normothermia may suppress acute and chronic phases of the injury by exerting metabolic and immunomodulatory effects [8, 9]. Yet hypothermia has a strict therapeutic window, has been shown to be ineffective in severe HIE cases, and 40%–50% of treated cases are still fatal or result in neurological impairment [8, 10]. Thus, there is a need to identify novel therapeutic strategies that have a broad therapeutic window and target the underlying pathophysiology of HI to protect infants from progressive injury and long-term disability.
Neuroinflammation is a major contributor to many CNS-targeted diseases and injuries. HI insult induces robust pro-inflammatory cascades in response to innate immune receptor activation [1, 11]. Under physiological conditions, microglia play important neurodevelopmental roles in synaptic pruning and neurogenesis. HI conditions, however, may induce rapid classical/M1 activation and the production of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-8, and tumor necrosis factor (TNF)-α, all of which are upregulated in the cerebrospinal fluid of early postnatal infants with HIE, and correlated with injury severity and neurological outcome [12,13,14]. Significantly increased microglial infiltration in the hippocampal dentate gyrus has also been reported in post-mortem clinical studies of HIE infants <9 months of age [15], and a rat model of neonatal HI has shown microglial activation and upregulated pro-inflammatory gene expression within 2 h of the insult [16]. Microglial activation may also lead to matrix metalloproteinase release and breakdown of the blood-brain barrier, prompting peripheral leukocyte influx and exacerbating the inflammatory state. Further, activated astrocytes may aggravate injury via secretion of cytokines, chemokines, and reactive oxygen species [17]. Immunomodulatory interventions in rodent neonatal HI models such as administration of an IL-1 receptor antagonist or toll-like receptor-2 (TLR-2) KO, protects against myelin loss and functional deficits or injury-induced infarction, respectively [18, 19]. It is important to note that the early toxic effects of the microglial response are followed by activation of the M2 phenotype, which promotes anti-inflammatory and recovery signaling [12]. However, if acute neuroinflammation is not resolved, it may lead to chronic inflammation, which increases the susceptibility of the developing brain to injury. Thus, regulating the early-destructive and late-protective inflammatory responses to HI injury represents a promising direction for therapy.
AD-16 (GIBH-130) is a novel anti-inflammatory compound recently identified by phenotypic screening assays in an LPS-induced microglial line [20]. While its molecular target remains unknown, AD-16 was found to inhibit pro-inflammatory cytokine production in LPS-stimulated microglia, and protect two separate rodent models of Alzheimer’s disease from memory deficits and cognitive decline. Further, oral administration of AD-16 increases anti-inflammatory IL-4 and decreases pro-inflammatory IL-6 concentrations while preserving neuronal morphology in Aβ1-42-injected mice [20]. With a bioavailability of 75%, the ability to penetrate the blood-brain barrier (AUC Brain/Plasma = 0.21), and recent approval by the China Drug and Food Administration for clinical trials, AD-16 is a promising pharmacological agent to treat HI brain injury via targeting neuroinflammation. In the present study, we investigated the therapeutic efficacy of AD-16 on primary astrocytes and neurons under OGD and in neonatal mouse pups subjected to HI brain injury. We demonstrated for the first time that AD-16 treatment preserved cell viability in vitro and reduced infarction volume with a therapeutic window of up to 6 h in vivo. AD-16 promoted behavioral recovery, and chronic administration was associated with improved survival and whole-brain morphology 21 days post-HI. The molecular mechanisms underlying the neuroprotective effects of AD-16 are associated with the inhibition of apoptotic and pro-inflammatory signaling, and regulation of TREM2 (transmembrane receptor triggering receptor expressed on myeloid cells 2).
Materials and Methods
Animals
All applicable national and institutional guidelines for the care and use of animals were followed. All procedures and protocols were performed in accordance with the Canadian Council on Animal Care guidelines and approved by the University of Toronto Animal Care Committee. Timed-pregnant CD1 mice were purchased from Charles River Laboratories (Sherbrooke, Quebec, Canada) and housed at an ambient temperature of 20 ± 1°C and a 12-h light/dark cycle with the standard laboratory chow diet and water fed ad libitum. Mouse pups of either sex at postnatal day 7 (P7) were used in all experiments. All experiments were performed in a blinded manner.
Drug Administration
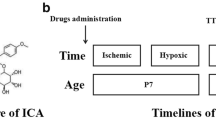
The chemical structure of AD-16 is shown in Fig. 1A (left) [20]. For in vivo experiments, AD-16 was dissolved in 2% DMSO and diluted in saline. P7 CD1 mouse pups were randomly assigned to either sham, vehicle, or AD-16 groups.

AD-16 preserves primary neuron and astrocyte viability after oxygen-glucose deprivation (OGD). A Chemical structure of AD-16 (left) and the experimental timeline of the in vitro OGD model (right). B Viability of primary astrocytes assessed 24 h after 90-min OGD. Viability is reduced by half compared to control (*P < 0.05, Ctrl: n = 5, OGD: n = 5). Compared to the vehicle-treated group, 10, 30, and 100 nmol/L AD-16 increase primary astrocyte viability after OGD (#P < 0.05, n = 5/group). C Viability of primary neurons assessed 24 h after 30-min OGD. Viability is reduced to ~68% of control. Compared to the vehicle-treated group, 1, 3, and 10 nmol/L AD-16 increases primary neuron viability (#P < 0.05, n = 3/group). All data are presented as the mean ± SEM; P < 0.05, one-way ANOVA followed by Tukey’s test for multiple comparisons.
To examine the acute effects, we first tested AD-16 along a pre-treatment timeline, where a single dose of AD-16 (0.1 mg/kg, 0.3 mg/kg, or 1 mg/kg) was administered intraperitoneally at 30 min prior to HI. For the post-treatment 1-, 3-, and 6-h timelines, a single dose of AD-16 (1 mg/kg) was administered subcutaneously at the stated time-point following HI. To assess the chronic effects, we administrated subcutaneous injections of AD-16 (1 mg/kg) every day for 21 days following HI. An equal volume of vehicle (saline with 2% DMSO) was administered for the vehicle group. The treatment timelines are illustrated in Fig. 2A.

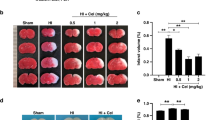
A single dose of AD-16 prior to or after the onset of hypoxia-ischemia (HI) reduces brain infarct volume in vivo. A Experimental timeline of in vivo neonatal mouse model of HI (7.5% O2, 60 min). B, C Representative 2,3,5-triphenyl tetrazolium chloride (TTC) images and summary of infarct volume assessed 24 h post-HI using the pre-treatment timeline. Administration of AD-16 (1 mg/kg) at 30 min prior to HI significantly reduces infarct volume. D–G Representative TTC images and summary of infarct volume assessed 24 h post-HI using the post-treatment timelines. Administration of 1 mg/kg AD-16 reduces infarct volume with a therapeutic window of up to 6 h post-injury. Numbers of mice tested are indicated in the bar graphs. All data are presented as the mean ± SEM; #P < 0.05, ##P < 0.01 vs vehicle; Student’s t test or one-way ANOVA followed by Tukey’s test for multiple comparisons. (A) created with BioRender.com
Neonatal Hypoxic-ischemic (HI) Brain Injury Model
The Rice Vannucci model of HI brain injury was applied with modifications. In brief, P7 CD1 mouse pups were randomly assigned to one of three groups: sham control, vehicle + HI, or AD-16 + HI. A fourth group, sham + AD-16, was included in subsequent experiments once the most efficacious dose of AD-16 had been determined. Pups were anesthetized with isoflurane (3.0% for induction and 1.5% for maintenance). The pup’s right common carotid artery was exposed and occluded using a bipolar electrical coagulation device (Vetroson). The duration of the surgery was controlled to <5 min. Body temperature was monitored and maintained using a heating pad (~37°C) throughout the surgical procedure. Pups were then returned to their dam with a heating pad and allowed to recover for 90 min. Pups were placed in an O2-deprived chamber (7.5% O2 balanced with 92.5% N2; Biospherix, USA) for 60 min. The temperature of the hypoxia chamber was maintained at 37°C with a homoeothermic blanket control unit. The right common carotid artery was exposed in the sham control group but not occluded; hypoxia was not induced.
Infarction Volume and Morphological Assessment
Brain infarction volume was assessed at 24 h post-surgery. Brains were removed, and 4 coronal brain sections were obtained for each animal. The sections were stained with 2,3,5-triphenyl tetrazolium chloride (TTC), a redox indicator that differentiates between metabolically active and inactive tissues. The infarct volume was quantified using ImageJ and calculated according to the following formula: Corrected infarct volume (%) = [contralateral hemisphere volume – (ipsilateral hemisphere volume – infarct volume)] / contralateral hemisphere volume × 100% [21].
Whole-brain tissue was collected and imaged after neurobehavioral tests on post-HI day 21 to assess the morphological changes. The ipsilateral liquefaction volume was quantified using ImageJ software and calculated according to the following formula: infarct volume (%) = infarct volume / contralateral hemisphere volume × 100% [21].
Neurobehavioral Assessment
Four neurobehavioral tests, the righting reflex, negative geotaxis, cliff avoidance, and grip test, were conducted on post-surgery day 1 as previously described [21]. These tests assess the sensorimotor functions of the animal at the early stages of development.
To assess the righting reflex, pups were placed in a supine position, and the time for them to turn over to a prone position with all 4 paws touching the surface was recorded. Negative geotaxis is an automatic, stimulus-bound orientation movement. Pups were placed head down in the middle of a plane inclined at a 45° angle and the latency to make a 180° turn was recorded. In the cliff avoidance test, pups were placed on the edge of a platform and the latency to avoid the edge, either by backing off or turning away, was recorded. These reflex tests were used to determine whether AD-16 treatment improves vestibular and proprioceptive function, as well as the integration of exteroceptive input and locomotor output. Finally, in the grip test, pups were suspended by their forepaws on a horizontal wire, and the latency to fall was recorded to indicate strength and fatigability.
The body weights and survival rates of the sham, vehicle, and AD-16 groups were also recorded daily until 21 days post-HI.
Cell Culture
Primary cultures of cortical neurons and astrocytes were made using P0 CD1 mice as described previously [22]. In brief, cortices were dissected and digested with 0.25% trypsin at 37°C for 30 min. Cell density was calculated using an Improved Neubauer hemocytometer, with viability >95%. Cells were then plated on 0.1 mg/mL poly-D-lysine-coated plates or dishes.
For primary cortical neurons, the cell medium was replaced with serum-free Neurobasal medium supplemented with 2% B27 and 1% Glutamax (Gibco) after incubation at 37°C for 4 h. Cells were cultured for 7 days in vitro (DIV) prior to OGD, with half of the medium changed every 3 days. One μmol/L cytarabine was added to the medium on DIV 3 to inhibit the growth of non-neuronal cells.
For primary astrocytes, the medium was replaced with DMEM with 10% FBS after incubation at 37°C for 24 h. Cells were cultured for 18–21 DIV prior to OGD, with the medium changed every 3 days. All cultures were maintained at 37°C in a humidified atmosphere of 5% CO2. One day prior to OGD, the cells were re-plated on poly-D-lysine-coated 96-well plates at a density of 1 × 104 per well.
In vitro Oxygen Glucose Deprivation (OGD)
The experimental timeline is shown schematically in Fig. 1A (right). OGD experiments were conducted at 7 DIV on primary cortical neurons and between 18 and 21 DIV on primary astrocytes, at which point cells had reached confluence and the maximum sensitivity to OGD-induced cell death [23]. In brief, AD-16 or vehicle (0.1% DMSO) was added to the culture medium prior to OGD induction. Various concentrations of AD-16 were used to determine the dose-response effects. The cells were incubated in glucose-free and FBS-free DMEM medium under anaerobic conditions (5% CO2 and 95% N2 at 37°C) with or without AD-16. The duration was set to 30 min for primary neurons and 90 min for primary astrocytes. The cells were subsequently incubated in an oxygenated, glucose-containing solution under normoxic conditions with or without AD-16 for an additional 24 h [14].
Cellular Viability Assay
According to the manufacturer’s protocols, the number of viable cells was assessed using Cell Counting Kit 8 (Beyotime, China). Briefly, 24 h after OGD, CCK8 solution (10 μL/100 μL culture medium) was added to each well. The plates were then placed in a humidified incubator (5% CO2, 37°C) for 3 h. Spectrophotometric absorbance, which is directly proportional to cell viability, was measured at a wavelength of 450 nm using a microplate reader.
Western Blot
Cells were harvested in RIPA buffer (50 mmol/L Tris, 150 mmol/L NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) with protease inhibitor cocktail (Sigma, USA) 24 h after OGD. Brain samples were collected on dry ice on post-HI day 1. Proteins from the ipsilateral hemisphere were extracted in RIPA buffer with a protease inhibitor cocktail (Santa Cruz Biotechnology, USA).
Protein concentrations were determined by a Pierce BCA Protein Assay Kit. Protein samples of equivalent amounts were separated on a 10% or 12% SDS-PAGE gel and electrophoretically transferred to nitrocellulose membranes (200 mA per gel, 60 min). Non-specific binding was blocked with 5% non-fat milk in PBS at pH 7.4. Membranes were then probed overnight at 4°C with specific primary antibodies against Bcl-2 (1:500), cleaved caspase-3 (1: 250), p-Akt (1:1000), Akt (1:1000), p-ERK (1:1000), ERK (1:1000), p-STAT-3 (1:500), STAT-3 (1:1000), NF-κB (1:500), p-NF-κB (1:300), IL-1β (1:500), myxovirus resistance MX dynamin-like GTPase 1 (MX1) (1:600), nuclear factor E2-related factor 2 (Nrf2) (1:500), and TREM2 (1:500). GAPDH (1:10,000) was used as a control for protein loading. The membranes were then washed and incubated with appropriate HRP-conjugated secondary anti-mouse or anti-rabbit IgG antibodies (1:7500, Chemicon) for 1 h at room temperature. Bands were detected with the ECL system (Perkin Elmer, USA), and protein expression was quantified by densitometry with ImageJ software and normalized to that of GAPDH.
Statistical Analysis
All statistical analyses were performed with GraphPad Prism 6 (GraphPad Software, CA, USA). Data are presented as means ± SEM. Student’s t-test was used to assess the statistical significance of the differences between vehicle- and AD-16-treated groups. One-way ANOVA with Tukey’s test was used for comparison between multiple groups. Statistical significance was defined as P <0.05.
Results
AD-16 Preserves Primary Astrocyte and Neuron Viability after OGD in vitro
We first tested whether AD-16 protects primary astrocytes and neurons from OGD-induced cell injury in vitro. As shown in the timeline in Fig. 1A (right), primary astrocytes and neurons were pre-treated with vehicle (0.1% DMSO) or AD-16 at various concentrations and subjected to OGD (5% CO2 and 95% N2, in glucose-free, FBS-free medium at 37°C). The duration of OGD was set to 90 min for astrocytes and 30 min for primary neurons, and cell viability was assessed 24 h post-OGD.
As shown in Fig. 1B, 90-min OGD reduced the viability of primary astrocytes by half (Ctrl: 0.98% ± 3.43%, n = 5; OGD + DMSO: 0.50% ± 2.83%, n = 5). Cell viability was significantly rescued to 0.64% ± 0.17%, 0.61% ± 1.90%, and 0.61% ± 1.78% with 10, 30, and 100 nmol/L AD-16, respectively (*P <0.05, one-way ANOVA followed by Tukey’s test for multiple comparisons, n = 5 per group). The viability of astrocytes was not significantly affected by either 3 or 300 nmol/L AD-16 (3 nmol/L: 0.57% ± 1.47%; 300 nmol/L: 0.50% ± 1.36%). Similar results were obtained in primary cortical neurons subjected to 30-min OGD (Fig. 1C). AD-16 at 1, 3, and 10 nmol/L significantly increased cell viability to 1.04% ± 8.49%, 1.22% ± 6.13%, and 1.07% ± 8.85%, respectively, compared to the vehicle-treated OGD neurons (0.68% ± 4.47%) (*P <0.05, one-way ANOVA followed by Tukey’s test for multiple comparisons, n = 3 per group). Furthermore, the 30 nmol/L-treated group showed an increasing trend of neuronal viability (P = 0.0678), while 100 nmol/L AD-16 did not affect neuronal viability following 30-min OGD (30 nmol/L: 0.98% ± 8.64%; 100 nmol/L: 0.78% ± 4.57%). Our in vitro results suggested that AD-16 at 10–100 nmol/L protects primary astrocytes and 1–10 nmol/L protects cortical neurons from OGD-induced injury.
A Single Dose of AD-16, Either Prior to or After HI Onset, Reduces Brain Infarction Volume in vivo
A single dose of AD-16 or vehicle (saline with 2% DMSO) was administered according to the timelines shown in Fig. 2A. AD-16 was first tested using the pre-treatment timeline, where three drug concentrations, 0.1, 0.3, and 1 mg/kg body weight, were administered 30 min prior to HI induction. Brain infarct volume was then assessed by TTC staining 24-h post-HI. Compared to the vehicle-treated group, the 0.1 and 0.3 mg/kg AD-16-treated groups showed a trend of reducing the corrected infarction volume (vehicle: 46.35% ± 3.33%, n = 12; 0.1 mg/kg: 33.85% ± 3.60%, n = 15, P = 0.187; 0.3 mg/kg: 32.55% ± 6.22%, n = 12, P = 0.155), while 1 mg/kg AD-16 resulted in a significantly reduced infarct volume in the ipsilateral hemisphere (1 mg/kg: 24.71% ± 4.47%, n = 11, P = 0.0102). Representative TTC staining of the brains of vehicle- and AD-16-pre-treated pups are shown in Fig. 2B, and the summary of infarction volume assessed 24 h after HI using the pre-treatment timeline is shown in Fig. 2C. As 1 mg/kg AD-16 had the most significant effect in reducing brain damage, this concentration was selected for our post-treatment timeline assessments.
The post-treatment 1-, 3-, and 6-h groups were administered either vehicle or AD-16 (1 mg/kg) at the stated time-point following HI. All AD-16-treated groups showed a significant reduction in corrected infarct volume 24 h after injury, compared with time-matched vehicle-treated groups (post-treatment 1 h vehicle: 47.05% ± 5.03%, n = 10, 1 mg/kg AD-16: 28.65% ± 3.53%, n = 10, P <0.05; post-treatment 3 h vehicle: 45.93% ± 3.44%, n = 10; 1 mg/kg AD-16: 28.96% ± 5.34%, n = 10, P <0.05; post-treatment 6 h vehicle: 41.00% ± 3.69%, n = 8; 1 mg/kg AD-16: 20.80% ± 4.59%, n = 7, P <0.05). Representative TTC staining of the brains of vehicle- and AD-16-post-treated pups are shown in Fig. 2D, and the summary of infarct volume assessed 24 h after HI using the post-treatment timelines are shown in Fig. 2E–G. These results demonstrated that AD-16 at 1 mg/kg has protective effects against HI-induced brain damage, with a therapeutic window up to 6 h post-injury.
A Single Dose of AD-16 after HI Onset Promotes Functional Recovery in Neonates
Short-term neurobehavioral tests were applied 24 h following HI insult to assess the effect of AD-16 on recovery outcomes. The frequently used assessments of reflex development (i.e. righting reflex, geotaxis reflex, cliff avoidance, and grip strength) were selected as abnormal early reflexes, and sensorimotor function may predict neurodevelopmental disability later in life. The neurobehavioral outcome was assessed in the post-treatment 1- and 3-h groups.
The post-treatment 1 h groups were administered either vehicle or AD-16 (1 mg/kg) 1 h post-HI, and behavior was assessed in vehicle-treated sham (n = 10), AD-16-treated sham (n = 10), vehicle-treated HI (n = 15), and AD-16-treated HI (n = 13) groups. Compared to the vehicle- and AD-16-treated sham groups, pups in the vehicle-treated HI group exhibited impaired functioning as measured by all tests (Fig. 3A–D). Specifically, vehicle-treated HI pups had a significantly longer righting reflex time compared to sham pups, and this was significantly improved by AD-16 treatment (sham: 0.79 ± 0.04 s; sham + AD-16: 0.73 ± 0.02 s; vehicle: 1.41 ± 0.13 s; AD-16: 0.93 ± 0.05 s; P <0.05). Vehicle-treated HI pups also exhibited a significantly longer geotaxis reflex time, as well as significantly reduced grip strength compared to the sham pups, while AD-16-treated pups showed a trend of improvement in both tests (geotaxis reflex: sham: 1.56 ± 0.05 s; sham + AD-16: 1.62 ± 0.10 s; vehicle: 2.23 ± 0.12 s; AD-16: 1.89 ± 0.10 s; P <0.05; grip strength: sham: 5.55 ± 0.53 s; sham + AD-16: 5.99 ± 0.74 s; vehicle: 3.63 ± 0.21 s; AD-16: 5.11 ± 0.48 s; P <0.05). The cliff-avoidance reflex was also significantly impaired in vehicle-treated pups compared to sham pups, yet was not affected by AD-16 treatment (sham: 1.56 ± 0.05 s; sham + AD-16: 1.17 ± 0.11 s; vehicle: 2.23 ± 0.12 s; AD-16: 1.89 ± 0.10 s). Among all neurobehavioral tests, no statistical differences were found between the sham and sham + AD-16 groups.

A single dose of AD-16 after the onset of hypoxia-ischemia (HI) promotes functional recovery in neonates. The righting reflex, negative geotaxis, cliff avoidance, and grip strength were assessed at 24 h following HI insult in the post-treatment 1- (A–D) and 3-h (E–H) timelines. Compared to sham and sham + AD-16 groups, HI significantly impairs the functional recovery of pups as measured by all four tests (*P <0.05). Compared to the vehicle-treated pups, those treated with 1 mg/kg AD-16 at 1-h post-HI show a significantly improved righting reflex. Compared to the vehicle-treated group, pups treated with 1 mg/kg AD-16 at 3-h post-HI show a significantly improved geotaxis reflex, cliff avoidance, and grip strength (#P <0.05). The numbers of animals tested are indicated in the bar graphs. All data are presented as the mean ± SEM; #,*P <0.05, Student’s t test or one-way ANOVA followed by Tukey’s test for multiple comparisons.
The functional outcome of the post-treatment 3 h groups administered either vehicle or AD-16 (1 mg/kg) 3 h post-HI, was assessed and compared between vehicle-treated HI (n = 16) and AD-16-treated HI (n = 16) pups (Fig. 3E–H). Compared to the vehicle-treated group, the AD-16-treated group exhibited a trend for reduced righting reflex latency (vehicle: 1.12 ± 0.13 s; AD-16: 0.89 ± 0.04 s; P = 0.111), while latency to complete the geotaxis reflex was significantly reduced (vehicle: 2.03 ± 0.14 s; AD-16: 1.64 ± 0.11 s; P <0.05). Furthermore, cliff-avoidance and grip strength were significantly improved in the AD-16-treated group compared to the vehicle-treated group (cliff avoidance: vehicle: 1.53 ± 0.11 s; AD-16: 1.264 ± 0.05 s; grip strength: vehicle: 3.62 ± 0.27 s; AD-16: 4.95 ± 0.40 s; P <0.05). These data suggest that 1 mg/kg AD-16 treatment improves the functional outcome and thereby promotes proper neurobehavioral development in neonates following HI, with a therapeutic window extending to 3 h post-injury.
Chronic Administration of AD-16 Improves Animal Survival Rate, General Well-being, and Whole-Brain Morphology 3 Weeks Post-HI Insult
To investigate its chronic effects, we repeatedly administered AD-16 (1 mg/kg) for 21 days following injury, with recovery parameters assessed every day in sham (n = 8), vehicle-treated HI (n = 11), and AD-16-treated HI (n = 17) mice. The fatality rate associated with neonatal rodent HI has been well-documented. In our study, the HI survival rate at the end of the 3-week assessment period was 0.647, most pups being lost at the 15-day mark (Fig. 4A-1 and -2). AD-16 treatment was found to improve the survival rate of HI pups to 0.944, with a survival percentage comparable to the sham group. The second recovery-related parameter measured was body weight – an indicator of general health and well-being. By day 21 post-HI, the mean body weight of the vehicle-treated HI group was lower than the sham group; however, as seen in Fig. 4B, AD-16-treated mice exhibited a trend toward a greater mean body weight every day over the 3-week recovery period compared to vehicle-treated mice (sham: 15.65 ± 1.21 g; vehicle: 12.97 ± 1.05 g; AD-16: 14.46 ± 1.1 g). No notable aversive side-effects were associated with chronic AD-16 administration over the 21-day recovery period. Finally, brains were extracted at the final time point to assess the potential effects of AD-16 on the whole brain morphological damage associated with HI. As seen in Fig. 4C and D, AD-16 significantly reduced the percentage liquefaction volume of the ipsilateral hemisphere compared to vehicle-treated animals (vehicle: 43.96% ± 3.35%, n = 8; AD-16: 32.79% ± 3.75%, n = 12, P <0.05). Together, these results indicate that chronic AD-16 administration preserves the survival, continued growth, and brain morphology of neonates exposed to HI, suggesting long-term recovery.

Chronic administration of AD-16 (1 mg/kg daily for 21 days) improves recovery parameters and preserves brain morphology 3 weeks after hypoxic-ischemic (HI) insult. Survival rates and body weights assessed daily until 21 days post-HI. Numbers of animals on the day of HI: sham (n = 8), vehicle-treated HI (n = 17), AD-16-treated HI (n = 18) and on post-HI day 21: sham (n = 8), vehicle-treated HI (n = 11) and AD-16-treated HI (n = 17). A, B Compared to the vehicle-treated pups, the AD-16-treated pups show an improved survival rate (A-1, A-2) and a trend of greater mean body weight (B). C–D Representative whole-brain images (C) and summary of whole-brain liquefaction volume (D) assessed at 3 weeks post-HI. AD-16-treated animals have a lower percentage liquefaction volume of the ipsilateral hemisphere than vehicle-treated animals. The numbers of animals tested are indicated in the bar graphs. All data are presented as the mean ± SEM; #P <0.05, Student’s t test or one-way ANOVA followed by Tukey’s test for multiple comparisons.
AD-16 Protects Against HI-Induced Brain Damage by Regulating Apoptosis, Neuronal Survival, and Neuroinflammation
To determine the potential molecular mechanism underlying the in vivo neuroprotection, we first assessed the effects of AD-16 on apoptotic and neuronal survival signaling 24 h after the HI insult. We found that HI brain injury significantly reduced the expression of Bcl-2, whereas mice pretreated with AD-16 (1mg/kg) exhibited a trend for Bcl-2 upregulation (Fig. 5A: Bcl-2/GAPDH normalized to sham group expression: sham + AD-16: 1.08 ± 0.08 (n = 5); vehicle + HI: 0.52 ± 0.17 (n = 3); HI + AD-16: 0.88 ± 0.13 (n = 3), P <0.05). Accordingly, the expression of cleaved caspase-3 was significantly increased after HI, while AD-16 pre-treatment showed a trend of restoring the expression level (Fig. 5B: cleaved caspase-3/GAPDH normalized to sham group expression: sham + AD-16: 1.13 ± 0.17 (n = 3); vehicle + HI: 39.69 ± 13.33 (n = 3); HI + AD-16: 13.0 ± 3.17 (n = 3), P <0.05).

Biochemical assessment of neuronal survival and apoptotic signaling at 24 h following hypoxia-ischemia (HI) using the pre-treatment timeline. Representative Western blot images and summary of protein expression of Bcl-2, cleaved caspase-3, pAkt, Akt, pERK, and ERK. GAPDH was used as a loading control. A HI significantly reduces the expression of Bcl-2 whereas the AD-16 pre-treated (1 mg/kg) group exhibits a trend of Bcl-2 upregulation (P = 0.164) compared to the vehicle-treated group. B HI significantly increases the expression of cleaved caspase-3, while the AD-16 pre-treated group shows a trend of restoring the level (p = 0.094). C, D Expression ratios of p-Akt/t-Akt (C) and p-ERK/t-ERK (D) are significantly decreased by HI, but again, the relative expression shows an increasing trend with AD-16 pre-treatment (P = 0.106 for p-Akt/t-Akt, and 0.079 for p-ERK/t-ERK). The numbers of animals tested are indicated in the bar graphs. All data are presented as the mean ± SEM; *P <0.05, **P <0.01 vs sham and sham + AD-16 groups, one-way ANOVA followed by Tukey’s test for multiple comparisons.
Next, we calculated the p-Akt/t-Akt and p-ERK/t-ERK expression ratios and found that while both were significantly reduced in the vehicle-treated group following HI insult, the relative expressions showed an increasing trend with AD-16 pre-treatment (Fig. 5C, D: p-Akt/t-Akt normalized to sham group expression: sham + AD-16: 1.16 ± 0.18 (n = 6); vehicle + HI: 0.55 ± 0.10 (n = 8); HI + AD-16: 0.89 ± 0.10 (n = 8), P <0.05; p-ERK/t-ERK normalized to sham group expression: sham + AD-16: 1.09 ± 0.14 (n = 5); vehicle + HI: 0.51 ± 0.10 (n = 10); HI + AD-16: 0.83 ± 0.12 (n = 10), P <0.05).
Finally, we sought to assess whether AD-16 exerted neuroprotection by regulating neuroinflammatory signaling. At 24 h post-HI, we found an increase in the expression of pro-inflammatory signaling proteins including significant upregulation of p-STAT3/STAT3 and p-NF-κB/NF-κB ratios, both of which were significantly reduced by AD-16 pre-treatment (Fig. 6A, B: p-STAT3/STAT3 normalized to sham group expression: sham + AD-16: 0.74 ± 0.21 (n = 7); vehicle + HI: 66.78 ± 21.88 (n = 9); HI + AD-16: 16.01 ± 5.68 (n = 9), P <0.05; p-NF-κB/NF-κB normalized to sham group expression: sham + AD-16: 0.71 ± 0.22 (n = 2); vehicle + HI: 3.05 ± 0.08 (n = 2); HI + AD-16: 0.05 ± 0.01 (n = 2), P <0.05). In addition, compared to the sham and sham + AD-16 groups, IL-1β expression exhibited a trend of upregulation in mice subjected to HI. However, no significant difference was found between the vehicle- and AD-16-treated HI groups (Fig. 6C: IL-1β/GAPDH normalized to sham group expression: sham + AD-16: 1.05 ± 0.17 (n = 2); vehicle + HI: 13.84 ± 8.24 (n = 3); HI + AD-16: 10.29 ± 6.40 (n = 3).

Biochemical assessment of neuroinflammatory signaling at 24 h following hypoxic-ischemic (HI) insult using the pre-treatment timeline. Representative Western blot images and summary of protein expression of pSTAT3, STAT3, p-NF-κB, NF-κB, IL-1β, MX1, Nrf2, and TREM2. GAPDH was used as a loading control. A, B Relative expression of p-STAT3/STAT3 (A) and p-NF-κB/NF-κB (B) are significantly upregulated after HI, and these levels are significantly reduced with AD-16 pre-treatment. C IL-1β expression exhibits a trend of upregulation after HI, however, there was no significant difference between the vehicle- and AD-16-treated groups. D MX1 expression is significantly downregulated after HI and significantly restored with AD-16 pre-treatment. E Nrf2 expression is also significantly downregulated after HI, however it is not mitigated by AD-16. F TREM2 upregulation only occurs in the vehicle-treated group following HI, with no protein detected in the sham, sham + AD-16 or AD-16 + HI groups. The numbers of animals tested are indicated in the bar graphs. All data are presented as the mean ± SEM; *P <0.05 vs sham and sham + AD-16 groups and #P <0.05 vs vehicle + HI group, one-way ANOVA followed by Tukey’s test for multiple comparisons.
MX1, Nrf2, and TREM2 have recently been proposed to mediate neuroinflammation after acute ischemic stroke, however their involvement in neonatal HI brain injury had yet to be demonstrated [24,25,26,27,28,29]. Interestingly, we found that the expression of MX1 was significantly downregulated following HI, but was restored with AD-16 pre-treatment (Fig. 6D: MX1/GAPDH normalized to sham group expression: sham + AD-16: 1.15 ± 0.17 (n = 3); vehicle + HI: 0.20 ± 0.04 (n = 6); HI + AD-16: 0.66 ± 0.06 (n = 6), P <0.05). HI insult also induced a significant downregulation of Nrf2 expression, with levels unchanged by AD-16 (Fig. 6E: Nrf2/GAPDH normalized to sham group expression: sham + AD-16: 0.93 ± 0.08 (n = 4); vehicle + HI: 0.32 ± 0.02 (n = 3); HI + AD-16: 0.42 ± 0.05 (n = 3), P <0.05). Finally, TREM2 upregulation only occurred in the vehicle-treated group following HI, with no protein detected in the sham, sham + AD-16, or HI + AD-16 groups (Fig. 6F: TREM2/GAPDH normalized to sham group expression: vehicle + HI: 6.85 ± 1.76 (n = 2); P <0.05).
Discussion
In a neonatal mouse model of HI brain injury, we evaluated a novel, potent, anti-neuroinflammatory compound, AD-16. Using both in vitro and in vivo approaches, we demonstrated that (1) AD-16 reduced OGD-induced injury in both primary astrocytes and cortical neurons; (2) pre-treatment with AD-16 reduced infarction volume 24 h post-HI; (3) repeat administration of AD-16 reduced the mortality rate while preserving whole-brain morphology at 21 days post-HI; (4) post-treatment with AD-16 reduced infarct volume with a therapeutic window of up to 6 h, and improved neurobehavioral outcomes following HI; and (5) AD-16 attenuated brain injury potentially through the regulation of neuronal survival, apoptosis, and neuroinflammatory signaling.
The pyridazine derivative AD-16 (GIBH-130) was first identified through microglia-based phenotypic screening and shown to potently inhibit the LPS-induced production of pro-inflammatory and neurotoxic mediators, including NO, TNF-α, and especially IL-1β (IC50 = 3.4 nmol/L) [20]. The in vivo therapeutic effects of AD-16 have been evaluated in the β-amyloid-induced and APP/PS1 transgenic mouse models of AD. In both cases, AD-16 significantly reduced pro-inflammatory cytokines in a dose-dependent manner and improved cognitive and functional recovery; surprisingly, comparable memory restoration effects at approximately one-50th doses of the currently available anti-AD drugs, donepezil and memantine, were reported with AD-16 treatment. Its anti-neuroinflammatory effects were further confirmed in vivo using the LPS-induced IL-1β-Luc transgenic mouse model [30]. A similar compound, AD-110, has also shown considerable neuroprotection against acute ischemic injury by enhancing the anti-inflammatory cytokine IL-10 in a rat transient middle cerebral artery occlusion (MCAO) model [31]. Together, these studies suggest that pyridazine derivatives have potential in vivo anti-neuroinflammatory effects in both AD and stroke models.
In the present study, we sought to determine whether the outlined neuroprotective effects of AD-16 were translatable to a model of neonatal HI, first through the use of a proof-of-principle pre-treatment paradigm. Of the three doses tested, that of 1 mg/kg conferred the greatest reduction in infarct volume at 24 h post-HI, and was thus selected for further in vivo testing. Clinically, the HI-related pathology HIE has a high mortality rate, ranging from 10% to 60% depending on the severity. In our study, daily administration of AD-16 at 1 mg/kg reduced the mortality of mice over 3 weeks following HI, with no notable side-effects. Further, we provided evidence that AD-16 treatment either before or after the injury onset reduced brain injury and improved functional recovery with a therapeutic window of up to 6 h. Clinically, the onset of HI insults can rarely be precisely determined, and treatment is limited to mild hypothermia [4, 32,33,34]. However, hypothermia has a strict therapeutic window and is most effective when initiated immediately after moderate HI, after which point effectiveness decreases linearly with time [8, 35]. Accordingly, studies have shown that asphyxiated newborns treated with hypothermia within 3 h of birth have significantly improved neurodevelopmental outcomes compared to those who are treated between 3 and 6 h [34]. Given the limitations of hypothermia, neuroprotective agents with a broader therapeutic window, such as AD-16, are of great clinical relevance. These findings, together, demonstrate that AD-16 is a promising treatment for HI and its associated injury in neonates.
Next, we investigated whether the molecular mechanisms underlying the protective effects of AD-16 were related to the regulation of caspase-mediated cell death and mitochondria-dependent apoptotic pathways. Caspase-3 cleavage and activation following HI injury are prominent in the developing brain. The pro-survival, anti-apoptotic Bcl-2 protein also controls mitochondrial outer-membrane permeabilization and participates in HI-induced injury [36,37,38]. In agreement with our previously published data, we showed that HI reduces Bcl-2 while upregulating cleaved caspase-3. Pre-treatment with AD-16, however, resulted in a trend for rescued Bcl-2 expression and attenuated cleaved caspase-3 at 24 h post-HI, suggesting potential regulation of apoptotic signaling by AD-16. In addition, the PI3K/Akt and MAPK/ERK signaling cascades have been closely implicated in neuroprotection in ischemic brain injury with Akt and ERK phosphorylation associated with neuronal survival [39]. In accordance with the literature, we found significant downregulation of the p-Akt/Akt and p-ERK/ERK ratios following HI insult, while increasing relative expressions were found with AD-16 treatment. These results demonstrate that activation of the pro-survival PI3K/Akt and MAPK/ERK signaling may contribute to AD-16-mediated neuroprotection.
As noted, the robust neuroinflammatory response to HI plays a substantial role in injury progression, with transcriptional and translational upregulation of various pro-inflammatory cytokines, particularly IL-1β, occurring as early as 3 h after HI in neonatal rats [40]. Consistent with previous findings, we found significant activation of the transcription factor NF-κB (p65 subunit), a central regulator of inflammation known to regulate >500 inflammation-related genes [41, 42], as well as the release of the pro-inflammatory cytokine IL-1β. Activation of JAK2/STAT3 by IL-6 has also been implicated in the post-ischemic inflammatory response, while inhibition of STAT3 phosphorylation or cell-specific deletion of STAT3 has been shown to reduce inflammation and tissue loss post-HI [43]. Accordingly, we found a significant upregulation of the p-STAT3/STAT3 ratio in HI-induced mice. Treatment with AD-16, however, attenuated both the p-NF-κB/NF-κB and p-STAT3/STAT3 ratios, highlighting its anti-inflammatory effects.
In recent years, TREM2, a single-pass transmembrane receptor of the immunoglobulin-like superfamily, has been identified and proposed to be involved in microglia-mediated neuroinflammation. TREM2 is a shared gene in both AD and ischemic stroke and is thought to mediate the phagocytic clearance of apoptotic/necrotic cells in brain pathologies, suggesting its role is likely protective [44]. TREM2 expression is upregulated in experimental models of ischemic stroke, peaking at 7 days post-injury, and TREM2 deficiency is associated with exacerbated injury and a reduction in microglial phagocytotic activity [25, 26, 45]. While TREM2 overexpression suppresses the inflammatory response and neuronal apoptosis, silencing TREM2 by siRNA increases infarction volume and apoptosis [26]. The proposed mechanism is through the suppression of TLR4/NF-κB signaling as well as activating the PI3K/Akt pathway [45, 46]. Although one controversial study has shown an attenuated inflammatory response in TREM2-KO mice following stroke, no difference in infarction volume was found between WT and TREM2-KO mice. These findings were perhaps due to the reperfusion and increased involvement of macrophages in the transient MCAO model, leading to more infiltration of blood-borne immune cells [27].
Despite being increasingly studied in adult ischemic stroke, the involvement of TREM2 had yet to be demonstrated in neonatal models. Here, we report for the first time that TREM2 is involved in neonatal HI, with significantly upregulated expression in the ischemic penumbra as early as 24 h post-injury. The upregulation of TREM2, however, was associated with an increased infarct volume and neuroinflammation in the vehicle-treated group, while the lack of expression in AD-16-treated mice correlated with neuroprotective effects. The conflicting roles of TREM2 in adult ischemic stroke and our model of neonatal HI brain injury may be attributed to its developmental profile; TREM2 has been proposed to be required for synaptic pruning, specifically the microglial instruction of astrocytic engulfment during neurodevelopment [47]. With TREM2-dependent synaptic loss persisting until 1 month in mice, the expression, function, and regulation of TREM2 in the P7 pups used in this study could differ from that of mature animals. Furthermore, a limitation in this study is that the TREM2 level was only assessed at 24 h after HI insult – whether TREM2 participates in resolution and repair at later time points after the injury remains to be elucidated.
In ischemic injury, dynamic phenotypic polarization of microglia to the classic M1 and the alternative M2 phenotypes has often been reported. M1 microglia have toxic effects on the surrounding neurons and glia by releasing pro-inflammatory cytokines, proteases, and reactive oxygen species, while M2 microglia release neurotrophic factors and anti-inflammatory cytokines and are involved in clearance and tissue repair [48]. M2 is rapidly activated in adult ischemic stroke and gradually transforms into M1 in peri-infarct regions [48, 49]. In neonatal HI brain injury, however, both M1 and M2 have been shown to be rapidly activated as early as 24 h after the insult, suggesting that the time course of microglia polarization after an ischemic insult is also development-dependent [50]. Similarly, astrocytes also undergo polarization and adopt a neurotoxic A1 and a protective A2 phenotype following ischemic insults [51]. Liddelow et al. showed that activated microglia strongly induce A1 astrocytes, MX1 being an A1-specific marker and Nrf2 an A2 marker [24]. Accordingly, activation and nuclear translocation of Nrf2 has been suggested to protect from secondary injury following ischemic stroke by modulating the neuroinflammatory response [28, 47].
As A1 astrocytes contribute to neuronal death, we postulated that A1-specific markers would be upregulated after HI but surprisingly found a significant downregulation of MX1 24 h post-HI injury. AD-16 treatment reversed these effects while also reducing the neuroinflammatory response. Our result is consistent with a previous study demonstrating that astrocytic MX1 upregulation via IFN-I/AHR signaling suppresses CNS inflammation [29]. As the transcriptional and translational levels of MX1 did not change in astrocytes after OGD in vitro (Fig. S1), the downregulation we found in vivo is unlikely to be mediated by astrocytes alone, but rather complex cell-cell interactions. Similar to MX1, we found downregulated Nrf2 expression after HI, yet there was no difference between the vehicle- and AD-16 treated groups. However, as Nrf2 alone is not a specific marker of A2 astrocytes, further studies are required to investigate the pathophysiological profile of A2-specific polarization in neonatal HI. RNA microarray and proteomic analysis may be considered in the future to identify the specific molecular targets and mechanism of AD-16.
Nevertheless, our results suggest that the anti-neuroinflammatory effects of AD-16 are potentially mediated through the regulation of MX1-dependent astrocyte polarization. However, further studies are required to delineate the contribution of astrocytic polarization and molecular regulation in neonatal brain injury. Moreover, this study provides evidence of the neuroprotective effects of AD-16 in a mouse model of HI brain injury, with a therapeutic window of up to 6 h. AD-16 treatment reduced brain infarct volume, improved the neurobehavioral outcome, and reduced the mortality rate in vivo. Neuroprotection is potentially mediated via the inhibition of apoptotic and neuroinflammatory signaling, and the activation of neuronal survival. Given the pressing need for effective clinical intervention for neonatal HI brain injury, AD-16 is a promising pharmacological agent for the prevention and treatment of HI and its related brain disorders.
References
Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS. The role of inflammation in perinatal brain injury. Nat Rev Neurol 2015, 11: 192–208.
Chalak LF, Rollins N, Morriss MC, Brion LP, Heyne R, Sánchez PJ. Perinatal acidosis and hypoxic-ischemic encephalopathy in preterm infants of 33 to 35 weeks’ gestation. J Pediatr 2012, 160: 388–394.
Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 2010, 86: 329–338.
Davidson JO, Wassink G, van den Heuij LG, Bennet L, Gunn AJ. Therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy - Where to from here? Front Neurol 2015, 6: 198.
Li B, Concepcion K, Meng X, Zhang L. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog Neurobiol 2017, 159: 50–68.
Azzopardi D, Wyatt JS, Cady EB, Delpy DT, Baudin J, Stewart AL, et al. Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy. Pediatr Res 1989, 25: 445–451.
Shankaran S, Pappas A, McDonald SA, Vohr BR, Hintz SR, Yolton K, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 2012, 366: 2085–2092.
Wood T, Osredkar D, Puchades M, Maes E, Falck M, Flatebø T, et al. Treatment temperature and insult severity influence the neuroprotective effects of therapeutic hypothermia. Sci Rep 2016, 6: 23430.
Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci 2012, 13: 267–278.
Edwards AD, Brocklehurst P, Gunn AJ, Halliday H, Juszczak E, Levene M, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 2010, 340: c363.
Li B, Dasgupta C, Huang L, Meng XM, Zhang LB. MiRNA-210 induces microglial activation and regulates microglia-mediated neuroinflammation in neonatal hypoxic-ischemic encephalopathy. Cell Mol Immunol 2020, 17: 976–991.
Liu FD, McCullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin 2013, 34: 1121–1130.
Martín-Ancel A, García-Alix A, Pascual-Salcedo D, Cabañas F, Valcarce M, Quero J. Interleukin-6 in the cerebrospinal fluid after perinatal asphyxia is related to early and late neurological manifestations. Pediatrics 1997, 100: 789–794.
Saliba E, Henrot A. Inflammatory mediators and neonatal brain damage. Biol Neonate 2001, 79: 224–227.
Del Bigio MR, Becker LE. Microglial aggregation in the dentate gyrus: A marker of mild hypoxic-ischaemic brain insult in human infants. Neuropathol Appl Neurobiol 1994, 20: 144–151.
Chen CY, Sun WZ, Kang KH, Chou HC, Tsao PN, Hsieh WS, et al. Hypoxic preconditioning suppresses glial activation and neuroinflammation in neonatal brain insults. Mediators Inflamm 2015, 2015: 632592.
Rocha-Ferreira E, Hristova M. Antimicrobial peptides and complement in neonatal hypoxia-ischemia induced brain damage. Front Immunol 2015, 6: 56.
Girard S, Sébire H, Brochu ME, Briota S, Sarret P, Sébire G. Postnatal administration of IL-1Ra exerts neuroprotective effects following perinatal inflammation and/or hypoxic-ischemic injuries. Brain Behav Immun 2012, 26: 1331–1339.
Stridh L, Smith PL, Naylor AS, Wang X, Mallard C. Regulation of toll-like receptor 1 and -2 in neonatal mice brains after hypoxia-ischemia. J Neuroinflammation 2011, 8: 45.
Zhou W, Zhong GF, Fu SH, Xie H, Chi TY, Li LY, et al. Microglia-based phenotypic screening identifies a novel inhibitor of neuroinflammation effective in Alzheimer’s disease models. ACS Chem Neurosci 2016, 7: 1499–1507.
Sun HS, Xu BF, Chen WL, Xiao AJ, Turlova E, Alibraham A, et al. Neuronal K(ATP) channels mediate hypoxic preconditioning and reduce subsequent neonatal hypoxic-ischemic brain injury. Exp Neurol 2015, 263: 161–171.
Turlova E, Bae CYJ, Deurloo M, Chen WL, Barszczyk A, Horgen FD, et al. TRPM7 regulates axonal outgrowth and maturation of primary hippocampal neurons. Mol Neurobiol 2016, 53: 595–610.
Juurlink BH, Hertz L, Yager JY. Astrocyte maturation and susceptibility to ischaemia or substrate deprivation. Neuroreport 1992, 3: 1135–1137.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541: 481–487.
Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci 2015, 35: 3384–3396.
Wu R, Li X, Xu P, Huang L, Cheng J, Huang X, et al. TREM2 protects against cerebral ischemia/reperfusion injury. Mol Brain 2017, 10: 20.
Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz-Gerlach B, Neumann H, et al. Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock-out mice following stroke. PLoS One 2013, 8: e52982. https://doi.org/10.1371/journal.pone.0052982.
Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, et al. The flavanol (-)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab 2010, 30: 1951–1961.
Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 2016, 22: 586–597.
Sun P, Yue H, Xing Q, Deng W, Ou Y, Pan G, et al. Compound AD16 reduces amyloid plaque deposition and modifies microglia in a transgenic mouse model of Alzheimer’s disease. ACS Pharmacol Transl Sci 2020, 3: 1100–1110.
Sun P, Zhou W, Yue H, Zhang C, Ou YT, Yang ZJ, et al. Compound AD110 Acts as therapeutic management for Alzheimer’s disease and stroke in mouse and rat models. ACS Chem Neurosci 2020, 11: 929–938.
Soul JS, Robertson RL, Tzika AA, du Plessis AJ, Volpe JJ. Time course of changes in diffusion-weighted magnetic resonance imaging in a case of neonatal encephalopathy with defined onset and duration of hypoxic-ischemic insult. Pediatrics 2001, 108: 1211–1214.
Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 2005, 353: 1574–1584.
Thoresen M, Tooley J, Liu X, Jary S, Fleming P, Luyt K, et al. Time is brain: Starting therapeutic hypothermia within three hours after birth improves motor outcome in asphyxiated newborns. Neonatology 2013, 104: 228–233.
Sabir H, Scull-Brown E, Liu X, Thoresen M. Immediate hypothermia is not neuroprotective after severe hypoxia-ischemia and is deleterious when delayed by 12 hours in neonatal rats. Stroke 2012, 43: 3364–3370.
Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008, 9: 47–59.
Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science 1998, 281: 1322–1326.
Zhu C, Hallin U, Ozaki Y, Grandér R, Gatzinsky K, Bahr BA, et al. Nuclear translocation and calpain-dependent reduction of Bcl-2 after neonatal cerebral hypoxia-ischemia. Brain Behav Immun 2010, 24: 822–830.
Xiong T, Tang J, Zhao J, Chen H, Zhao F, Li J, et al. Involvement of the Akt/GSK-3β/CRMP-2 pathway in axonal injury after hypoxic-ischemic brain damage in neonatal rat. Neuroscience 2012, 216: 123–132.
Hagberg H, Gilland E, Bona E, Hanson LA, Hahin-Zoric M, Blennow M, et al. Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatr Res 1996, 40: 603–609.
Shih RH, Wang CY, Yang CM. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci 2015, 8: 77.
Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell 2004, 6: 203–208.
Hristova M, Rocha-Ferreira E, Fontana X, Thei L, Buckle R, Christou M, et al. Inhibition of signal transducer and activator of transcription 3 (STAT3) reduces neonatal hypoxic-ischaemic brain damage. J Neurochem 2016, 136: 981–994.
Wei CJ, Cui P, Li H, Lang WJ, Liu GY, Ma XF. Shared genes between Alzheimer’s disease and ischemic stroke. CNS Neurosci Ther 2019, 25: 855–864.
Rosciszewski G, Cadena V, Murta V, Lukin J, Villarreal A, Roger T, et al. Toll-like receptor 4 (TLR4) and triggering receptor expressed on myeloid cells-2 (TREM-2) activation balance astrocyte polarization into a proinflammatory phenotype. Mol Neurobiol 2018, 55: 3875–3888.
Liu AH, Chu M, Wang YP. Up-regulation of Trem2 inhibits hippocampal neuronal apoptosis and alleviates oxidative stress in epilepsy via the PI3K/Akt pathway in mice. Neurosci Bull 2019, 35: 471–485.
Jay TR, von Saucken VE, Muñoz B, Codocedo JF, Atwood BK, Lamb BT, et al. TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 2019, 67: 1873–1892.
Hu XM, Li PY, Guo YL, Wang HY, Leak RK, Chen S, et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43: 3063–3070.
Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci 2017, 18: E2135.
Hellström Erkenstam N, Smith PL, Fleiss B, Nair S, Svedin P, Wang W, et al. Temporal characterization of microglia/macrophage phenotypes in a mouse model of neonatal hypoxic-ischemic brain injury. Front Cell Neurosci 2016, 10: 286.
Wang YZ, Tian D, Zhao YS, Qu MY, Pan Y, Wei CW, et al. Propofol protects regulatory T cells, suppresses neurotoxic astrogliosis, and potentiates neurological recovery after ischemic stroke. Neurosci Bull 2021, 37: 725–728.
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR PJT-153155) to ZPF and a Natural Sciences and Engineering Research Council of Canada Discovery Grant (NSERC RGPIN-2016-04574) to HSS.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, Z., Luo, Z., Ovcjak, A. et al. AD-16 Protects Against Hypoxic-Ischemic Brain Injury by Inhibiting Neuroinflammation. Neurosci. Bull. 38, 857–870 (2022). https://doi.org/10.1007/s12264-021-00816-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-021-00816-3