
Summary
This short review aims at summarizing the current standards of lymphoma diagnostics and some novelties in the recent WHO classification. The importance of close collaboration between clinicians and pathologists to render the correct diagnosis and to find the most appropriate treatment for each individual patient is highlighted. In lymphomas, the diagnostic evaluation of histopathology, immune phenotype and genetics are puzzle pieces that have to be put into a broader context with the help of the information given by the clinical colleagues, such as patient’s age and sex, location of the lesion, previous medical history and medication. An excision of the affected lymph node is always preferable to fine needle biopsies, as—in many instances—only the evaluation of the whole specimen allows for reliable diagnosis, grading and additional investigations.
The new WHO classification entailed many changes in the category of diffuse large B‑cell lymphoma and high-grade B‑cell lymphoma. The obligatory specification of the cell of origin in diffuse large B‑cell lymphoma is obtained via additional immunohistochemical stainings. The identification of high-grade B‑cell lymphomas with genetic double/triple hits, requiring a more aggressive management, can only be achieved by the detection of chromosomal translocations (MYC, BCL2 and/or BCL6). Significant changes in the classification of T‑cell lymphomas have occurred due to the recognition of the follicular T‑helper cell origin in some instances, and sharpening diagnostic borders of intestinal T‑cell- and Epstein-Barr-virus-associated proliferations. Finally, the discovery of disease-defining and/or prognostically relevant mutations makes the introduction of proper routine molecular testing mandatory.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lymphomas are the 5th to 7th most frequently occurring malignant diseases in both sexes. It is not completely understood why there is still an increasing incidence of mature B‑cell lymphomas, yet a link to the broad use of immunomodulatory drugs, the increased prevalence of immunoderegulatory diseases and the exposure to environmental pesti-/herbicides is suspected [1, 2]. Fortunately, the progress in lymphoma therapy is noticeable: survival rates of patients continuously increase, at least when referring to the most common nodal lymphomas [1].
Here, we summarize the current standards in lymphoma diagnostics from the pathologist’s point of view, with a special focus on a multidisciplinary approach.
Gain of material
Histopathology is the keystone in lymphoma diagnostics, since this is the procedure of choice to distinguish between benign and malignant lymph node (LN) changes [3]. Importantly, a substantial number of benign LN disorders may both clinically and morphologically mimic lymphomas. Furthermore, genetic aberrations detectable in lymphomas as well as clonal outgrowths may be observed in reactive lymphoid processes, emphasizing the role of specifically trained histopathologists with regard to this type of diagnostics.
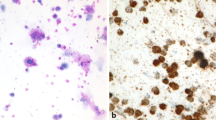
The trend towards less invasive procedures in the past years did not stop in front of hemato-oncology, leading to widespread use of fine needle biopsies (FNB) for diagnostic purposes. Though being informative in approximately 85% of cases (Fig. 1 and Table 1), there are several objective limitations and risks arising from the general use of FNB. For instance the distinction between angioimmunoblastic T‑cell lymphoma (AITL) and (e.g. drug-induced) paracortical hyperplasia [3] can only be achieved by the assessment of the total LN architecture, including the capsule and the surrounding soft tissue. These features cannot be evaluated on FNB as they merely contain a small detail of the LN. For obvious reasons, partial LN involvements by any processes are hardly possible to be diagnosed on FNB. In follicular lymphoma (FL), a statement about the grading is required, since grade 3B and—probably—grade 3A require a more aggressive clinical management, but this is barely obtainable on FNB. Additionally, some entities, such as Hodgkin lymphoma (HL) and AITL, go along with a very small amount of diagnostic tumor cells, which can easily be missed in a thin core cylinder (Fig. 1). Last but not least, lymphoma diagnostics require in almost all cases additional work-up, which sometimes cannot be applied to FNB due to lack of material or a too small amount of tumor cells. This also implies that there will be usually no material available for scientific questions after a diagnostic FNB work-up.

Lymphadenectomy specimen of a patient suffering from nodular lymphocyte predominant Hodgkin lymphoma with hyperplastic follicles and progressively transformed germinal centers. Note that the so called lymphocytic and histiocytic (L&H) tumor cells are only focally present in the respective zoomed-in zone (upper right part of the figure). A previous core needle biopsy of the patient (shown in the lower right part of the figure) was of no diagnostic yield; H&E stain
For the reasons stated above, we strongly recommend—whenever possible—a total LN excision for lymphoma diagnostic purposes in order to avoid misinterpretations and unnecessary re-biopsies. If a positron emission tomography (PET) is preformed prior to the excisional procedure, the LN with the highest PET activity should be extirpated [4]. FNB should remain a tool for exceptional situations, e.g. in difficult surgically accessible regions such as the mediastinum and retroperitoneum. To avoid material waist in such circumstances, careful histotechnical handling is advisable, particularly production of sufficient amounts (at least 15) of unstained sections on adhesive glass slides for subsequent in situ histopathologic (immunohistochemistry, in situ hybridization) and in vitro molecular (gene expression profiling, sequencing on material scrapped-off those slides) analyses. In addition and minimizing the risk of expiry, parafilm wrapped unstained sections can be stored for a long time for scientific questions.
Processing of the specimen
Optimally, the gained material should be fixed immediately. If specific queries such as frozen section examination arise—which must be regarded as an exception in that consideration—the fresh specimen should be immediately brought to the histopathology lab and the remaining LN tissue timely fixed. Usually, buffered formalin (final formaldehyde concentration 4%) is used and should be applied in a specimen:formalin ratio of at least 1:10. The fixed material is best stored at a temperature between 4 and 8 °C. Due to crystallization artefacts, storage below 0 °C should be avoided. Depending on the size, sufficient fixation is obtained after 4 to 12 h. If frozen section is considered inevitable and immediate delivery for histopathologic examination is not feasible but manageable within 3 h, the material should be sent in a dry container for respective examination by means of a courier transport, in our opinion—taxi. Contact of the tissue with any fluids, such as saline, would induce considerable freezing procedure artifacts, and is therefore not recommended. If transportation within 3 h is not possible, a fast-track formalin/paraffin technical processing (fixation/dehydration) should be preferred to frozen section examination. If submission of unfixed material is considered for other requests, such as flow cytometry or cytogenetics, one part of the obtained tissue can be put into Roswell Park Memorial Institute (RPMI) medium and used for these examinations, while the other should be submitted in formalin and utilized for morphological diagnostics.
Clinical information
The delivery of sufficient clinical information is crucial for lymphoma diagnostics. Indeed, as indicated in Table 2, several lymphoma entities cannot be diagnosed outside specific clinical settings. In the following, we list the minimum clinical key-facts for rendering a correct diagnosis.
-
1.
Age: Several lymphoma entities are defined by the patient’s age as they almost exclusively occur in young individuals and are thought to be associated with the immaturity of the immune system: e.g. pediatric type follicular lymphoma (PTFL), pediatric nodal marginal zone lymphoma and EBV-positive T‑cell and NK-cell lymphoproliferative diseases of childhood [5]. PTFL is an impressive example how the correct pathological classification influences the following treatment: in contrast to patients with conventional FL, who are often treated with multimodal chemotherapy, surgical excision alone seems to be sufficient for most PTFL patients [6, 7]. Information on sex and especially on ethnicity may be of upmost diagnostic importance in some virally driven lymphomas such as adult T‑cell lymphoma/leukemia [5].
-
2.
Location: Some lymphomas manifest at characteristic locations and information about the site can be an important diagnostic component. For example, primary cutaneous follicle center lymphomas occur most often at the head or trunk, have an excellent prognosis and mostly require only localized therapy; without the knowledge of the clinical appearance, the differential diagnosis to cutaneous involvement by conventional FL might be difficult if not impossible [5]. The same applies to CD30+ cutaneous lymphoproliferations with respect to the differential diagnosis of cutaneous involvement by systemic anaplastic T‑cell lymphoma (ALCL) [5]. The rare entity of large B‑cell lymphoma with IRF4 rearrangement (Fig. 2) is almost exclusively found in the Waldeyer ring or head and neck region of younger individuals. It has to be distinguished from PTFL, on the one hand, as it probably needs a more aggressive treatment, and from DLBCL or FL grade 3B, on the other, as the latter two will need much more aggressive treatment [8].
Special characteristics of the site of involvement can play an important diagnostic role. The presence of chronic inflammatory conditions such as chronic pyothorax, vascular and joint prosthesis, chronic skin ulcers or chronic osteomyelitis will render the correct diagnosis of a DLBCL associated with chronic inflammation [9, 10]. These cases should be recognized since they benefit from adjuvant surgical treatment of the underlying inflammatory condition and, despite being associated with Epstein–Barr virus (EBV) infection and displaying MYC amplifications and TP53 mutations, seem not to run a very aggressive course. Unquestionably, almost all patients with breast implant-associated ALCL, which is also a nice example of an integrative diagnostic entity, will be in need of excision alone [11].
-
3.
Previous illnesses: Detailed anamnestic information about pre-existing illnesses are indispensable. For example, the diagnosis of post-transplant lymphoproliferative disorders cannot be set without appropriate background. The same applies for lymphoproliferative diseases associated with (primary) immunodeficiencies, such as e.g. Wiskott-Aldrich syndrome or ataxia-teleangiectasia [5]. Furthermore, the correct classification of DLBCL arising in/transforming from chronic lymphocytic leukemias (CLL), marginal zone lymphomas and FL can only be made with the clinical note of a previously existing small B‑cell lymphoma (SBCL), in case the low grade compound is not found in the diagnostic LN. This is of importance since DLBCL, high-grade B‑cell lymphoma (HGBCL) or HL transformed from SBCL show a more aggressive behavior than their de novo counterparts [12].
-
4.
Medication: Considering the above-mentioned examples, it becomes also obvious that information about the patient’s previous medication is essential for the correct diagnosis of iatrogenic immunodeficiency-associated lymphoproliferative disorders, which not only virologically and genetically differ from their immunoproficient equivalents, but also often require a different management [5, 13, 14]. A number of applied drugs (e.g. carbamazepine, allopurinol, sulfonamides, methimazol, lamotrigine, gabapentin, nevirapine) and vaccinations against hepatitis B virus and smallpox can induce profound morphologic LN changes mimicking AITL, DLBCL or HL [3], the misdiagnosis of which will be most likely avoided in the setting of proper medication history knowledge.

Large B‑cell lymphoma (Giemsa stain) with IRF4 rearrangement (insert). Without application of fluorescent in situ hybridization (FISH) this consultational case has elsewhere been diagnosed as diffuse large B‑cell lymphoma, illustrating that the sole possibility establishing this integrative genetico-pathologic diagnosis is specific testing for the respective recurrent aberration. The insert shows the typical appearance of a translocation/chromosomal break with a break-apart FISH probe with fused yellow-orange IRF4 locus signals corresponding to the non-rearranged allele at 6p25 and free red and green signals corresponding to the rearranged allele
Morphology and phenotype
After paraffin embedding of the LN specimen, conventional histotechniques will mostly allow the correct differentiation between benign and malignant lesions or at least establishing a few differential diagnoses. The proper lymphoma classification concerning B‑ versus T‑cell origin, developmental stage of the lymphoma cells and specific entity assignment is mostly done via immunohistochemical stainings. The current WHO classification brought important changes in this regard, two of which will be exemplified here. Inevitably, now the “cell of origin” specification has to be given in all DLBCL, not otherwise specified: distinguishing these deriving from germinal center B‑cells from those displaying a program of activated B‑cells [15]. There are several techniques and algorithms to tackle the cell of origin in DLBCL, such as gene expression profiling and immunohistochemistry, each with different costs, prerequisites, feasibility and validity. No special technique has currently been recommended by the WHO [5], yet data suggest that stratification based on immunohistochemical algorithms for guiding therapy should be viewed very cautiously [16]. Nonetheless, it appears that stratification according to the so-called “Tally” algorithm, based on the evaluation of CD10, GCET1, FOXP1, MUM1p and LMO2, avoiding BCL6, may give the best results [17].
Also by immunophenotyping, a more precise classification of mature T‑cell lymphomas is now possible. Next to AITL, further entities of follicular T‑helper cell (TFH) origin have been identified (nodal peripheral T‑cell lymphoma with TFH phenotype (mostly T‑zone lymphoma), and follicular T‑cell lymphoma) and can be diagnosed using proper markers: PD1, BCL6, ICOS, CXCL13 and CD10 [18]. The detection of EBV by in situ hybridization has become extremely sensitive [19] and should be broadly applied to identify EBV-associated lymphomas and related lymphoproliferations [5, 19].
Genotype
Several lymphomas bear characteristic, to a part entity-specific, chromosomal aberrations (Table 2). This is also reflected in the recent WHO classification with the introduction of several genetically defined entities such as HGBCL with MYC and BCL2 and/or BCL2 rearrangements, large B‑cell lymphoma with IRF4 rearrangement, Burkitt-like lymphoma with 11q aberration, and ALK-positive lymphomas. Importantly, the (over)emphasis of genetics over other diagnostic parameters is particularly noticeable in HGBCL with MYC and BCL2 and/or BCL6 rearrangements, which are defined only by the presence of the respective rearrangements [20, 21]. Put into practice, this means that all mature blastic B‑cell lymphomas have to be tested for those translocations, e.g. by FISH, irrespective of their double-expressor score, since protein recognition by antibodies for MYC and, occasionally, of BCL2 may be abrogated due to somatic (hyper-)mutation of the respective genes [22]. Since interphase FISH with histopathological material is performed on sliced cells, cut-off scores to judge cases positive for rearrangements are needed. These scores depend on the probes utilized, the exact slide thickness and the size of the (tumor) cells studied, and are—in the case of commonly used break-apart FISH probes for MYC, BCL2 and BCL6 applied to 4 μm slides of DLBCL—4, 3 and 1.5% cells with break-apart signals, respectively [23].
Besides structural chromosomal anomalies, point mutations play an important role in the development of lymphomas and can be used for diagnostic or—yet to be broadly proven—theranostic purposes. For instance, diagnosing a lymphoplasmacytic lymphoma (LPL) used to be difficult and not always doubtlessly possible. Since the characteristic MYD88 L265P mutation (especially in combination with CXCR4 mutation) has been detected, the correct diagnosis can easily be set in many cases [24]. Importantly, in the setting of LPL and DLBCL (particularly of the ABC subtype) this MYD88 mutation seems to predict sensitivity towards ibrutinib [25]. The same applies for the BRAF V600E-mutation in hairy cell leukemia: although the mutation is not specific for this entity, its detection in the context of mature leukemic B‑cell lymphomas provides not only an important diagnostic tool, but also predicts sensitivity towards vemurafenib [26, 27]. Furthermore, quantification of both described mutations is valuable for monitoring disease progression or detection of minimal residual disease. Alterations in the TP53 gene are found in many malignant diseases, including different lymphomas. Identifying such mutations allows in small lymphocytic B‑cell lymphoma (B-CLL) very precise risk stratification, prediction of lack of response to fludarabine-containing therapy and sensitivity towards ibrutinib [28, 29]. In B‑CLL, the landscape of predictive mutations steadily grows (e.g. NOTCH1, ibrutinib-resistant BTK mutations), and we expect that similar data will soon emerge for other lymphomas [30, 31].
Finally, genetic testing is helpful in the diagnostics of recurrent disease: e.g. the differentiation between true late relapsing DLBCL or HL and metachronous de novo lymphoma can only be answered by proving a clonal relationship between both; same applies for the distinction of true SBCL transformation into e.g. DLBCL versus independent metachronous DLBCL [32,33,34].
Conclusion
The close collaboration between clinical oncologists and pathologists is indispensable for high standard lymphoma diagnostics. Equally, the pathologists depend on the information given by the clinical colleagues, as the clinician is dependent on a correct diagnosis provided by the pathologist. In the last few years, an enormous increase of knowledge considering tumorigenesis in general and lymphomagenesis in particular, has been recorded. This progress will make individualized, patient- and disease-tailored and, thus, more efficient and effective treatment possible.
Take-home message
For rendering a correct lymphoma diagnosis, a minimum of information provided by the clinician is needed: patient’s age (sex, ethnicity), tumor location, medical history and medication.
Besides accurate diagnostics, progress in molecular pathology makes statements on prognosis and theranostics in lymphomas possible.
References
Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. US lymphoid malignancy statistics by World Health Organization subtypes. Ca Cancer J Clin. 2016; https://doi.org/10.3322/caac.21357.
Schinasi L, Leon ME. Non-Hodgkin lymphoma and occupational exposure to agricultural pesticide chemical groups and active ingredients: a systematic review and meta-analysis. Int J Environ Res Public Health. 2014;11(4):4449–527.
Tzankov A, Dirnhofer S. A pattern-based approach to reactive lymphadenopathies. Semin Diagn Pathol. 2018;35(1):4–19.
Ansell SM, Armitage JO. Positron emission tomographic scans in lymphoma: convention and controversy. Mayo Clin Proc. 2012;87(6):571–80.
Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon: IARC; 2017.
Attarbaschi A, Beishuizen A, Mann G, Rosolen A, Mori T, Uyttebroeck A, et al. Children and adolescents with follicular lymphoma have an excellent prognosis with either limited chemotherapy or with a “watch and wait” strategy after complete resection. Ann Hematol. 2013;92(11):1537–41.
Louissaint A Jr., Schafernak KT, Geyer JT, Kovach AE, Ghandi M, Gratzinger D, et al. Pediatric-type nodal follicular lymphoma: a biologically distinct lymphoma with frequent MAPK pathway mutations. Blood. 2016;128(8):1093–100.
Salaverria I, Philipp C, Oschlies I, Kohler CW, Kreuz M, Szczepanowski M, et al. Translocations activating IRF4 identify a subtype of germinal center-derived B‑cell lymphoma affecting predominantly children and young adults. Blood. 2011;118(1):139–47.
Copie-Bergman C, Niedobitek G, Mangham DC, Selves J, Baloch K, Diss TC, et al. Epstein-Barr virus in B‑cell lymphomas associated with chronic suppurative inflammation. J Pathol. 1997;183(3):287–92.
Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B‑cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832–6.
Miranda RN, Aladily TN, Prince HM, Kanagal-Shamanna R, de Jong D, Fayad LE, et al. Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol. 2014;32(2):114–20.
Fong D, Steurer M, Greil R, Gunsilius E, Spizzo G, Gastl G, et al. Hodgkin lymphoma in Tyrol—a population-based study. Ann Hematol. 2009;88(5):449–56.
Menter T, Juskevicius D, Alikian M, Steiger J, Dirnhofer S, Tzankov A, et al. Mutational landscape of B‑cell post-transplant lymphoproliferative disorders. Br J Haematol. 2017;178(1):48–56.
DeStefano CB, Desai SH, Shenoy AG, Catlett JP. Management of post-transplant lymphoproliferative disorders. Br J Haematol. 2018; https://doi.org/10.1111/bjh.15263.
Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B‑cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–11.
Gutiérrez-García G, Cardesa-Salzmann T, Climent F, González-Barca E, Mercadal S, Mate JL, et al. Gene-expression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B‑cell lymphoma treated with immunochemotherapy. Blood. 2011;117(18):4836–43.
Meyer PN, Fu K, Greiner TC, Smith LM, Delabie J, Gascoyne RD, et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B‑cell lymphoma treated with rituximab. J Clin Oncol. 2011;29(2):200–7.
Lunning MA, Vose JM. Angioimmunoblastic T‑cell lymphoma: the many-faced lymphoma. Blood. 2017;129(9):1095–102.
Weiss LM, Chen YY. EBER in situ hybridization for Epstein-Barr virus. Methods Mol Biol. 2013;999:223–30.
Howlett C, Snedecor SJ, Landsburg DJ, Svoboda J, Chong EA, Schuster SJ, et al. Front-line, dose-escalated immunochemotherapy is associated with a significant progression-free survival advantage in patients with double-hit lymphomas: a systematic review and meta-analysis. Br J Haematol. 2015;170(4):504–14.
Aukema SM, Siebert R, Schuuring E, van Imhoff GW, Kluin-Nelemans HC, Boerma EJ, et al. Double-hit B‑cell lymphomas. Blood. 2011;117(8):2319–31.
Hoeller S, Tzankov A, Stenner F, Dirnhofer S. When and how to test for C‑MYC in aggressive B cell lymphomas. J Hematop. 2015;8(1):13–20.
Tzankov A, Xu-Monette ZY, Gerhard M, Visco C, Dirnhofer S, Gisin N, et al. Rearrangements of MYC gene facilitate risk stratification in diffuse large B‑cell lymphoma patients treated with rituximab-CHOP. Mod Pathol. 2014;27(7):958–71.
Swerdlow SH, Kuzu I, Dogan A, Dirnhofer S, Chan JK, Sander B, et al. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch. 2016;468(3):259–75.
Wang YL. MYD88 mutations and sensitivity to Ibrutinib therapy. J Mol Diagn. 2018;20(2):264–6.
Turakhia S, Lanigan C, Hamadeh F, Swerdlow SH, Tubbs RR, Cook JR. Immunohistochemistry for BRAF V600E in the differential diagnosis of hairy cell leukemia vs other splenic B‑cell lymphomas. Am J Clin Pathol. 2015;144(1):87–93.
Pettirossi V, Santi A, Imperi E, Russo G, Pucciarini A, Bigerna B, et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood. 2015;125(8):1207–16.
Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, et al. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B‑cell leukemias. Blood. 1995;85(6):1580–9.
Buccheri V, Barreto WG, Fogliatto LM, Capra M, Marchiani M, Rocha V. Prognostic and therapeutic stratification in CLL: focus on 17p deletion and p53 mutation. Ann Hematol. 2018;97(12):2269–78.
Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127(17):2122–30.
Bottoni A, Rizzotto L, Lai TH, Liu C, Smith LL, Mantel R, et al. Targeting BTK through microRNA in chronic lymphocytic leukemia. Blood. 2016;128(26):3101–12.
Obermann EC, Mueller N, Rufle A, Menter T, Mueller-Garamvoelgyi E, Cathomas G, et al. Clonal relationship of classical Hodgkin lymphoma and its recurrences. Clin Cancer Res. 2011;17(16):5268–74.
Juskevicius D, Lorber T, Gsponer J, Perrina V, Ruiz C, Stenner-Liewen F, et al. Distinct genetic evolution patterns of relapsing diffuse large B‑cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia. 2016;30(12):2385–95.
Montoto S. Treatment of patients with transformed lymphoma. Hematology Am Soc Hematol Educ Program. 2015;2015:625–30.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M.M. Gerlach and A. Tzankov declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Gerlach, M.M., Tzankov, A. Current lymphoma diagnostic standards: the pathologists’ view. memo 12, 17–23 (2019). https://doi.org/10.1007/s12254-019-0472-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12254-019-0472-y