
Abstract
Epilepsy is a chronic and severe neurological disorder that has negative effects on the autonomous activities of patients. Functionally, Trem2 (triggering receptor expressed on myeloid cells-2) is an immunoglobulin receptor that affects neurological and psychiatric genetic diseases. Based on this rationale, we aimed to assess the potential role of Trem2 integration with the PI3K/Akt pathway in epilepsy. We used microarray-based gene expression profiling to identify epilepsy-related differentially-expressed genes. In a mouse hippocampal neuron model of epilepsy, neurons were treated with low-Mg2+ extracellular fluid, and the protein and mRNA expression of Trem2 were determined. Using a gain-of-function approach with Trem2, neuronal apoptosis and its related factors were assessed by flow cytometry, RT-qPCR, and Western blot analysis. In a pilocarpine-induced epileptic mouse model, the malondialdehyde (MDA) and 8-hydroxy-2′-deoxyguanosine (8-OHdG) content and superoxide dismutase (SOD) and glutathione-peroxidase (GSH-Px) activity in the hippocampus were determined, and the protein expression of Trem2 was measured. In addition, the regulatory effect of Trem2 on the PI3K/Akt pathway was analyzed by inhibiting this pathway in both the cell and mouse models of epilepsy. Trem2 was found to occupy a core position and was correlated with epilepsy. Trem2 was decreased in the hippocampus of epileptic mice and epileptic hippocampal neurons. Of crucial importance, overexpression of Trem2 activated the PI3K/Akt pathway to inhibit neuronal apoptosis. Moreover, activation of the PI3K/Akt pathway through over-expression of Trem2 alleviated oxidative stress, as shown by the increased expression of SOD and GSH-Px and the decreased expression of MDA and 8-OHdG. The current study defines the potential role of Trem2 in inhibiting the development of epilepsy, indicating that Trem2 up-regulation alleviates hippocampal neuronal injury and oxidative stress, and inhibits neuronal apoptosis in epilepsy by activating the PI3K/Akt pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy is a chronic disorder of the brain featuring epileptic seizures caused by a continuous tendency for, or triggered by, overstimulation and super-synchrony of neurons [1, 2]. The latest statistics state that ~ 65 million people worldwide are suffering from epilepsy. The daily life of patients is badly affected by the loss of autonomous activities, and patients are commonly discriminated against or misunderstood [3]. Furthermore, epilepsy has high incidence and prevalence rates. Although some antiepileptic drugs are capable of controlling seizures, the limited supply and quality of these drugs are obstacles to effective treatment [4]. Although antiepileptic drugs are considered to be the fundamental therapeutic method [5], seizures still occur in > 20% of patients after optimal drug treatment. The alleviation of seizures can presumably be achieved by gene therapy targeted to the epileptogenic zone, allowing modification of the neuronal activity at the seizure focus without excision of the area or pathway transection [6]. Accordingly, it is essential to find effective genetic markers to improve the therapeutic effect for patients suffering from epilepsy.
Trem2 (triggering receptor expressed in myeloid cells-2) is an immune receptor and functions as a trigger of phagocytosis and a modulator of the inflammatory response in microglia [7]. The pathogenic role of Trem2 mutations in Alzheimer’s disease (a typical neurodegenerative disease characterized by chronic inflammatory responses) has been demonstrated in several studies [8, 9]. In addition, Trem2 depletion in mouse microglia results in low phagocytic activity which impairs the clearance of apoptotic neurons, while Trem2 elevation inhibits inflammatory responses [10]. Moreover, loss of Trem2 in mice may contribute to delay of neurological recovery and exacerbation of ischemic damage [11]. The phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway has been reported to be crucially involved in human cancers [12]. Strikingly, activation of this pathway assists the regeneration of adult central neurons [13]. The relationship between Trem2 and the PI3K/Akt pathway has been investigated, demonstrating that a Trem2-mediated anti-bacterial effect through activation of the PI3K/Akt pathway facilitates the intracellular killing of Pseudomonas aeruginosa by macrophages [14]. So, we hypothesized that Trem2 may inhibit neuronal apoptosis and injury and alleviate oxidative stress in epilepsy via the PI3K/Akt pathway. We therefore investigated the physiological role of Trem2 during epilepsy both in vivo and in vitro.
Materials and Methods
Ethics Statement
The current study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal experiments were approved by the Institutional Animal Care Committee of Xuanwu Hospital, Capital Medical University. The experimental procedures were performed in accordance with the “three Rs” principle (reduction, replacement, and refinement) for minimal suffering of the animals.
Microarray-Based Gene Expression Profiling of Epilepsy
Epilepsy-related chip data were searched on the GEO database (https://www.ncbi.nlm.nih.gov/geo/). Differential analysis of data from patients with epilepsy and healthy controls was conducted using the LIMMA package of the R language. A |logFC (fold changes)| value > 2 and a P value < 0.05 were the screening criteria for differentially-expressed genes (DEGs). The ‘pheatmap’ package was used to establish the heatmap of DEGs.
Gene Ontology (GO) Analysis
The WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) (http://www.webgestalt.org/option.php), a web tool for gene functional enrichment analysis, was used for GO analysis of the epilepsy-related DEGs.
Gene-Gene Interaction Analysis
The gene-gene interactions of epilepsy-related DEGs were analyzed using the STRING database (https://string-db.org/) and an interaction network was constructed using Cytoscape (http://js.cytoscape.org/).
Screening of Trem2-Related Pathways
Trem2-related pathway information was obtained using the multifaceted pathway database, WikiPathways (https://www.wikipathways.org/index.php/WikiPathways) [15].
A Hippocampal Neuron Model of Epilepsy
Mouse hippocampal neurons (Shanghai Kang Lang Biotech Co., Ltd, Shanghai, China) were cultured in neuronal culture medium containing 98% Neurobasal medium (Gibco, Carlsbad, CA), 2% B27 supplement (Gibco), 0.2 mol/L L-glutamine, 1 × 105 U/L penicillin and 1 × 105 U/L streptomycin at 37 °C under 5% CO2. The medium was discarded when establishing the epilepsy model, and the neurons were rinsed in low-Mg2+ extracellular fluid (ECF) containing (in mmo1/L) 145 NaCl, 12.5 KCl, 10 HEPES (pH 7.2), 2 CaCl2, 10 glucose, and 0.002 glycine, and stored at 4 °C until use. The hippocampal neuron model of epilepsy was constructed using low-Mg2+ ECF culture [16]. The neurons were cultured in the low-Mg2+ ECF (2 mL per well) at 37 °C under 5% CO2. After 3 h, the neurons were rinsed twice with neuronal culture medium, and then cultured in neuronal culture medium at 2 mL per well. Normally-cultured hippocampal neurons were used as controls. Whole-cell voltage-clamp recordings were performed using an extracellular patch clamp (EPC)-10 system (HEKA Electronik, Lambrecht, Germany). The recording electrode was controlled by a microelectrode control instrument (Axon, Molecular Devices, LLC, CA). As expected, hippocampal neurons lacking Mg2+ for 3 h generated spontaneous recurrent epileptiform discharges and showed the same electropherogram characteristics as human epilepsy [17].
Cell Transfection
Hippocampal neurons from mice were assigned into a normal group (normal neurons), a blank group (epileptic neurons without transfection), a pcDNA3 group (epileptic neurons transfected with pcDNA3 vector), a pcDNA3-Trem2 group (epileptic neurons transfected with pcDNA3-Trem2 plasmid), an LY294002 group (epileptic neurons treated with the PI3K/Akt pathway inhibitor LY294002 at 36 μmol/L for 36 h), and an LY294002 + pcDNA3-Trem2 group (epileptic neurons treated with LY294002 after transfection with pcDNA3-Trem2 plasmid). All plasmids used for transfection were synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China).
To a 1.5 μL sterile Eppendorf (EP) tube were added 1.5 μg packaged mixed plasmids, 0.5 μg expression plasmids, and 250 μL serum-free medium, which were gently mixed and kept at room temperature for 5 min. To another 1.5 μL sterile EP tube were added Liposome 2000 in 250 μL serum-free medium (9:1), and these were gently mixed and kept at room temperature for 5 min. The DNA and liposome solutions were mixed gently and incubated at room temperature for 20 min. Then, 293T cells were treated with trypsin, counted, and re-suspended in serum-containing medium. A total of 1 mL serum-containing medium per well was added to the six-well plate, and supplemented with the DNA-liposome mixture. Next, 1 mL of the re-suspended 293T cells (1 × 106 cells/mL) were added to the plate and incubated overnight at 37 °C in a CO2 incubator. Then the culture medium was replaced. The viral supernatant was harvested 48 h–72 h after transfection, centrifuged at 3000 rpm for 20 min, and then the precipitate was removed.
Twenty-four hours prior to transfection, mouse hippocampal neurons were inoculated in a six-well plate. Transfection was conducted when cells reached 80%–90% confluence.
A Mouse Model of Epilepsy
Fifty clean adult male ICR mice (6 weeks old, weighing 25 g–30 g) were provided by Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The mice were fed in separate cages under 12 h light/dark cycles at 22 °C–25 °C. Stereotaxic injection was performed as previously described [19]. The mice were assigned to three groups: the epilepsy (EPI) + Ad-Trem2 group (injected with Trem2 over-expressing lentiviral vector), the EPI + Ad-NC group (injected with negative control lentiviral vector), and the EPI + Ad-Trem2 + LY294002 group (simultaneously injected with Ad-Trem2 and 5 μL LY294002). The lentiviral vectors were from Shanghai Genechem Co., Ltd. (Shanghai, China). Mice in each group were anesthetized by intraperitoneal injection of 2.5% sodium pentobarbital at 10 mg/kg. After mice exhibited even breathing and signs of anesthesia, they were fixed on a stereotaxic frame. The cranial skin was disinfected with 75% alcohol, and a 0.5 cm incision was made, after which the skin was sterilized with 0.05% hydrogen peroxide, and finally the bregma was exposed. Based on the mouse brain atlas position (– 2.18, – 2.6, – 2.5) (PMID: 21275977), the cranium was opened with a cranial drill and 2 holes were drilled, each of 0.8 mm in diameter. The vectors were then injected into each side using a 2 μL microsyringe at 0.5 μL/min, and the needle was left in place for 6 min. The scalp was sutured, the mice were kept warm, and returned to their cages when they could act autonomously. The mice were further maintained for two weeks, after which models were constructed [19]. After stereotaxic injections with lentiviral vectors, the mice were pretreated with intraperitoneal injection of 127 mg/kg lithium chloride. After 20 h, 30 mg/kg pilocarpine was injected intraperitoneally (1 mg/kg scopolamine hydrobromide was injected subcutaneously 30 min before pilocarpine injection to antagonize the peripheral cholinergic responses induced by pilocarpine). In the normal group, physiological saline was injected instead of pilocarpine 20 h after intraperitoneal injection of lithium chloride. Behavioral changes were observed, and the seizure activity was assessed on the basis of Racine’s stages [21]. Mice graded level IV or V were used as models. If some mice were excluded, the corresponding number of mice was added to balance the sample size of each group. After mice were in status epilepticus for 60 min, 10 mg/kg diazepam was injected intraperitoneally to terminate the seizures. Twenty-four hours after the onset of epilepsy, the mice were anesthetized with 2.5% sodium pentobarbital, sacrificed by decapitation, and the brain rapidly removed. The bilateral hippocampi were quickly isolated on ice. The fresh hippocampal tissue was stored at – 80 °C until assessment of malondialdehyde (MDA), superoxide dismutase (SOD), glutathione-peroxidase (GSH-Px), and determination of DNA 8-hydroxy-2′-deoxyguanosine (8-OHdG). At the same time, some of the hippocampal tissue was dehydrated to wax for 23 h and embedded in paraffin for hematoxylin-eosin (HE) staining and terminal deoxynucleotidyl transferase (TDT)-mediated dUTP-biotin nick end-labeling (TUNEL) staining.
Observation of Animal Behavior
The seizure activity of epileptic mice was graded into 6 levels based on Racine’s stages: 0, no seizure; I, facial clonus, including rhythmic chewing, blinking, and stereotypical mouthing; II, nodding-dominated neck muscle twitching; III, unilateral forelimb myoclonic jerks, convulsions, but no hindlimb standing; IV, convulsions in bilateral forelimbs and body rearing; V, comprehensive rigidity, clonic seizures, bilateral hindlimb rigidity, body dorsiflexion, and falling. Kindling-induced epilepsy in mice was assessed as follows: mice exhibiting seizures of levels IV–V after three consecutive intraperitoneal injections of pilocarpine, indicative of a successful kindling model of epilepsy, were used for subsequent experiments. Latent period was defined as the time from pilocarpine injection to the first occurrence of level IV–V seizures. Seizure period was defined as the time from the onset of level IV–V seizures within 30 min after pilocarpine injection until the termination of seizures. The data from all mice that died during the experiment were excluded.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA from hippocampal neurons and tissue was extracted using a TRIzol kit (Invitrogen Inc., Carlsbad, CA). cDNA was synthesized using a reverse transcription system which was prepared according to the instructions with the TRIzol kit. The reaction mixture (25 μL) was as follows: 2 × Super Real Premix 12.5 μL, 2 μL cDNA template, 0.75 μL forward primer (10 μmol/L), 0.75 μL reverse primer (10 μmol/L), and 9 μL RNase-free ddH2O. The conditions consisted of pre-denaturation at 94 °C for 15 min, and 40 cycles of denaturation at 94 °C for 10 s, and annealing and extension at 66 °C for 30 s. The melting curve was plotted after reaction. The β-actin gene served as an internal reference. The relative mRNA expression of the target gene was calculated using the 2−ΔΔCt method. Experiments were repeated three times in an independent manner. The primer sequences are listed in Supplementary Table S1.
Western Blot Analysis
Total protein of hippocampal neurons and tissue was extracted using a protein extraction kit (Bestbio, Shanghai, China). The protein concentration of each sample was determined using a bicinchoninic acid kit (23225, Pierce, Rockford, IL), and adjusted to the same concentration with deionized water. Next, 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel (P0012A, Beyotime Biotechnology Co., Shanghai, China) was prepared. To each lane were added 50 μg protein samples, and 2-h electrophoresis was conducted at 80 V and 120 V. Subsequently, the proteins were transferred onto a polyvinylidene fluoride membrane (ISEQ00010, Millipore, Billerica, MA) using the wet transfer method at a constant current of 250 mA for 90 min. The membrane was blocked for 2 h with Tris-buffered saline Tween-20 (TBST) containing 5% skim milk powder. After the blocking fluid was discarded, the membrane was rinsed with TBST. After that, the membrane was incubated overnight at 4 °C with the primary antibodies (all from Abcam Inc., Cambridge, UK): rabbit anti-Trem2 (dilution ratio 1:500, ab86491), rabbit anti-p-PI3K (1:1000, ab40776), rabbit anti-PI3K (1:1000, ab127617), rabbit anti-p-Akt (1:500, ab38449), rabbit anti-Akt (1:500, ab8805), rabbit anti-Bcl-2 (1:500, ab692), rabbit anti-Bax (1:1000, ab32503), rabbit anti-caspase3 (1:500, ab13847), and rabbit anti-β-actin (1:500, ab8226). Then, the membrane was rinsed three times with TBST (10 min each), and incubated with the horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG (1:2000, ab6721, Abcam) at room temperature for 1 h. Then, the membrane was rinsed three times with PBST (10 min each). Next, the membrane was developed with enhanced chemiluminescent solution (WBKLS0100, Millipore), exposed in the dark, and visualized. β-actin served as an internal reference, and the ratio of the gray value of the target band to the internal reference band was considered to be the relative expression of the protein.
Assessment of Oxidative Stress
The MDA content and SOD and GSH-Px activity were determined in accordance with the instructions with each of the detection kits (all from Nanjing Jiancheng Bioengineering Institute, Nanjing, China). The MDA content was determined using the thiobarbituric acid colorimetric method, SOD activity was assessed using the WST-1 method and GSH-Px activity using the UV colorimetric assay.
Enzyme-Linked Immunoassay (ELISA)
The total DNA of hippocampal neurons or hippocampal tissue was extracted according to the instructions with the TRIzol kit (Qiagen Co., Hilden, Germany). The 8-OHdG content in the DNA of hippocampal neurons was determined using the 8-OHdG ELISA kit (Japan Institute for Control of Aging, Shizuoka, Japan) which has high sensitivity. Finally, the optical density (OD) was measured at 450 nm, and each experiment was repeated three times. The results were calculated based on a linear calibration curve plotted from 8-OHdG standard solutions. The 8-OHdG content is expressed as picograms per microgram of DNA.
Flow Cytometry
Following the instructions with the Annexin V apoptosis detection kit (Beijing Biosea Biotechnology Co., Ltd., Beijing, China), hippocampal neurons were treated with 0.01, 0.10, and 1.0 nmol/L lidazyme for 48 h and collected after centrifugation. Then, the neurons were rinsed 2–3 times with PBS and centrifuged at 120 × g at 4 °C for 10 min. After the supernatant was discarded, another 150 μL of binding buffer was added for cell resuspension. Finally, 10 μL Annexin V-fluorescein isothiocyanate and 5 μL propidium iodide were gently mixed, and added to the neurons at room temperature without light exposure. Afterwards, a further 150 μL of binding buffer was added, and a flow cytometer (Coulter Electronics, Hialeah, FL) was used to measure the fluorescence intensity. The data were processed using FACScan software, and the result was expressed as the apoptosis rate.
TUNEL Staining
Apoptotic hippocampal tissue was labeled according to the procedure with the in situ apoptosis detection kit. Next, the tissue was cut into sections, which were routinely dewaxed to water, and immediately placed in a beaker with 200 mL citrate buffer (0.1 mol/L, pH 6.0) pre-heated to 90 °C–95 °C. Then, the sections were irradiated using a 680 W microwave (80% power) for 1 min, rapidly cooled in 80 mL double-distilled water (20 °C–25 °C), moved into phosphate buffer (20 °C–25 °C), and rinsed three times with phosphate buffer (5 min each). Next, the sections were incubated with 20% normal bovine serum at room temperature for 30 min. Subsequently, they were treated with 50 μL TUNEL reaction mixture, and incubated at 37 °C for 90 min (negative control sections were incubated without the TUNEL reaction mixture). Then the sections were rinsed three times with phosphate buffer (5 min each), blocked in 3% H2O2/methanol solution at room temperature for 10 min, and further incubated at 37 °C for 90 min, followed by another incubation at 37 °C for 90 min with the addition of HRP solution (50 μL/section). After rinsing three times with phosphate buffer (5 min each), the sections were stained with diaminobenzidine (DAB)/H2O2. Finally, the sections were lightly counter-stained with hematoxylin, dehydrated, cleared, and sealed with neutral gum. Next, 2 sections from each mouse were observed under a microscope (× 400). A total of 5 individual fields on each section were randomly observed in the CA1 and CA3 regions. The number of TUNEL-stained cells per unit area was calculated (positive cells with nuclei stained dark brown).
HE Staining and Immunohistochemical Assay
Serial coronal sections (10 μm) were cut on a microtome, floated in a warm water bath, and then placed on clean slides coated with gelatin-chrome alum. The sections were baked in an oven for 2 h and then stored at 4 °C for later use. Two of every 5 sections were taken to form one set of 8–10 sections, and were kept for HE staining. The morphology and distribution of neurons in the hippocampal CA1 and CA3 regions of stained sections were observed under an optical microscope. Hippocampal tissue was obtained from normal and acute epileptic mice, and 4-μm paraffin sections were prepared, routinely dewaxed, and hydrated, followed by microwave repair. After endogenous enzymes were inactivated with H2O2 and blocked in normal goat serum, the sections were incubated with Trem2 antibody (1:100, Cell Signaling Technology Inc., Danvers, MA) at 4 °C for 24 h. Then they were incubated with HRP-labeled goat anti-rabbit IgG antibody (1:200, KPL, Gaithersburg, MD) for 50 min at room temperature. The sections were rinsed three times with PBS (pH 7.4) (5 min each) before incubation with the above reagents. Then, the sections were developed with DAB (Dako Denmark A/S, Glostrup, Denmark); the positive samples exhibited brown-yellow coloration. Next, the sections were lightly counterstained with hematoxylin, routinely dehydrated, cleared, and mounted. The primary antibody was replaced with normal goat serum as a negative control. The integrated OD values of 10 cortical fields per group were measured under a microscope (400 ×) using Image-Pro Plus software (Version 6.0, Olympus, Tokyo, Japan). The criteria were based on the literature [21]. Cells with brown-yellow protein were evaluated as positive. The protein-positive cells were assessed in 5 randomly-selected high-power fields (200 ×) on each section, with 100 cells in each field. First, staining intensity was scored as follows (1–4 points): 1, weak positive staining; 2, moderate positive staining; 3, strong positive staining; 4, strongest positive staining. Then, the percentage of positive cells among the total cells was scored as follows (0–4 points): 0, negative cells; 1, ≤ 25% positive cells, 2, 25%–50% positive cells; 3, 51%–75% positive cells; and 4, > 75% positive cells. Finally, the sum of the two scores was used to identify protein-positive cells as follows: 0, negative (–); 1–4, weak positive (+); and 5–8, strong positive (++). Weak positive cells were classified as negative.
Statistical Analysis
The SPSS 21.0 software package (IBM Corp. Armonk, NY) was used for statistical analysis. Data are displayed as mean ± standard deviation. Enumeration data were analyzed using the χ2 test, and comparisons between two groups were analyzed using the independent t-test. Multiple groups were compared with one-way analysis of variance (ANOVA). A value of P < 0.05 was considered to be statistically significant.
Results
Trem2 has Potential to Protect Against Epilepsy Through the PI3K/Akt Pathway
The data from GSE77578 chip were obtained from the GEO database, and 67 DEGs were obtained after differential analysis of the epilepsy and control samples. Subsequently, a heatmap of the top 50 DEGs was constructed (Fig. 1A). The 67 DEGs were mainly implicated in “biological regulation”, “membrane”, and “protein binding” according to the results of GO functional analysis (Fig. 1B). This finding indicated that biological processes are closely associated with the occurrence and development of epilepsy. Next, gene-gene interactions of the top 50 DEGs were analyzed (Fig. 1C), and this revealed that Trem2 and Tyrobp were in core positions. Interestingly, previous studies have proposed that Trem2 plays an important role in epilepsy [22, 23], and the PI3K/Akt pathway has also been reported to be a crucial regulator in epilepsy [24,25,26]. Moreover, several studies have demonstrated a target relationship between Trem2 and the PI3K/Akt pathway [14, 27], indicating that Trem2 could modulate the PI3K/Akt pathway as shown in Fig. 1D. The above results indicate that Trem2 has the potential to affect the progression of epilepsy by regulating the PI3K/Akt pathway.

Epilepsy chip GSE77578 was obtained, and 67 DEGs were associated with epilepsy by microarray-based gene expression profiling. A Heatmap of the top 50 epilepsy-related DEGs (abscissa, sample number; ordinate, genes). The upper tree diagram shows the clustering of sample types, and the left tree diagram shows the clustering of gene expression. Each small square represents the expression of one gene in one sample. Upper right, color scale. B GO analysis of these DEGs showing that they were mainly implicated in “biological regulation”, “membrane”, and “protein binding”. The three histograms represent three clustering results for biological processes, cellular components, and molecular function (abscissa, GO items; ordinate, number of genes). C Gene-gene interactions of epilepsy-related DEGs analyzed using the STRING database. Each circle represents a gene and lines between circles indicate direct or indirect interactions between two genes. D Trem2-related pathway information analyzed using WikiPathways, suggestive of a relationship between Trem2 and the PI3K/AKT pathway. DEGs, differentially-expressed genes; GO, Gene Ontology; Trem2, triggering receptor expressed on myeloid cells 2.
Down-Regulated Protein Expression of Trem2 in Epileptic Hippocampal Tissue
Immunohistochemistry was used to determine the protein expression of Trem2 in epileptic hippocampal tissue. The protein expression rate of Trem2 was 76.71% in the normal group, but 15.49% in the EPI group (P < 0.05, Fig. 2). These results demonstrated that the protein expression of Trem2 is down-regulated in epileptic hippocampal tissue.

Immunohistochemistry showed that the protein expression of Trem2 was down-regulated in hippocampal tissue from epileptic (EPI) mice. A Trem2-positive cells (brown) in hippocampal tissue from mice in the normal and EPI groups (scale bars, 50 μm). B Trem2 protein expression was lower in EPI hippocampal tissue (*P < 0.05 vs normal group, independent t-test.
Trem2 Elevation Attenuates Apoptosis of Hippocampal Neurons by Activating the PI3K/Akt Pathway
We used RT-qPCR and Western blot to investigate the function of Trem2 in the epileptic hippocampal neuron model, and to determine its mRNA and protein expression as well as PI3K/Akt pathway-related genes. We found that the mRNA and protein expression of Trem2, as well as the extent of PI3K and Akt phosphorylation, were significantly lower in the EPI group than in the normal group (Fig. 3A–C), while no significant differences were found between the normal and EPI groups for PI3K and Akt expression. As for the epileptic hippocampal neurons with Trem2 over-expression or LY294002, Trem2 was significantly higher in the pcDNA3-Trem2 and LY294002 + pcDNA3-Trem2 groups, but lower in the pcDNA3 and LY294002 groups than in the normal group. Compared to the blank group, the levels of p-PI3K/PI3K and p-Akt/Akt were significantly higher in the pcDNA3-Trem2 group but lower in the LY294002 group, with no differences in Trem2 expression. Moreover, compared to the LY294002 group, the LY294002 + pcDNA3-Trem2 group demonstrated increased expression of Trem2, p-PI3K/PI3K, and p-Akt/Akt (P < 0.05). All these data demonstrated the role played by Trem2 elevation to activate the PI3K/Akt pathway in epileptic hippocampal neurons. In addition, the expression of apoptosis-related genes revealed its effect in epileptic hippocampal neurons. Compared to the blank group, markedly higher mRNA and protein expression of Bcl-2 but lower Bax and caspase3 were found in the pcDNA3-Trem2 group (P < 0.05), while the LY294002 group presented opposite results (P < 0.05). In comparison with the LY294002 group, the mRNA and protein expression of Bcl-2 was increased, in addition to the decreased Bax and caspase3 in the LY294002 + pcDNA3-Trem2 group (Fig. 4, P < 0.05). All these findings revealed that Trem2 elevation suppresses the apoptosis of hippocampal neurons by activating the PI3K/Akt pathway.

Trem2 up-regulation activated the PI3K/Akt pathway. A RT-qPCR determination of the mRNA expression of Trem2 revealed that Trem2 was lower in the EPI group. B–C Protein expression of Trem2, p-PI3K/PI3K, and p-Akt/Akt determined by Western blot suggesting that Trem2 upregulation activates the PI3K/Akt pathway (*P < 0.05 vs normal group, t-test; each experiment was repeated three times).

Trem2 up-regulation inhibited hippocampal neuronal apoptosis in epilepsy. A mRNA expression of Trem2, Bcl-2, Bax, and caspase3 determined by RT-qPCR, suggesting that Trem2 upregulation inhibits hippocampal neuronal apoptosis. B–C Protein expression of Trem2, Bcl-2, Bax, and caspase3 and the extent of PI3K and Akt phosphorylation determined by Western blot, indicating that Trem2 upregulation and PI3K/Akt pathway activation inhibit hippocampal neuronal apoptosis (mean ± standard deviation; each experiment repeated three times; &P < 0.05 vs normal group, *P < 0.05 vs blank group, #P < 0.05 vs LY294002 group, one-way ANOVA).
Trem2 Up-Regulation Alleviates Oxidative Stress and Inhibits Hippocampal Neuronal Apoptosis
Flow cytometry was used to determine hippocampal neuronal apoptosis (Fig. 5A, B). In comparison with the normal and pcDNA3-Trem2 groups, significantly higher neuronal apoptosis rates were found in the other four groups. Compared to the LY294002 group, the blank, pcDNA3, and pcDNA3-Trem2 + LY294002 groups exhibited lower neuronal apoptosis rates (P < 0.05). There were no significant differences between the normal and pcDNA3-Trem2 groups, as well as no evident differences among the blank, pcDNA3, and pcDNA3-Trem2 + LY294002 groups. These data revealed that neuronal apoptosis is inhibited by up-regulating Trem2, which leads to activation of the PI3K/Akt pathway.

Oxidative stress and hippocampal neuronal apoptosis were alleviated by up-regulation of Trem2. A Flow cytometry scatter plot showing that neuronal apoptosis was inhibited by Trem2 elevation. B The neuronal apoptosis rate was decreased by Trem2 upregulation. C SOD activity was increased by Trem2 elevation (WST-1 method). D MDA content was decreased by Trem2 upregulation (thiobarbituric acid colorimetry). E GSH-Px activity was increased by Trem2 elevation (UV colorimetry). F 8-OHdG content was decreased by Trem2 elevation (ELISA). *P < 0.05 vs EPI group, #P < 0.05 vs LY294002 group, one-way ANOVA. EPI, epileptic hippocampal neurons in mice. The experiments were repeated three times.
In addition, we measured the levels of the oxidative stress-related factors MDA (by thiobarbituric acid colorimetry), SOD (using WST-1), and GSH-Px (by UV colorimetry) in hippocampal neurons. Compared with the normal and pcDNA3-Trem2 groups, the activity of SOD and GSH-Px was significantly lower in the blank, pcDNA3, pcDNA3-Trem2 + LY294002 and LY294002 groups, but the MDA level was higher in these groups (Fig. 5C–E). Compared with the LY294002 group, SOD and GSH-Px activity was higher in the blank, pcDNA3, pcDNA3-Trem2 + LY294002, and LY294002 groups, while the MDA level was lower; there were no significant differences between the normal and pcDNA3-Trem2 groups, as well as among the blank, pcDNA3, and pcDNA3-Trem2 + LY294002 groups. The expression of DNA 8-OHdG in neurons from each group was determined using ELISA (Fig. 5F). Compared with the normal and pcDNA3-Trem2 groups, the 8-OHdG content was significantly higher in the blank, pcDNA3, pcDNA3-Trem2 + LY294002, and LY294002 groups. However, the 8-OHdG levels in the blank, pcDNA3, and pcDNA3-Trem2 + LY294002 groups were lower than that in the LY294002 group. There were no significant differences between the normal and pcDNA3-Trem2 groups, as well as among the blank, pcDNA3, and pcDNA3-Trem2 + LY294002 groups. These findings indicated that elevation of Trem2 alleviates oxidative stress and inhibits hippocampal neuronal apoptosis.
Trem2 Up-Regulation Inhibits Pilocarpine-Induced Seizures in Mice
With the aim of elucidating the role of Trem2 in epileptic mice, the mice were given lithium chloride-pilocarpine to establish the epileptic model. The seizure behavior in each group was graded according to Racine’s stages, and the results are shown in Table 1. There was no apparent seizure behavior of mice in the normal group, which was classified as level 0, and the seizure behavior of mice in the EPI, EPI + Ad-NC, and EPI + Ad-Trem2 + LY294002 groups was classified as levels III–V, most presenting level IV–V seizures. In the EPI + Ad-Trem2 group, the seizure behavior was classified as levels 0–II, primarily level 0. This evidence revealed that pilocarpine-induced seizures in mice are inhibited by elevation of Trem2.
Trem2 Overexpression Alleviates Hippocampal Neuronal Injury via the PI3K/Akt Pathway
HE staining was used to observe histopathological changes in the hippocampal tissue of mice (Fig. 6A). A large number of dense pyramidal cells were aligned in the CA1 and CA3 regions of the normal and EPI + Ad-Trem2 groups. The cellular structure was clear, complete, and normal with clear edges, and chromatin was uniformly distributed with rich Nissl bodies in the cytoplasm. However, in the EPI, EPI + Ad-NC, and EPI + Ad-Trem2 + LY294002 groups, there showed distinct hippocampal neuronal injury and some neuron loss. The cells were arranged in a disorderly manner. The structure of neurons was incomplete, the cytoplasm and nucleus had shrunk, together with chromatin condensation, margination as well as fewer Nissl bodies.

Trem2 over-expression alleviated hippocampal neuronal injury. A Representative images of HE staining of CA1 and CA3 in the hippocampus (scale bars, 50 μm). In the EPI, EPI + Ad-NC, and EPI + Ad-Trem2 + LY294002 groups, hippocampal neurons showed distinct injuries; neurons were lost, and their arrangement was disordered. The structure of neurons was incomplete, and there showed a serial of shrunk cytoplasm and nucleus, and chromatin condensation margination as well as fewer Nissl bodies. B Representative images of TUNEL staining of CA1 and CA3 in the hippocampus (scale bars, 25 μm). Positive granulosa cells were absent from the normal and EPI + Ad-Trem2 groups, while TUNEL- positive cells were evident in the CA1 and CA3 regions in the EPI, EPI + Ad-NC and EPI + Ad-Trem2 + LY294002 groups. C The apoptosis rate of hippocampal neurons in CA1 and CA3 was decreased by Trem2 overexpression (*P < 0.05 vs normal group, one-way ANOVA).
TUNEL staining was used to assess the apoptosis rate in hippocampal tissue from mice (Fig. 6B, C). Positive granulosa cells were not found in the normal group, and TUNEL-positive cells were significantly increased in the EPI group. However, no significant difference was found in TUNEL-positive cells between the EPI + Ad-NC and EPI groups. In addition, TUNEL-positive cells in the EPI + Ad-Trem2 group were less than that in the EPI group. These results suggested that over-expression of Trem2 reduces injury or apoptosis of mouse hippocampal neurons, and its mechanism is related to the PI3K/Akt pathway.
Trem2 Up-Regulation Alleviates Oxidative Stress via the PI3K/Akt Pathway
Oxidative stress in hippocampal tissue was assessed in the EPI mice (Fig. 7A–C). Compared with the normal and EPI + Ad-Trem2 groups, the SOD and GSH-Px activity was significantly lower in the EPI, EPI + Ad-NC, and EPI + Ad-Trem2 + LY294002 groups, while the MDA levels were higher. There were no significant differences between the normal and EPI + Ad-Trem2 groups, as well as among the EPI, EPI + Ad-NC, and EPI + Ad-Trem2 + LY294002 groups.

Trem2 over-expression alleviated oxidative stress. A WST-1 data suggesting that SOD activity in hippocampal tissue was increased by Trem2 overexpression. B Thiobarbituric acid colorimetric data showing that the MDA content in hippocampal tissue was decreased by Trem2 overexpression. C UV colorimetric data showing that the GSH-Px activity in hippocampal tissue was increased by Trem2 overexpression. D ELISA data showing that the 8-OHdG content in hippocampal tissue was decreased by Trem2 overexpression. *P < 0.05 vs normal group, one-way ANOVA.
Furthermore, we used UV colorimetry to determine the expression of DNA 8-OHdG in hippocampal tissue from each group (Fig. 7D). Compared with the normal and EPI + Ad-Trem2 groups, the 8-OHdG content was higher in the EPI, EPI + Ad-NC, and EPI + Ad-Trem2 + LY294002 groups. There were no significant differences between the normal and EPI + Ad-Trem2 groups, as well among the EPI, EPI + Ad-NC, and EPI + Ad-Trem2 + LY294002 groups. Accordingly, the evidence suggested that elevation of Trem2 alleviates oxidative stress and inhibits neuronal apoptosis through the PI3K/Akt pathway.
Discussion
Epilepsy is mainly characterized by recurrent seizures and a series of neurobehavioral comorbidities including cognitive disorders and abnormal social-adaptive behavior, both representing major sources of disability in children and adults [28]. Interestingly, a recent study has suggested that Trem2 plays an important role in ischemic brain injury, and is thought to be an attractive target for the treatment of ischemic stroke, exhibiting a protective action against neuronal injury and apoptosis via the NF-κB/ERK pathway [29]. Therefore, we investigated the regulation of epilepsy by Trem2 through the PI3K/Akt pathway in both a hippocampal neuron model and a mouse model of epilepsy. Of crucial importance, our results showed that over-expression of Trem2 activated the PI3K/Akt pathway, thereby inhibiting the apoptosis and injury of hippocampal neurons as well as alleviating oxidative stress.
We found that Trem2 expression was down-regulated in the epileptic hippocampal neuron model. Using a gain-of-function approach, we hypothesized that up-regulation of Trem2 contributes to the inhibition of neuronal apoptosis and injury in mice. Interestingly, another study has revealed that Trem2 interacts with endogenous ligands on neurons, and Trem2 ligands that link microglia with apoptotic neurons may contribute to the repair of apoptotic neurons [30]. A previous study reported that over-expression of Trem2 alleviates the neurotoxicity and neuroinflammation in Parkinson’s disease, providing a potential therapeutic strategy [31]. In addition, elevation of Trem2 has been found to alleviate neuropathology and the spatial cognitive impairment of a mouse model of Alzheimer’s disease [32]. Moreover, Takahashi et al. showed that Trem2 gene transfer has anti-inflammatory properties to repair injured central nervous system tissue and further improve the phagocytosis of apoptotic neurons [33]. However, a previous study reported that reduction of Trem2 may be favorable to improvement, and might increase the possibility of neurodegeneration [34]. Ercegovac et al. further reported that enhancement of protein, lipid, and DNA oxidation and increased antioxidant enzyme activity are manifestations of the oxidative stress in epilepsy, and an increment of 8-OHdG was further noted [35]. In a preventive approach, decreased MDA and 8-OHdG content along with increased SOD and GSH-Px activity has been found in epileptic mice with Trem2 over-expression, which demonstrated an amelioration of oxidative stress in epilepsy. Partially coinciding with our findings, the elevation of SOD and GSH-Px activity as well as downregulation of MDA content induced by electroacupuncture has been reported to inhibit the oxidative stress of Parkinson’s disease, thereby protecting against dopaminergic neuronal apoptosis [36]. Importantly, our results indicated that over-expression of Trem2 has therapeutic potential to inhibit the progression of epilepsy, and to improve the quality of life of patients suffering from epilepsy.
Subsequently, we also demonstrated that the elevation of Trem2 resulted in increased levels of p-PI3K/PI3K and p-Akt/Akt, demonstrating the activation of the PI3K/Akt pathway, which is thought to prevent the apoptosis and injury of hippocampal neurons and oxidative stress. In addition, our results indicated that there were declines in Bax and caspase3, along with an increase of Bcl-2 after up-regulation of Trem2, while the opposite results were found after inactivation of the PI3K/Akt pathway. Similarly, another study revealed that activation of the PI3K/Akt pathway is involved in the development of ischemic stroke, and increased phosphorylation of Akt contributes to the inhibition of neuronal apoptosis [37]. Along the same lines, a previous study showed that activation of the PI3K/Akt pathway is protective against neuronal apoptosis [38]. Interestingly, a previous report noted that the Bcl-2, Bax, and caspase-3 proteins play a role in the process of neuronal injury, and elevation of caspase-3 and Bax as well as down-regulation of Bcl-2 may be associated with neuronal apoptosis in cerebral ischemia/reperfusion injury that leads to neurological impairment [39]. In addition, Mao et al. reported that an increased proportion of Bcl-2/Bax favors the inhibition of neuronal apoptosis, and CXCL12 is involved in many neurodegenerative and neuroinflammatory diseases [40]. In our study, suppression of the apoptosis-related genes Bax and caspase3 and elevation of Bcl-2 as well as activation of the PI3K/Akt pathway were induced by Trem2 over-expression, and these findings may be therapeutically useful in inhibiting neuronal apoptosis.
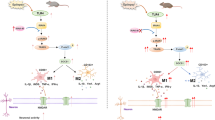
In summary, we demonstrated that up-regulation of Trem2 inhibits neuronal apoptosis and injury, and alleviates oxidative stress in epilepsy through activation of the PI3K/Akt pathway (Fig. 8). Therefore, our results provide evidence that over-expression of Trem2 may be used to develop alternative therapeutic strategies for epilepsy. To explore the potential therapeutic action of Trem2 in epilepsy, further research is warranted to evaluate its effectiveness and safety in the complex cascade of alterations in molecular, cellular, and neuronal networks.

Trem2 inhibits the progression of epilepsy via the PI3K/Akt pathway. In the epileptic hippocampal neuron model, neurons were exposed to low-Mg2+ extracellular fluid. In the epileptic mouse model, epilepsy was induced by pilocarpine. In both in the hippocampal neuron and mouse models of epilepsy, down-regulation of Trem2 had an inhibitory effect on activation of the PI3K/Akt pathway. This resulted in a decrease of Bcl-2 and increase of Bax and caspase3, eventually contributing to neuronal apoptosis and injury. On the contrary, epilepsy-induced neuronal apoptosis and injury was inhibited by up-regulating Trem2 along with increases in the levels of PI3K and Akt phosphorylation, which increased Bcl-2 while decreasing Bax and caspase3. Trem2, triggering receptor expressed on myeloid cells 2; Bcl-2, B-cell lymphoma-2; Bax, BCL2-associated X.
References
Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 2014, 55: 475–482.
Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci 2013, 36: 174–184.
Moshe SL, Perucca E, Ryvlin P, Tomson T. Epilepsy: new advances. Lancet 2015, 385: 884–898.
Newton CR, Garcia HH. Epilepsy in poor regions of the world. Lancet 2012, 380: 1193–1201.
Jacobs J, Staba R, Asano E, Otsubo H, Wu JY, Zijlmans M, et al. High-frequency oscillations (HFOs) in clinical epilepsy. Prog Neurobiol 2012, 98: 302–315.
Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 2012, 4: 161ra152.
Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet 2014, 23: 5838–5846.
Benitez BA, Cooper B, Pastor P, Jin SC, Lorenzo E, Cervantes S, et al. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging 2013, 34: 1711 e1715–1717.
Fu Y, Zhu H, Wu W, Xu J, Chen T, Xu B, et al. Clinical significance of lymphoid enhancer-binding factor 1 expression in acute myeloid leukemia. Leuk Lymphoma 2014, 55: 371–377.
Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 2005, 201: 647–657.
Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci 2015, 35: 3384–3396.
Davies MA. The role of the PI3K-AKT pathway in melanoma. Cancer J 2012, 18: 142–147.
Kretz A, Happold CJ, Marticke JK, Isenmann S. Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol Cell Neurosci 2005, 29: 569–579.
Zhu M, Li D, Wu Y, Huang X, Wu M. TREM-2 promotes macrophage-mediated eradication of Pseudomonas aeruginosa via a PI3K/Akt pathway. Scand J Immunol 2014, 79: 187–196.
Slenter DN, Kutmon M, Hanspers K, Riutta A, Windsor J, Nunes N, et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res 2018, 46: D661–D667.
Xie N, Wang C, Wu C, Cheng X, Gao Y, Zhang H, et al. Mdivi-1 protects epileptic hippocampal neurons from apoptosis via inhibiting oxidative stress and endoplasmic reticulum stress in vitro. Neurochem Res 2016, 41: 1335–1342.
Whalley BJ, Stephens GJ, Constanti A. Investigation of the effects of the novel anticonvulsant compound carisbamate (RWJ-333369) on rat piriform cortical neurones in vitro. Br J Pharmacol 2009, 156: 994–1008.
Theofilas P, Brar S, Stewart KA, Shen HY, Sandau US, Poulsen D, et al. Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 2011, 52: 589–601.
Tao H, Zhao J, Liu T, Cai Y, Zhou X, Xing H, et al. Intranasal delivery of miR-146a mimics delayed seizure onset in the lithium-pilocarpine mouse model. Mediators Inflamm 2017, 2017: 6512620.
Ma DL, Qu JQ, Goh EL, Tang FR. Reorganization of basolateral amygdala-subiculum circuitry in mouse epilepsy model. Front Neuroanat 2015, 9: 167.
Kelkar MG, Thakur B, Derle A, Chatterjee S, Ray P, De A. Tumor suppressor protein p53 exerts negative transcriptional regulation on human sodium iodide symporter gene expression in breast cancer. Breast Cancer Res Treat 2017, 164: 603–615.
Wyatt SK, Witt T, Barbaro NM, Cohen-Gadol AA, Brewster AL. Enhanced classical complement pathway activation and altered phagocytosis signaling molecules in human epilepsy. Exp Neurol 2017, 295: 184–193.
Le Ber I, De Septenville A, Guerreiro R, Bras J, Camuzat A, Caroppo P, et al. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol Aging 2014, 35: 2419 e2423–2419 e2425.
Wei H, Duan G, He J, Meng Q, Liu Y, Chen W, et al. Geniposide attenuates epilepsy symptoms in a mouse model through the PI3K/Akt/GSK-3beta signaling pathway. Exp Ther Med 2018, 15: 1136–1142.
Guo XQ, Cao YL, Hao F, Yan ZR, Wang ML, Liu XW. Tangeretin alters neuronal apoptosis and ameliorates the severity of seizures in experimental epilepsy-induced rats by modulating apoptotic protein expressions, regulating matrix metalloproteinases, and activating the PI3K/Akt cell survival pathway. Adv Med Sci 2017, 62: 246–253.
Xiao Z, Peng J, Gan N, Arafat A, Yin F. Interleukin-1beta plays a pivotal role via the PI3K/Akt/mTOR signaling pathway in the chronicity of mesial temporal lobe epilepsy. Neuroimmunomodulation 2016, 23: 332–344.
Thompson S, Kelly CA, Griffiths ID, Turner GA. Abnormally-fucosylated serum haptoglobins in patients with inflammatory joint disease. Clin Chim Acta 1989, 184: 251–258.
Zhao X, Chen Y, Qiu G, Xiao M, Zhong N. Reducing preterm births in China. Lancet 2012, 380: 1144–1145; author reply 1145.
Wu R, Li X, Xu P, Huang L, Cheng J, Huang X, et al. TREM2 protects against cerebral ischemia/reperfusion injury. Mol Brain 2017, 10: 20.
Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 2009, 109: 1144–1156.
Ren M, Guo Y, Wei X, Yan S, Qin Y, Zhang X, et al. TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson’s disease. Exp Neurol 2018, 302: 205–213.
Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39: 2949–2962.
Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med 2007, 4: e124.
Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 2014, 6: 243ra286.
Ercegovac M, Jovic N, Simic T, Beslac-Bumbasirevic L, Sokic D, Djukic T, et al. Byproducts of protein, lipid and DNA oxidative damage and antioxidant enzyme activities in seizure. Seizure 2010, 19: 205–210.
Li J, Wang LN, Xiao HL, Li X, Yang JJ. Effect of electroacupuncture intervention on levels of SOD, GSH, GSH-Px, MDA, and apoptosis of dopaminergic neurons in substantia Nigra in rats with Parkinson’s disease. Zhen Ci Yan Jiu 2014, 39: 185–191.
Kilic U, Caglayan AB, Beker MC, Gunal MY, Caglayan B, Yalcin E, et al. Particular phosphorylation of PI3K/Akt on Thr308 via PDK-1 and PTEN mediates melatonin’s neuroprotective activity after focal cerebral ischemia in mice. Redox Biol 2017, 12: 657–665.
Zhuang Z, Zhao X, Wu Y, Huang R, Zhu L, Zhang Y, et al. The anti-apoptotic effect of PI3K-Akt signaling pathway after subarachnoid hemorrhage in rats. Ann Clin Lab Sci 2011, 41: 364–372.
Liu G, Wang T, Wang T, Song J, Zhou Z. Effects of apoptosis-related proteins caspase-3, Bax and Bcl-2 on cerebral ischemia rats. Biomed Rep 2013, 1: 861–867.
Mao W, Yi X, Qin J, Tian M, Jin G. CXCL12 inhibits cortical neuron apoptosis by increasing the ratio of Bcl-2/Bax after traumatic brain injury. Int J Neurosci 2014, 124: 281–290.
Acknowledgments
We sincerely appreciate the reviewers’ critical comments on this article. This work was supported by Beijing Key Laboratory of Neuromodulation (BZ0098), the Precision Medicine Project of the Ministry of Science and Technology of China (2016YFC0904400).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest in this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, AH., Chu, M. & Wang, YP. Up-Regulation of Trem2 Inhibits Hippocampal Neuronal Apoptosis and Alleviates Oxidative Stress in Epilepsy via the PI3K/Akt Pathway in Mice. Neurosci. Bull. 35, 471–485 (2019). https://doi.org/10.1007/s12264-018-0324-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-018-0324-5