
Abstract
Increasing evidence suggests that spinal microglia regulate pathological pain in males. In this study, we investigated the effects of several microglial and astroglial modulators on inflammatory and neuropathic pain following intrathecal injection in male and female mice. These modulators were the microglial inhibitors minocycline and ZVEID (a caspase-6 inhibitor) and the astroglial inhibitors L-α-aminoadipate (L-AA, an astroglial toxin) and carbenoxolone (a connexin 43 inhibitor), as well as U0126 (an ERK kinase inhibitor) and D-JNKI-1 (a c-Jun N-terminal kinase inhibitor). We found that spinal administration of minocycline or ZVEID, or Caspase6 deletion, reduced formalin-induced inflammatory and nerve injury-induced neuropathic pain primarily in male mice. In contrast, intrathecal L-AA reduced neuropathic pain but not inflammatory pain in both sexes. Intrathecal U0126 and D-JNKI-1 reduced neuropathic pain in both sexes. Nerve injury caused spinal upregulation of the astroglial markers GFAP and Connexin 43 in both sexes. Collectively, our data confirmed male-dominant microglial signaling but also revealed sex-independent astroglial signaling in the spinal cord in inflammatory and neuropathic pain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Numerous studies suggest that spinal glial cells, such as microglia and astrocytes, play a critical role in the development and maintenance of inflammatory and neuropathic pain via neuronal-glial and glial-glial interactions [1,2,3,4]. Upon activation by painful stimuli or injury, microglia release pro-inflammatory and pro-nociceptive factors, such as tumor necrosis factor (TNF), interleukin-1-beta and interleukin-18 (IL-1β and IL-18), and brain-derived growth factor (BDNF), via activation of p38 MAP kinase, to directly modulate synaptic transmission by interacting with nociceptive neurons [5,6,7,8,9]. Astrocytes are the most abundant cells in the spinal cord and contact synapses very closely [10, 11]. Compared to the rapid reaction of microglia, especially after nerve injury, astrocytic activation has been found in a variety of pain-related injury states, usually occurring several days after the injury and much longer-lasting [2, 12]. Activated astrocytes can also release multiple inflammatory mediators and neuromodulators such as cytokines (IL-1β) [13] and chemokines (CCL2 and CXCL1), via upregulation of the hemichannel and gap junction protein connexin43 (Cx43) and activation of c-Jun N-terminal kinase (JNK), to enhance and extend persistent pain states [10, 14, 15]. Activation of extracellular signal-regulated kinases (ERK) in astrocytes has also been implicated in the maintenance of neuropathic pain [16, 17]. Since it was not expected that these cellular and molecular signaling pathways would differ between the sexes, previous animal studies primarily focused on males to avoid confounding sex-hormone fluctuations in females (but see [18, 19]).
However, it is apparent that for most pathologic states, females suffer at a higher rate [20,21,22]. The 2009 U.S. National Health Interview Survey found that women report greater amounts of low back pain, neck pain, and orofacial pain, with an almost 2-fold increase in women reporting migraines or severe headaches, or pain in the face or jaw [23]. A review of experimentally-induced pain in humans also provided support for the observation that females are more sensitive to pressure and thermal pain than males, which suggests differences in nociception between the sexes [21]. Several mechanisms have been proposed to explain these differences, including psychosocial factors, familial factors, and sex hormones [21, 24,25,26]. Intriguingly, recent studies have demonstrated a male-dominance of microglial signaling in neuropathic pain, although both sexes show identical morphological reactivity of microglia (i.e. microgliosis) after nerve injury [27, 28]. Previous results from our group suggest that secreted protease caspase-6 (CASP6) released from axonal terminals induces microglial activation and releases TNF via activation of p38 MAP kinase [29]. Our recent results also suggested that sex-dependent p38 activation and signaling occur in spinal microglia in some inflammatory and neuropathic pain conditions [30]. Although microglial signaling presents a clear sex dichotomy in pain, whether astrocyte signaling is also sex-dependent in the modulation of pain is still unknown.
Thus, in this study, we used an approach involving intrathecal (i.t.) administration of microglial and astroglial modulators to investigate the signaling of microglia and astrocytes in inflammatory pain and neuropathic pain in both male and female mice.
Materials and Methods
Reagents and Administration
Formaldehyde solution, carbenoxolone (CBX, a gap-junction inhibitor), L-α-aminoadipate (L-AA, an astroglial toxin) [31], and minocycline (a microglial inhibitor) [32] were from Sigma (St. Louis, MO). Recombinant full-length active human caspase-6 protein (CASP6) was from Abcam (Cambridge, MA), the CASP6 inhibitor Z-V-E(OMe)-I-D(OMe)-FMK (ZVEID) and TNF were from R&D Systems (Minneapolis, MN). SB 225002 (a CXCR2 antagonist) was from Tocris (Ellisville, MO), and U0126 (an MEK inhibitor) was from Calbiochem (Darmstadt, Germany). D-JNKI-1 (a JNK inhibitor) was kindly provided by Dr. Christopher Bonny, University of Lausanne, Switzerland. For i.t. injection, spinal cord puncture was made with a 30G needle between the L5 and L6 levels to deliver reagents (10 μL) into the cerebrospinal fluid [33]. The doses of inhibitors/antagonists/recombinant proteins used in this study were from our previous reports [14, 34,35,36].
Animals and Surgery
Adult CD1 mice (8–10 weeks old, 25–30 g) of both sexes were from Charles River Laboratories (Wilmington, MA) and used for the majority of experiments. In selected experiments, adult knockout mice lacking Casp6 (Casp6 –/–), from The Jackson Laboratory (Bar Harbor, ME), were used. All animals were housed under a 12-h light/dark cycle with food and water available ad libitum. To induce inflammatory pain, diluted formalin (5%, 20 μL) was injected into the plantar surface of a hindpaw. Neuropathic pain was produced by chronic constriction injury (CCI) of the sciatic nerve [14]. In brief, animals were anesthetized with isoflurane, then the left sciatic nerve was exposed and three ligatures (LOOK® 6-0 Silk, #SP102, Surgical Specialities Co., Reading, PA) were placed around the nerve proximal to the trifurcation with 1 mm between ligatures. The ligatures were gently tightened until a short flick of the ipsilateral hind limb was observed. All animal procedures performed in this study were approved by the Animal Care Committee of Duke University Medical Center.
Behavioral Testing
Animals were habituated to the testing environment for at least 2 days before testing. All behaviors were assessed in a blinded manner. The time course of formalin-induced licking and flinching behavior was monitored and recorded for 45 min and analyzed every 5 min. For assessing mechanical sensitivity after formalin injection and CCI, the hindpaw was stimulated with a series of von Frey hairs of logarithmically increasing stiffness (0.02–2.56 g, Stoelting, Wood Dale, IL), presented perpendicularly to the central plantar surface. Based on Dixon’s up-down method [37], 6 von Frey tests were performed in each animal and the 50% paw-withdrawal threshold was determined.
Immunohistochemistry
As we reported previously [14], mice were deeply anesthetized with isoflurane and perfused through the ascending aorta with PBS, followed by 4% paraformaldehyde. After perfusion, the L4–L5 spinal segments were removed and postfixed in the same fixative overnight. Spinal cord sections (30 μm, free-floating) were cut in a cryostat, blocked with 2% goat serum for 1 h at room temperature, and then incubated overnight at 4 °C with anti-GFAP (mouse, 1:1000, Millipore, Colorado, MA) and anti-Connexin 43 (rabbit, 1:1000, Sigma, St. Louis, MO) primary antibodies, followed by cyanine 3(Cy3)- and/or FITC-conjugated secondary antibodies (1:400; Jackson ImmunoResearch, West Grove, PA) for 2 h at room temperature. The stained sections were examined under a Nikon fluorescence microscope, and images were captured with a CCD Spot camera. We collected 5 sections from each mouse for quantification of immunofluorescence. The intensity of fluorescence in the superficial dorsal horn was analyzed using ImageJ software (NIH, Bethesda, MD).
Statistical Analyses
All data are expressed as mean ± SEM. Behavioral data were analyzed using Student’s t-test (two groups) or one-way and two-way ANOVA followed by the Bonferroni post-hoc test. The criterion for statistical significance was P < 0.05.
Results
Minocycline Inhibits Inflammatory Pain in Males while L-AA Has no Effect on Inflammatory Pain in Mice of Both Sexes
To assess sex-dependent glial signaling in inflammatory pain, male and female mice were administered the microglial inhibitor minocycline (10 and 50 µg, ~20 and 100 nmol) and the astroglial toxin L-AA (50 nmol) via the i.t. route 30 min prior to intraplantar injection of 5% formalin. The time course of formalin-induced licking and flinching behavior are shown in Fig. 1A. Data are also presented as phase I (0–10 min after formalin injection, related to peripheral nerve activity) and phase II (10–45 min after formalin injection, related to central sensitization) (Fig. 1B). We found no difference in phase I or phase II behavior between vehicle- and L-AA-treated males and females, showing no effect of L-AA on formalin-induced spontaneous pain (phase I, P = 0.3090 in males and P = 0.1892 in females; phase II, P > 0.9999 in males and females, one-way ANOVA). The results suggest that astrocyte signaling does not play a role in this model. Intrathecal minocycline at a low dose of 10 µg reduced both phase I and phase II behavior in males but not in females, while a higher dose of 50 µg reduced both phase I and phase II behavior in males and females (P < 0.05, one-way ANOVA, Fig. 1A, B). Regardless of the dose, the analgesic effect was much stronger in males than in females. There were also no significant differences in phase I and phase II behavior in males with either 10 or 50 µg minocycline (phase I, P = 0.9561; phase II, P > 0.9999, one-way ANOVA,). Although this suggests a role of microglial signaling in males and females, the effect appeared more pronounced in males. Furthermore, as minocycline also has other unrelated actions at higher doses, such as antibacterial [38], neuroprotective [39], and anti-inflammatory effects [40], this may be a reason why only a high dose of minocycline reduced formalin-induced spontaneous pain behavior in females. Above all, these results show a sex-dependence of the role of microglial signaling in formalin-induced spontaneous pain.

Effects of spinal injection of microglial inhibitor and astroglial toxin on formalin-induced pain in male and female mice. A Time-course of licking and flinching behavior following intraplantar injection of 5% formalin. B Formalin-induced Phase I (1–10 min) and Phase II (10–45 min) responses. Intrathecal injection of vehicle (PBS), the microglial inhibitor minocycline (10 and 50 µg), and the astroglial toxin L-AA (50 nmol), 30 min prior to intraplantar injection of formalin. Mean ± SEM; *P < 0.05 versus corresponding control (Vehicle); # P < 0.05 vs opposite sex; n.s., not significant vs different doses; one-way ANOVA, n = 5 mice per group.
Mechanical Hypersensitivity is Induced in Both Naïve Male and Female Mice by Intrathecal TNF but only in Naïve Male Mice by Intrathecal CASP6
Previous results from our lab had shown that CASP6, a cysteine protease released from spinal axonal terminals, modulates synaptic plasticity and inflammatory pain [29]. I.t. injection of recombinant human CASP6 protein induces mechanical hypersensitivity via releasing TNF from microglia in naïve male mice [29, 35]. To test whether the CASP6 signaling pathway in naïve mice is sex-dependent, males and females were given human active CASP6 (5 U, i.t.)[29], which shares ~84% of its sequence with mouse CASP6 (checked by protein blast). Compared with the vehicle group, CASP6 elicited rapid (<2 h) and persistent (>24 h) mechanical hypersensitivity only in naïve males (Fig. 2A) but not in females (Fig. 2B). Since TNF is mainly produced by spinal microglia and acts as a main trigger for the pathogenesis of pain [15, 29, 41, 42] via modulation of spinal synaptic plasticity [43,44,45] and astrocyte activation [15], we tested the effect of i.t. injection of TNF in males and females, and found that it induced similar levels of rapid and persistent mechanical hypersensitivity in both (Fig. 2A, B). These results imply that the spinal neuronal and astrocytic responses to TNF are similar in males and females and that the sex-dependent CASP6 signaling in naïve mice is due to the sex-dependent release of TNF from microglia.

Effects of spinal injection of CASP6 and TNF on naïve male and female mice. A Intrathecal injection of CASP6 (5 U) and TNF (20 ng) both elicited rapid and persistent mechanical hypersensitivity in naïve males. B Intrathecal injection of TNF but not CASP6 elicited rapid and persistent mechanical hypersensitivity in naïve females. Mean ± SEM; *P < 0.05, n.s., not significant, two-way ANOVA; n = 4 mice per group; BL, baseline. Arrows indicate drug injection.
CASP6 Inhibition or Casp6 Deletion in the Formalin Model Reduces Inflammatory Pain and Delays Neuropathic Pain in Male but not in Female Mice
To test the sex-dependent CASP6 signaling in inflammatory pain, males and females were given the CASP6 inhibitor ZVEID [29] (30 µg, i.t.) 30 min prior to intraplantar injection of 5% formalin. ZVEID reduced both formalin-induced phase I and phase II behavior in males but only phase I in females (P < 0.05, one-way ANOVA; Fig. 3A, B). No sex difference was found in phase I pain behavior with ZVEID treatment (P = 0.837, two-tailed Student’s t-test), but phase II in males with ZVEID was reduced (P = 0.039, two-tailed Student’s t-test). We have previously shown that after vehicle injection, formalin-induced pain is comparable in males and females [30]. We have also shown that the baseline thresholds in WT and CASP6-knockout mice are comparable in both sexes [35]. Here, we further compared the formalin-induced pain and mechanical hypersensitivity in Casp6 –/– males and females. Compared with Casp6 –/– females, formalin-induced phase II pain in males was also reduced (P = 0.002, two-tailed Student’s t-test Fig. 3C, D). Formalin induces not only acute inflammatory pain but also delayed neuropathic pain, due to nerve injury. As a result, intraplantar formalin injection causes marked morphological activation of microglia (microgliosis) in the spinal dorsal horn after 24 h [46]. Interestingly, formalin-induced persistent mechanical hypersensitivity on days 5 and 7 was also reduced in Casp6 –/– males (Fig. 3E). These results indicate sex-dependent CASP6 signaling in formalin-induced acute inflammatory pain and persistent neuropathic pain.

CASP6 contributes to formalin-induced pain and late-phase mechanical hypersensitivity in male mice. A Time course of formalin-induced pain in males and females with intrathecal vehicle or the CASP6 inhibitor ZVEID (30 µg). B ZVEID (30 µg, i.t.) reduced both formalin-induced phase I and phase II pain in males but only phase I in females. *P < 0.05, two-tailed Student’s t-test, n = 5 mice per group. C, D Formalin-induced pain was reduced in male but not female Casp6 –/– mice. Mean ± SEM; *P < 0.05, two-tailed Student’s t-test, n = 4–5 mice per group. E Formalin-induced mechanical hypersensitivity in the late phase was lower in male than in female Casp6 –/– mice. Mean ± SEM; *P < 0.05, two-way ANOVA; n = 4–5 mice per group.
Microglial Inhibition Reduces CCI-Induced Mechanical Hypersensitivity in Male but not Female Mice While Astroglial Inhibition Shows No Sexual Dimorphism
Microglia and astrocytes play distinct roles in the induction and maintenance of neuropathic pain. After nerve injury, the time course of spinal glial activation depends upon the cell type. The microglial reaction (microgliosis) peaks in the first week [12, 47], but astrogliosis is more prominent at a later stage (10–21 days) [14, 48]. To test sex-dependent glial signaling in neuropathic pain, males and females were given the microglial inhibitor minocycline (50 µg, i.t.) 7 days after CCI, and the results showed that mechanical hypersensitivity was not significantly different between males and females at 7 days after surgery (Fig. 4A). Consistent with previous reports, minocycline rapidly (<1 h) and completely reversed the mechanical hypersensitivity for 3 h in males. This inhibitory effect faded after 5 h. In contrast, minocycline only partially reversed the mechanical hypersensitivity in females (Fig. 4A). The effects of minocycline at 7 days after CCI also differed in males and females (P < 0.0001, two-way ANOVA; Fig. 4A). Next, males and females were given the astroglial toxin L-AA (50 nmol, i.t.) 12 days after CCI, and this had comparable effects in males and females (P > 0.05, two-way ANOVA; Fig. 4B). These results indicate sex-dependent microglial but not astroglial signaling in the spinal cord in CCI-induced neuropathic pain.

In the CCI model, microglial inhibition reduces mechanical hypersensitivity in male but not female mice, while astroglial inhibition shows no sexual dimorphism. A CCI-induced mechanical hypersensitivity at 7 days was fully reversed by spinal injection of minocycline (50 µg = 100 nmol) in males and it was only partially reversed in females. B CCI-induced mechanical hypersensitivity at 12 days was partially reversed by L-AA (50 nmol), but there was no difference between males and females. Mean ± SEM; *P < 0.05 vs corresponding CCI 7, one-way ANOVA; # P < 0.05, n.s., not significant vs opposite sex, two-way ANOVA; n = 8 mice per group; BL, baseline. Arrows indicate drug injection.
Sex-Independent Astrocytic Signaling in the Spinal Cord in Neuropathic Pain
The mitogen-activated protein kinase (MAPK) family, which consists of three major members (p38, ERK, and JNK) is activated in spinal glial cells after nerve injury and contributes to pain sensitization [49]. After nerve injury, p38 is activated in microglia, ERK is activated sequentially in microglia and astrocytes, and JNK is activated persistently in astrocytes [50]. Our recent paper demonstrated sex-dependent p38 signaling in spinal microglia under inflammatory and neuropathic pain conditions [30, 35]. Here, we investigated the roles of JNK and ERK in male and female mice in the CCI model. Intrathecal injection of the JNK inhibitor D-JNKI-1 (4 nmol) or the ERK kinase inhibitor U0126 (10 µg = 25 nmol) at 15 or 14 days both reversed the CCI-induced mechanical hypersensitivity in males and females equally (P > 0.05, two-way ANOVA; Fig. 5A, B).

Spinal inhibition of JNK, ERK, Cx43 hemichannels, and CXCR2 reduces mechanical hypersensitivity in both males and females equally in mice with chronic constriction injury. A–C CCI-induced mechanical hypersensitivity was reduced at 15 days by the JNK inhibitor D-JNKI-1 (4 nmol) (A), and at 14 days by the ERK inhibitor U0126 (25 nmol) (B); it was fully reversed at 17 days by CBX (5 µg = 8 nmol) (C), and reduced at 13 days by the CXCR2 antagonist SB225002 (20 µg = 56 nmol) (D) in males and females. E CCI-induced mechanical hypersensitivity at 14 days was not changed by vehicle (PBS) injection, and males and females did not significantly differ. Mean ±SEM; *P < 0.05 vs corresponding CCI, n.s., not significant, two-way ANOVA; n = 5–6 mice per group; BL, baseline. Arrows indicate drug injection.
Our previous experiments on male mice had shown that Connexin-43-mediated hemichannels control the astrocytic release of CXCL1 in the spinal cord, and that CXCL1 maintains spinal synaptic plasticity via activation of neuronal CXCR2 [14]. To further assess the sex-independence of astrocytic signaling in neuropathic pain, male and female mice were given the non-selective gap-junction inhibitor CBX [51] (5 µg = 8 nmol) and the CXCR2 antagonist SB225002 [52] (20 µg = 56 nmol) via the i.t. route at 13 or 17 days after CCI. Injection of CBX rapidly (<1 h) and completely reversed the mechanical hypersensitivity for >5 h in both males and females equally (P > 0.05, two-way ANOVA). This reversal faded after 24 h (Fig. 5C). SB225002 partially reversed the mechanical hypersensitivity in both males and females equally (P > 0.05, two-way ANOVA; Fig. 5D). As a control, vehicle injection in the CCI model did not change the paw-withdrawal threshold in males and females (Fig. 5E).
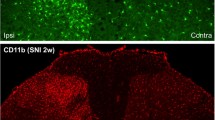
We have previously demonstrated that the microglial reaction (morphological activation, IBA-1 staining intensity, and proliferation) is identical in male and female mice after CCI [30]. Here, to determine whether nerve injury differentially activates spinal astrocytes in males and females, we examined the expression of astrocyte markers GFAP and the hemichannel Cx43 on lumbar cord sections from both sexes 14 days after CCI using double immunohistochemistry. We found that both males and females displayed increases in GFAP- and Cx43-immunoreactivity (IR) in the dorsal horn ipsilateral to the injury (Fig. 6A, B). Quantification of IR in the dorsal horn confirmed the upregulation of GFAP and Cx43 14 days after CCI on the ipsilateral side versus the contralateral side (Fig. 6C, D). However, no differences between males and females were found in the intensity and co-localization of GFAP and Cx43 in the ipsilateral dorsal horn (Fig. 6C–E). Collectively, these data suggest that spinal astrocytic signaling is sex-independent in CCI-induced neuropathic pain.

CCI increases GFAP and Cx43 expression in the spinal dorsal horn equally in mice of both sexes. A, B Low-magnification images of double immunofluorescent staining with GFAP (red) and connexin 43 (Cx43, green) in lumbar dorsal horn sections showing both ipsilateral (Ipsi.) and contralateral (Contra.) sides in a male (A) and a female (B) 14 days after CCI (scale bars, 100 µm). A’, B’ High-magnification images (from boxes in A, B) of the double staining in the ipsilateral dorsal horn (scale bars, 50 µm). C, D Quantification of GFAP (C) and Cx43 (D) immunofluorescence from lumbar sections showing a significant increase in fluorescence intensity in the ipsilateral dorsal horn in both males and females. E Co-localization ratios of GFAP and Cx43 IR staining in the ipsilateral dorsal horn in males and females 14 days after CCI. Mean ± SEM; *P < 0.05 between ipsilateral and contralateral dorsal horn of same sex, n.s. not significant, two-tailed Student’s t-test, n = 4 mice per sex per group, 5 sections per mouse.
Discussion
The overall goal of this study was to determine the sex differences in spinal microglial and astroglial signaling in inflammatory pain and neuropathic pain by testing several glial modulators. Our results showed that spinal microglial signaling is sex-dependent in the formalin-induced model of acute inflammatory pain and the CCI model of neuropathic pain. First, spinal injection of the microglial inhibitor minocycline reduced both the formalin-induced pain behavior and CCI-induced mechanical hypersensitivity in males with less or no response in females. Second, mechanical hypersensitivity was induced only in naïve males but not in females using the microglial activator CASP6. Third, spinal injection of the CASP6 inhibitor ZVEID reduced formalin-induced pain behavior only in males. Finally, in addition to formalin-induced early-phase inflammatory pain, we also examined the formalin-induced late-phase neuropathic pain in WT and Casp6-knockout mice. The latter showed a decrease in both pain behavior and subsequent mechanical hypersensitivity in males but not in females. Here, although the effects of glial modulators were compared between sexes and only one dose was tested in most conditions, we cannot exclude the possibility that the potency is different and a higher dose would also produce an effect in females, as for the data shown in Fig. 1. However, these results clearly demonstrated that male-specific and CASP6-mediated microglial signaling is involved in formalin-induced inflammatory pain and formalin- and CCI-induced neuropathic pain.
Conversely, our results showed that spinal astrocyte signaling plays a less important role in acute inflammatory pain, and spinal astrocytic and/or neuronal signaling is sex-independent in the CCI model of neuropathic pain. The evidence was obtained by multiple experimental approaches. First, there was no effect of spinal injection of the astroglial toxin L-AA on formalin-induced pain behavior in either male or female mice. Second, i.t. injection of TNF, which is released by activated microglia and acts on astrocytes and neurons, induced similar levels of mechanical hypersensitivity in both naïve males and females. Third, i.t. injection of the astroglial toxin L-AA reduced the nerve injury-induced mechanical hypersensitivity in both males and females equally. Fourth, inhibition of JNK and ERK, two MAP kinases important for astrocytic signaling, reduced the mechanical hypersensitivity in both males and females equally. Previous reports from our lab have shown that on day 10 after spinal nerve ligation (SNL), pERK is present in microglia in the deep dorsal horn (laminae III–V) and in astrocytes in the superficial dorsal horn (laminae I–II). However, in the late phase at 21 days after SNL, pERK is predominantly expressed in astrocytes. This may explain why the analgesic effect of the ERK kinase inhibitor U0126 showed no sex difference 14 days after CCI. Fifth, inhibition of astrocytic hemichannel function or CXCR2 activity reduced the mechanical hypersensitivity in both males and females equally. Finally, no difference between males and females was found in the staining intensity of GFAP and Cx43 in the dorsal horn ipsilateral to the injury14 days after CCI.
These results are critical, as clinical and human experimental data have identified sex differences in pain, including sensitivity to various painful stimuli and the response to some methods of pain treatment [20, 21, 23]. Recent data have shown that this sex difference is apparent in the different mechanisms underlying pain processing [27, 28, 30, 35]. For example, Sorge et al. found that Toll-like receptor 4 (TLR4) signaling in the spinal cord mediates inflammatory and neuropathic pain in male mice but not in females [28]. Their study suggested a possible sex-dependence of microglial signaling in the spinal cord, as TLR4 is known to regulate microglial activation and pain [53]. In a follow-up study, they showed that male and female mice process spinal nerve injury-induced neuropathic pain and complete Freund’s adjuvant-induced inflammatory pain through completely different immune cells – microglia in males and T cells in females – and described a sex-dependent response to microglia-targeted pain treatments [27]. Previous data from our lab also showed that there is sex-specific p38 activation and signaling in spinal microglia under inflammatory and neuropathic pain conditions, but no sex differences in the microglial reaction (morphological activation, IBA-1 staining intensity, and proliferation) after CCI [30]. However, it is noteworthy that sex-dependent microglial signaling may not occur in all pain conditions and in all phases of pain development. For example, Yang et al. demonstrated that spinal microglia play an important role in the maintenance of bone cancer pain in female rats [19].
In sharp contrast, astrocyte signaling appears to be independent of sex. A close association between synapses and astrocytic processes makes it possible for astrocytes to regulate synaptic transmission by releasing astrocytic mediators (e.g. ATP, cytokines, and growth factors) [2, 5, 54]. Under various pathological conditions, reactive astrocytes have been found to be associated with enhanced pain states [14, 15]. In most cases, the astrocytic reaction occurs after the microglial reaction and is thought to be led by the microglial reaction. Compared to the microglial reaction, the spinal astrocyte reaction is more widespread, evident, and longer-lasting after a painful injury [1, 55]. Mounting evidence has suggested that spinal astrocytes play an important role in the induction and maintenance of inflammatory and neuropathic pain as well as spinal microglia [2, 10]. For example, i.t. injection of astrocyte inhibitors, such as fluorocitrate, fluoroacetate, and L-AA, has been shown to reduce pain behaviors in male rodents with inflammatory pain and neuropathic pain [14, 56,57,58]. The expression of Cx43 is increased markedly in spinal astrocytes under various pain conditions, and inhibition of gap junction function by CBX or Cx43 blockers produces analgesia in different pain models [14, 59, 60]. In addition, astrocytes also express phosphorylated ERK, JNK, and JNK-1, as well as secreting IL-1β, TNF-α, CCL2, and CXCL1 in response to nerve injury or inflammation [1, 2, 10]. However, all these studies were experiments carried out on male rodents. We still do not know about the generalizability of these results across sexes, as the conclusions have not been tested in females. Our data do suggest though that JNK-, gap junction-, and CXCR2-mediated signaling in the spinal cord is independent of sex.
A limitation of our work is that we used relatively non-specific inhibitors/modulators of microglia and astrocytes. For example, the microglial inhibitor minocycline may have nonselective effects on neuronal activity and synaptic transmission at high concentrations (20–100 μmol/L) [29, 61]. This may explain why an effect of minocycline was noted in phase I of the formalin-induced inflammatory pain model but no effect of CASP6 inhibition or Casp6-knockout on phase I behavior was found. Interestingly, this effect of minocycline on phase I behavior was not found by two other groups who applied the drug via the systemic route in male mice and rats [62]. As we noted earlier, minocycline has many other mechanisms of action, including antimicrobial activity, strong metal chelation, and inhibition of matrix metalloprotease-9 [63]. Notably, neuronal modulation of central sensitization in the spinal cord appears to be sex-independent. Intrathecal delivery of an NMDA receptor antagonist is effective in suppressing the mechanical hypersensitivity after nerve injury in both male and female mice [27]. Consistently, i.t. TNF induced mechanical hypersensitivity in both sexes (Fig. 2A) by modulating synaptic plasticity and central sensitization [43] and astrocyte activation [15]. Activation of CXCR2 by the astrocyte-derived chemokine CXCL1 also plays an active role in facilitating synaptic transmission and central sensitization [14]. Consistently, i.t. administration of the CXCR2 antagonist SB22502 attenuated the CCI-induced mechanical hypersensitivity in both sexes (Fig. 5C). Thus, although the glial modulators are not very selective, the sex-dependent effects they produce may indicate whether these effects involve microglial signaling.
In addition to pain research, sex differences have been recognized in many other fields. To facilitate translational research, preclinical studies and therapeutic development are required to consider the biological differences between males and females according to the guidelines of the U.S. Food and Drug Administration (1993), the National Institutes of Health (1999, 2014), and the Institute of Medicine (2001). However, this pressure for applicability is at odds with the resource constraints of funding and the need to reduce animal usage. This was the major reason for designing this study, to help understand which aspects of pain-processing are sex-dependent. Although it is well-known that spinal microglia and astrocytes play important roles in the induction, development, and maintenance of pain, we were still not sure of the exact role of glia in pain conditions in females, since the majority of these experiments were performed in male animals. As a negative example [64], the results from nearly 100 animal studies showed that Dextromethorphan, an NMDA receptor antagonist, nonselective serotonin reuptake inhibitor, and sigma-1 receptor agonist, potentiates morphine analgesia at low doses, and attenuates it at high doses. However, a Phase II clinical trial failed because this effect is not apparent in females. This had not been realized, as all previous studies were performed only in male animals. To avoid such errors and save time and money in the future, researchers should pay attention to sex differences in pain studies.
References
Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain 2013, 154 Suppl 1: S10–28.
Old EA, Clark AK, Malcangio M. The role of glia in the spinal cord in neuropathic and inflammatory pain. Handb Exp Pharmacol 2015, 227: 145–170.
Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354: 572–577.
Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med 2010, 16: 1267–1276.
Ren K, Dubner R. Activity-triggered tetrapartite neuron-glial interactions following peripheral injury. Curr Opin Pharmacol 2016, 26: 16–25.
Wen YR, Tan PH, Cheng JK, Liu YC, Ji RR. Microglia: a promising target for treating neuropathic and postoperative pain, and morphine tolerance. J Formos Med Assoc 2011, 110: 487–494.
Taves S, Berta T, Chen G, Ji RR. Microglia and spinal cord synaptic plasticity in persistent pain. Neural Plast 2013, 2013: 753656.
Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci 2005, 28: 101–107.
Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 2004, 45: 89–95.
Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics 2010, 7: 482–493.
Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Prog Neurobiol 1998, 55: 1–26.
Mika J, Osikowicz M, Rojewska E, Korostynski M, Wawrzczak-Bargiela A, Przewlocki R, et al. Differential activation of spinal microglial and astroglial cells in a mouse model of peripheral neuropathic pain. Eur J Pharmacol 2009, 623: 65–72.
Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci 2007, 27: 6006–6018.
Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 2014, 137: 2193–2209.
Gao YJ, Zhang L, Ji RR. Spinal injection of TNF-alpha-activated astrocytes produces persistent pain symptom mechanical hypersensitivity by releasing monocyte chemoattractant protein-1. Glia 2010, 58: 1871–1880.
Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical hypersensitivity in this neuropathic pain model. Pain 2005, 114: 149–159.
Xu X, Chen H, Ling BY, Xu L, Cao H, Zhang YQ. Extracellular signal-regulated protein kinase activation in spinal cord contributes to pain hypersensitivity in a mouse model of type 2 diabetes. Neurosci Bull 2014, 30: 53–66.
Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur J Immunol 2008, 38: 448–458.
Yang Y, Li H, Li TT, Luo H, Gu XY, Lu N, et al. Delayed activation of spinal microglia contributes to the maintenance of bone cancer pain in female Wistar rats via P2X7 receptor and IL-18. J Neurosci 2015, 35: 7950–7963.
Mogil JS. Chapter 23 Sex, gender and pain. Handb Clin Neurol 2006, 81: 325–341.
Bartley EJ, Fillingim RB. Sex differences in pain: a brief review of clinical and experimental findings. Br J Anaesth 2013, 111: 52–58.
Bale TL, Epperson CN. Sex differences and stress across the lifespan. Nat Neurosci 2015, 18: 1413–1420.
Pleis JR, Ward BW, Lucas JW. Summary health statistics for U.S. adults: National Health Interview Survey, 2009. Vital Health Stat 10 2010: 1–207.
Kowalczyk WJ, Sullivan MA, Evans SM, Bisaga AM, Vosburg SK, Comer SD. Sex differences and hormonal influences on response to mechanical pressure pain in humans. J Pain 2010, 11: 330–342.
Mogil JS. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci 2012, 13: 859–866.
Fillingim RB. Biopsychosocial contributions to sex differences in pain. BJOG 2015, 122: 769.
Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 2015, 18: 1081–1083.
Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin JS, Ritchie J, et al. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci 2011, 31: 15450–15454.
Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lu N, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin Invest 2014, 124: 1173–1186.
Taves S, Berta T, Liu DL, Gan S, Chen G, Kim YH, et al. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav Immun 2016, 55: 70–81.
Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, et al. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical hypersensitivity after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci 2006, 26: 3551–3560.
Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 2003, 306: 624–630.
Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol 1980, 67: 313–316.
Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 2009, 29: 4096–4108.
Berta T, Qadri YJ, Chen G, Ji RR. Microglial signaling in chronic pain with a special focus on caspase 6, p38 MAP kinase, and sex dependence. J Dent Res 2016, 95: 1124–1131.
Chen G, Xie RG, Gao YJ, Xu ZZ, Zhao LX, Bang S, et al. beta-arrestin-2 regulates NMDA receptor function in spinal lamina II neurons and duration of persistent pain. Nat Commun 2016, 7: 12531.
Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 1980, 20: 441–462.
Qadri SM, Halim M, Ueno Y, Saldin H. Susceptibility of methicillin-resistant Staphylococcus aureus to minocycline and other antimicrobials. Chemotherapy 1994, 40: 26–29.
Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002, 417: 74–78.
Bastos LF, Merlo LA, Rocha LT, Coelho MM. Characterization of the antinociceptive and anti-inflammatory activities of doxycycline and minocycline in different experimental models. Eur J Pharmacol 2007, 576: 171–179.
Sommer C, Lindenlaub T, Teuteberg P, Schafers M, Hartung T, Toyka KV. Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res 2001, 913: 86–89.
Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical hypersensitivity after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 2003, 23: 2517–2521.
Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 2008, 28: 5189–5194.
Li J, Xie W, Zhang JM, Baccei ML. Peripheral nerve injury sensitizes neonatal dorsal horn neurons to tumor necrosis factor-alpha. Mol Pain 2009, 5: 10.
Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain 2011, 152: 419–427.
Fu KY, Light AR, Matsushima GK, Maixner W. Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 1999, 825: 59–67.
Cao H, Zhang YQ. Spinal glial activation contributes to pathological pain states. Neurosci Biobehav Rev 2008, 32: 972–983.
Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein-1 expression and spinal glial activation following peripheral nerve injury. J Neurochem 2006, 97: 772–783.
Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev 2009, 60: 135–148.
Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ. Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol 2007: 359–389.
Spataro LE, Sloane EM, Milligan ED, Wieseler-Frank J, Schoeniger D, Jekich BM, et al. Spinal gap junctions: potential involvement in pain facilitation. J Pain 2004, 5: 392–405.
White JR, Lee JM, Young PR, Hertzberg RP, Jurewicz AJ, Chaikin MA, et al. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J Biol Chem 1998, 273: 10095–10098.
Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A 2005, 102: 5856–5861.
Singh SK, Stogsdill JA, Pulimood NS, Dingsdale H, Kim YH, Pilaz LJ, et al. Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1alpha and NL1 via Hevin. Cell 2016, 164: 183–196.
Gosselin RD, Suter MR, Ji RR, Decosterd I. Glial cells and chronic pain. Neuroscientist 2010, 16: 519–531.
Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci 2003, 23: 1026–1040.
Obata H, Eisenach JC, Hussain H, Bynum T, Vincler M. Spinal glial activation contributes to postoperative mechanical hypersensitivity in the rat. J Pain 2006, 7: 816–822.
Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology 1994, 33: 1471–1478.
Chen MJ, Kress B, Han X, Moll K, Peng W, Ji RR, et al. Astrocytic CX43 hemichannels and gap junctions play a crucial role in development of chronic neuropathic pain following spinal cord injury. Glia 2012, 60: 1660–1670.
Ohara PT, Vit JP, Bhargava A, Jasmin L. Evidence for a role of connexin 43 in trigeminal pain using RNA interference in vivo. J Neurophysiol 2008, 100: 3064–3073.
Cho IH, Chung YM, Park CK, Park SH, Lee H, Kim D, et al. Systemic administration of minocycline inhibits formalin-induced inflammatory pain in rat. Brain Res. 2006, 1072: 208–214.
Bastos LF, Prazeres JD, Godin AM, Menezes RR, Soares DG, Ferreira WC, et al. Sex-independent suppression of experimental inflammatory pain by minocycline in two mouse strains. Neurosci Lett 2013, 553: 110–114.
Ji RR, Xu ZZ, Wang X, Lo EH. Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol Sci 2009, 30: 336–340.
IOM (Institute of Medicine). Sex Differences and Implications for Translational Neuroscience Research: Workshop Summary. Washington, DC: The National Academies Press, 2011: 10–11. ISBN: 0-309-16125-8.
Acknowledgements
This work was supported by NIH R01 grants DE17794, DE22743, and NS87988 to RRJ. YJQ was supported by NIH T32 2T32GM008600 and a Foundation of Anesthesia Education and Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, G., Luo, X., Qadri, M.Y. et al. Sex-Dependent Glial Signaling in Pathological Pain: Distinct Roles of Spinal Microglia and Astrocytes. Neurosci. Bull. 34, 98–108 (2018). https://doi.org/10.1007/s12264-017-0145-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-017-0145-y