
Abstract
Microarray, RT-qPCR based arrays and next-generation-sequencing (NGS) are available high-throughput methods for miRNA profiling (miRNome). Analytical and biological performance of these methods were tested in identification of biologically relevant miRNAs in non-functioning pituitary adenomas (NFPA). miRNome of 4 normal pituitary (NP) and 8 NFPA samples was determined by these platforms and expression of 21 individual miRNAs was measured on 30 (20 NFPA and 10 NP) independent samples. Complex bioinformatics was used. 132 and 137 miRNAs were detected by all three platforms in NP and NFPA, respectively, of which 25 were differentially expressed (fold change > 2). The strongest correlation was observed between microarray and TaqMan-array, while the data obtained by NGS were the most discordant despite of various bioinformatics settings. As a technical validation we measured the expression of 21 selected miRNAs by individual RT-qPCR and we were able to validate 35.1%, 76.2% and 71.4% of the miRNAs revealed by SOLiD, TLDA and microarray result, respectively. We performed biological validation using an extended number of samples (20 NFPAs and 8 NPs). Technical and biological validation showed high correlation (p < 0.001; R = 0.96). Pathway and network analysis revealed several common pathways but no pathway showed the same activation score. Using the 25 platform-independent miRNAs developmental pathways were the top functional categories relevant for NFPA genesis. The difference among high-throughput platforms is of great importance and selection of screening method can influence experimental results. Validation by another platform is essential in order to avoid or to minimalize the platform specific errors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are short, noncoding RNA molecules that posttranscriptionally regulate gene expression through RNA interference. They target mRNAs at the 3′, 5′ untranslated regions or even the coding sequence [1,2,3,4]. It has been shown that about 30-50% of all protein coding genes are regulated by miRNAs [5, 6], hence participating in the regulation of various physiological and pathophysiological cellular processes such as proliferation, differentiation, metabolism and apoptosis.
Pituitary adenomas represent the second most frequent (15.3%) central nervous system tumors following meningiomas [7]. Clinically non-functioning pituitary adenomas (NFPAs) constitute approximately 30% of all tumor types of the anterior pituitary [8] and the majority of them are gonadotrophic and null cell adenomas [9].
The role of miRNAs in pituitary adenomas have been evaluated extensively and thoroughly reviewed by Sivapragasam [10] and Li [11]. In the past few years several high-throughput techniques such as hybridization-based approaches (e.g. miRNA microarrays), reverse transcription PCR based arrays, or next generation sequencing (NGS) based methods become available for an initial screening to identify miRNome expression and to select specific miRNA candidates for further validation. However, detection of miRNAs can be more challenging than gene transcripts due to their special structure making difficult the assay and probe designs. Firstly, miRNAs are short RNA sequences hence it is hard to achieve high sensitivity assays. It has been proved that a certain mature miRNA can sometimes comprise a distribution of sizes of 15 − 23 nt (centered around 22 nt.) rather than having a single length [12] which is called sequence heterogeneity and the miRNA “variants” are called ‘isomiRs’. Occurrence of isomiRs has been attributed to posttranscriptional modifications at 3′ and 5′ ends that seems to affect miRNA stability and function (especially if it occurs at the 5′ influencing seed region [12]. Mature miRNAs lack of polyA tail (or other common sequence) making unavailable application of universal primers/probes in their expression studies. Additionally, members of the same miRNA family can differ only in 1-2 nucleotides making the discrimination difficult. Finally, among miRNA sequences the GC content show significant variance which results in difference in melting temperatures hence in multiplex assays it can create miRNA-specific biases [12].
Due to these difficulties it has been well known that miRNA expression profiles could be different using various platforms [13, 14]. Therefore our aim was to compare the results obtained by three high-throughput miRNA screening methods and by performing technical and biological validations to identify the differentially expressed miRNAs in NFPA compared to normal pituitary (NP). The functional relevance of differentially expressed miRNAs revealed by high-throughput methods was tested using pathway and network analysis.
Methods
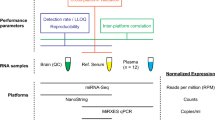
Samples and RNA Extraction
In high-throughput experiments 8 NFPAs and 4 normal pituitary tissues, in validation phase 20 NFPA and 10 NP specimens were examined. Adenoma tissues were removed by transsphenoideal surgery at the Hungarian National Institute of Neurosurgery. All adenoma samples were gathered with the permission of the Local Committee on Human Research. The diagnosis of NFPA was based on clinical findings, hormone levels and immunohistochemistry analysis of the removed tumor tissue. Routine immunohistochemical examination included immunostaining for anterior pituitary hormones (GH, PRL, ACTH, LH, FSH, SF1 and TSH) and staining for Ki-67 proliferation marker. All routine immunohistochemical studies were performed at the 1st Department of Pathology and Experimental Cancer Research, Semmelweis University, Budapest, Hungary as previously described [15, 16]. Normal pituitary samples were obtained by autopsy within 6 h of death from patients with no evidence of any endocrine disease (University Clinical Centre, Belgrade, Serbia) [15]. We performed gene expression measurements in all isolated RNA specimens to measure the expression of genes encoding anterior hormones and Pit1. This later gene has been demonstrated that is expressed exclusively in the Rathke’s pouch and adenohypophysis during the development of pituitary in both chicken and mouse embryos and not in the neuroectoderm (Proszkowiec-Weglarz et al. 2011). By using these expressions it could be concluded that all pituitary samples used as normal contained all five adenohypophophysis specific cell types (data not shown) [15,16,17,18]. Tumor tissue specimens were immediately frozen in liquid nitrogen after the adenoma removal, and stored at -80 °C until use. Total RNA was extracted with miRNeasy Mini Kit (Qiagen Inc., Chatsworth, CA). RNA integrity (RIN) and concentration was measured using Agilent Bioanalyzer 2100 System (Agilent Tech Inc., Santa Clara, USA), samples having RNA integrity number (RIN) >7 were included.
Next-Generation Sequencing and Data Analysis
MicroRNA expression of 8 NFPA and 4 NP tissue specimens were analyzed using SOLiD next-generation-sequencing in two pools. From total RNA small RNA enrichment was performed with miRNA Isolation Kit (Invitrogen). MicroRNA libraries were prepared according to the manufacturer’s protocol (Small RNA Expression Kit, Applied Biosystems). Briefly, 100 ng miRNA was hybridized and ligated overnight with adapter mix. cDNA was generated by reverse transcription from adaptors ligated to ends of the small RNA molecule. PCR products were cleaned and selected on agarose gels by size 105–150 bp. Template bead preparation, emulsion PCR were performed using the SOLiD V2 sequencing system (Applied Biosystems). Data analysis was carried out using Small RNA Analysis tool of CLCBio Genomics Workbench v5.0 using general settings for quality check and adapter trimming. We accepted reads having 15-23 nt in size as “expressed”. Then we aligned miRNA reads to miRBase mature miRNA sequences allowing 0 or 1 mismatches. Read numbers then were normalized for the total reads of each sample. Fold change was determined in NFPA samples compared to normal ones. The deep sequencing was performed at the Sequencing Platform, Institute of Biochemistry, Biological Research Centre of the Hungarian Academy of Sciences, Szeged, Hungary [19].
Microarray Experiment and Data Analysis
GeneChip® microRNA Galaxy Array v1 (Affymetrix, CA, USA), which comprises probe sets for 1015 human mature miRNAs was used for miRNA expression analysis. 500 ng pooled total RNA was processed for each group. Poly (A) tailing reaction and ligation of the biotinylated signal molecule to the target RNA sample was carried out using FlashTag Biotin HSR RNA Labeling Kit (Affymetrix, PN 703095) following the manufacturer’s instructions. Then, the labeled and biotinylated RNA and hybridization controls (GeneChip Eukaryotic Hybridization Control Kit, PN 900454) were hybridized to miRNA arrays for 16 h at 48 °C. Hybridization and staining was performed using GeneChip® Hybridization, Wash, and Stain Kit (PN 702731) and each array was washed and stained in a GeneChip Fluidics station 450 (Affymetrix) and scanned by a GeneChip 3000 scanner (Affymetrix) according to the manufacturer’s instructions.
Data analysis was performed by Genespring GX 12 Software (Agilent Tech Inc., Santa Clara, CA, USA) using standard settings. Briefly, raw data was filtered by percentile (lower cut-off: 20). Fold change filter was set to 2-fold, and then unpaired t-test was used to identify significant (p < 0.05) gene expression changes with multiple testing correction (Benjamini-Hochberg) to control the false discovery rate.
miRNA TaqMan Low Density Array and Data Analysis
We used our previously published miRNA expression dataset obtained using TaqMan Low Density Array (TLDA) Human MicroRNA Panel v.2 (Applied Biosystems, Foster City, CA) on 8 NFPA and 4 normal pituitary samples. Procedures are described in details by Butz [20].
Individual miRNA qPCR and Data Analysis
Expression level of following miRs: hsa-miR-128a (Assay ID: 4,395,327), hsa-miR-135a (Assay ID: 4,373,140), hsa-miR-135b (Assay ID: 4,395,372), hsa-miR-140-5p (Assay ID: 4,373,374), hsa-miR-155 (Assay ID: 4,395,459), hsa-miR-15a (Assay ID: 4,373,123), hsa-miR-16 (Assay ID: 4,373,121),hsa-miR-17-5p (Assay ID: 4,395,419), hsa-miR-20a (Assay ID: 4,373,286), hsa-miR-383 (Assay ID: 4,373,018), hsa-miR-422a (Assay ID: 4,395,408), hsa-miR-424 (Assay ID: 4,373,201), hsa-miR-486-3p (Assay ID: 4,395,204), hsa-miR-503 (Assay ID: 4,373,228), hsa-miR-516a-3p (Assay ID: 4,373,183), hsa-miR-542-3p (Assay ID: 4,378,101), hsa-miR-543 (Assay ID: 4,395,487), hsa-miR-582-3p (Assay ID: 4,395,510), hsa-miR-582-5p (Assay ID: 4,395,175), hsa-miR-93 (Assay ID: 4,373,302), hsa-miR-98 (Assay ID: 4,373,009), RNU44 (Assay ID: 4,373,384), RNU48 (Assay ID: 4,373,383), U6 snRNA (MammU6, Assay ID: 4,395,470) were determined in pituitary samples using individual TaqMan MicroRNA Assays (Applied Biosystems) followed the protocol provided by the supplier and was described earlier in details by Butz [20]. Expression level was calculated by ddCt method, and fold changes were obtained using the formula 2-ddCt [20].
Bioinformatics Analysis
Target prediction for miRNAs differentially expressed between NFPA and NP tissues was performed using Tarbase and miRecords databases. Only the experimentally observed miRNA targets were taken into consideration in order to increase the reliability of results. Then the target lists with the predicted expression alteration were submitted to Ingenuity Pathway analysis (http://www.ingenuity.com/ products/ipa). We also used miRNA-target interaction data obtained from each platform to build networks that were visualized by Cytoscape 3.1.0. software as previously described [21, 22]. Briefly, following network structure analysis node’s color and size were indicated by indegree (number or targeting miRNA). Correlations were performed using R, p values were considered to be significant at p < 0.05.
Results
miRNA Expression Profiles and Correlations Among Platforms
A final database was generated containing all miRNAs detected by all three platforms aligning miRNAs by sequences (MiRBase release 21). We included miRNAs expressed at least in one sample/platform and we filtered out miRNAs where sequences were not exact matches among the three platforms due to miRNA annotation differences among platforms. Therefore, in our merged database we compared 166, 718 and 440 miRNAs by SOLiD, microarray and TLDA, respectively in NP and 180, 718 and 440 miRNAs in NFPA samples, respectively. Among which 132 (in NP) and 137 (in NFPA) were common in all three datasets (Fig. 1a).

a Numbers of detected miRNAs by SOLiD, Microarray and TLDA platforms in normal pituitary (NP) and non-functioning pituitary adenoma (NFPA) b Multiplex correlation among samples and platforms. Numbers indicate correlation coefficient (R), scales show normalized expression. Above the diagonal line the scatter plots, below the diagonal line the correlation coefficients are indicated for each comparison. See the text for detailed explanation
After correlating miRNA expression profiles obtained by the different platforms significant correlations between platforms, with the best correlation (R = 0.75) between microarray and TLDA and the weakest between SOLiD-Microarray and SOLiD-TLDA data was observed (Fig. 1b). We performed cross-correlation of four datasets: TLDA-NP (normal pituitary), TLDA-NFPA (non-functioning adenoma), SOLiD-NP and SOLiD-NFPA. We found that expression values obtained by the same platform correlated better than expression values obtained by different platforms for the same samples (e.g R = 0.82 between TLDA-NP and TLDA-NFPA versus R = 0.44 between TLDA-NP and SOLiD-NP or R = 0.52 between TLDA-NFPA and SOLiD-NFPA). This phenomenon was observed in all comparison (Microarray vs. TLDA and Microarray vs. SOLiD as well). Based on this result we can conclude that platform effect is more significant than the group (tissue) effect (Fig. 1b).
SOLiD showed the weakest correlation with the other two platforms. In order to investigate whether these results might be related to the bioinformatical analysis we evaluated how the different minimum read numbers influenced the correlation. We determined the correlation coefficients using 3, 5 or 10 reads as a minimum expression cut-off, but changing the minimum read number cut-off had only a minor influence on correlation coefficients (Table 1). It also has to be mentioned that lower correlation was observed in normal tissues compared to NFPA samples (Table 1). Based on these results we considered a miRNA to be expressed using the cut-off = 5 reads.
In order to find out the reasons for the low correlation observed between data obtained by SOLiD deep sequencing and the other two platforms additional bioinformatical analysis were carried out. miRNAs that were detectable by both the TLDA and microarray platforms but not by SOLiD had expression values in the lower detection range compared to those which were detected by SOLiD (Fig. 2). In addition, a significant number of miRNAs (268 and 264 miRNA in NP and NFPA, respectively) were excluded from the statistical analysis because the reads mapping these miRNAs were either too short (<15 nt) or aligned to multiple positions in the miRNA reference database (Supplemental Table 1). Of these miRNAs we focused on those 28 miRNAs which were unidentified by SOLiD but were detected in NP and NFPA by both microarray and TLDA platforms. (Supplemental Table 2). No difference in the GC content of these miRNAs versus those detected was observed (data not shown). However we found some differences in the number of repeats in sequences: 39% and 31% of miRNAs contains more 3-6mer short repeats in the undetectable sequences compared to detectable ones by SOLiD in NP and NFPA, respectively (Supplemental Table 3). Regarding unbalanced nucleotide composition adenine or thymine at the first position in these miRNAs (21 out of 28 undetected miRNAs had A or T as the first base possibly influencing adapter ligation) may also be responsible for their failure through SOLiD system.

miRNAs that were not detected by SOLiD NGS but were identified by both microarray and TLDA, were in the low detection range. Columns represent mean expression, error bars indicating standard error (SE), *: p < 0.0001
Despite of low correlations, expression of 85 miRNAs changed in the same direction in all three high throughput methods (Supplementary Table 4). Of these, 25 miRNAs (12 downregulated and 13 upregulated in NFPA vs. NP) were identified whose expression significantly differed (>2 fold) in the same direction between NFPA and NP samples and their expression change occurred in the same direction in all the three platforms used (Table 2, Supplementary Figure 1).
Technical and Biological Validation
To reveal the impact of the differences in miRNAs profiles obtained by the different platforms, we validated the expression of 21 selected miRNAs by individual RT-qPCR using TaqMan miRNA Assays. These 21 miRNAs were chosen to cover miRNAs whom expression was concordant among platforms or discordant between SOLID and the other two platforms. Six out of 21 miRNAs (miR-17-5p, miR-20a, miR-543, miR-582-5p, miR-93 and miR-98) showed the same expression pattern in all the three high throughput methods, 9 miRNAs (miR-128a, miR-135a, miR-135b, miR-16, miR-422a, miR-486-3p, miR-516a-3p, miR-542-3p and miR-582-3p) were not detectable by SOLiD but measured by microarray and TLDA. MiR-135a was not detectable in the microarray experiment. During the technical validation, in the same samples as were used for high throughput analysis (8 NFPAs and 4 NPs) we were able to validate 35.1%, 76.2% and 71.4% of the miRNAs revealed by SOLiD, TLDA and microarray measurements (Fig. 3a). The technical validation was followed by biological validation using an extended number of samples (20 NFPAs and 8 NPs) (Fig. 3b). The expression values measured in this extended sample size correlated well with the expression values measured during the technical validation (p < 0.001; R = 0.96) (Fig. 3b and c).

Validation of selected 21 miRNAs’ expression a Technical validation shows the miRNA expression determined by individual RT-qPCR on the same samples as used for SOLiD, Microarray and TLDA experiment. b Technical vs. biological validation. Biological validation was performed on an extended number of samples (20 NFPAs and 10 NPs). c Correlation among different platforms (log2fold change) regarding biological validation. Red and green colours represent over- and underexpression in NFPAs compared to NPs, black indicates not identified miRNAs.
Pathway and Molecular Network Analysis for miRNAs Revealed by High Throughput Screening
In order to investigate the biological significance of high-throughput data we carried out a complex bioinformatical pathway and network analysis in order to reveal molecular function, signaling pathways and cellular processes affected by these miRNAs. We generated the differentially expressed gene lists from each platform applying a fold change cut-off 2.
Pathways for miRNAs identified by each, high throughput platform
A target prediction for each miRNA identified during expression analysis by high throughput methods were performed and the results were narrowed for the experimentally validated targets. Then, these miRNA target lists were subjected to pathway and network analysis. The pathway called “Molecular Mechanisms of Cancer” was the most significant pathway altered by miRNAs revealed by all three platforms. In addition, other five pathways were common among the miRNAs revealed by high throughput screening (Supplementary Figure 2A).
We also investigated the activation z-scores of pathways. The primary purpose of z-score is to infer the activation states of predicted pathway. A significantly altered pathway implies that the genes involved in the pathway (both activators and inhibitors) are significantly enriched in the gene set we used. Therefore z-score analysis can give a more thorough information about the pathway function and that can explain the difference between the result of pathway and z-score analysis (Supplementary Fig. 2a-b). However, no common signaling showing the same direction (activation or inhibition) by all three platforms was detected (Supplementary Figure 2B).
Network for miRNAs identified by each platform
Using Ingenuity Knowledge Database containing up-to-date information on molecular interactions we built networks of miRNAs and their experimentally validated target molecules. After analyzing network structure, remarkable differences according to the most significant miRNAs (defined as miRNAs with the highest number of targets) were observed. MiR-15a-5p, miR-98-5p and miR-155-5p were the most influential miRNAs having the most targets in SOLiD miRNA-target network, miR-124-3p, miR-1-3p and miR-497-5p in the microarray network and miR-506-3p, miR-206 and miR-424-5p in the TLDA network. Good overlap was found among the miRNA targets; PTEN, CDK6 and CCND1 being the most commonly influenced molecules by miRNAs in NFPA compared to NP. PTEN was targeted by 8, 10 and 7 miRNAs in SOLiD, Microarray and TLDA miRNA-networks. CDK6 was targeted by 7, 10 and 7 miRNAs, while CCND1 was targeted by 7, 9 and 8 miRNAs in the SOLiD, Microarray and TLDA networks (Fig. 4a, b and c).

Network analysis of miRNA-target interactions of different platforms (a SOLiD; b microarray; c TLDA; d 25 platform independent miRNAs). Node (molecule) size and colour represent the number of targeting miRNAs (indegree)
Pathways and molecular networks for common miRNAs among different high throughput methods
As we mentioned earlier 25 miRNAs were significantly differentially expressed in the same direction between NFPA and normal pituitary samples measured by all three platforms (Table 2, Supplementary Fig. 1). Investigating the potential pathways affected by the platform independent miRNAs we found a very similar result to those revealed by platform dependent miRNAs (Supplementary Figure 3). “Developmental biology”, “Wnt Signalling Pathway” and “TGF-beta Receptor Signaling Pathway” were the most significant signaling pathways (Supplementary Figure 3). We performed z-score analysis as well. In this analysis we presented the z-score of those pathways where the scores showed the same direction at least two studies. Interestingly, in case of “platform independent” miRNA analysis only the “Aryl Hydrocarbon Receptor Signaling” and “Cell Cycle: G1/S Checkpoint Regulation” showed significant alteration in z-scores whilst these two pathways did not have significant z-scores using the individual studies (Supplementary Fig. 2B).
The network analysis for these miRNAs revealed very similar results observed for platform dependent miRNAs. Of the 25 miRNAs miR-497, miR-34a and miR-429 were identified having the most targets in the network (Fig. 4b). GO analysis revealed the most important molecular functions and processes related to “RNA binding and transcription regulation”, “cell cycle” and “Wnt signaling Pathway” (Supplementary Figure 4).
Discussion
Various high throughput molecular biological methods in miRNA profiling are available for many tumor types including pituitary. However, miRNA profiling using deep sequencing has not been performed in non-functioning pituitary tumors. Our study was carried out in order to evaluate how the high throughput method would influence the results in miRNA expression studies. We showed that the number of detected miRNAs were very different among the three platforms used and globally the correlation among these methods vary significantly. The strongest correlation was found between microarray and TLDA cards; and correlations between deep sequencing performed by SOLiD System and microarray or TLDA were weaker. These significant but not remarkably strong correlations are in line with earlier data. Git et al. have found similar correlation between NGS read numbers and microarray hybridization intensity (correlation coefficient 0.66 ± 0.12) [14], while in a study by Wang et al. the inter-platform reproducibility correlation coefficients varied between 0.106 ± 0.039 and 0.48 ± 0.096 among LNA microarray, beads arrays and TLDA [23]. It is thoμght that the disagreement arises from nonspecific contributions, varying degrees of cross-hybridization of miRNA family members or reduced discrimination between unprocessed and mature forms of the miRNAs [23].
To choose the best high throughput screening method seems essential in identification of differentially expressed miRNAs but the method itself may bias the results. Each technique has pros and cons reviewed by Pritchard [12]. Hybridization-based methods are widely used techniques requiring ng-ug total input RNA hence the sensitivity is limited and the dynamic range for detection is not that wide compared to PCR-based approaches. Also, due to the short sequence length, end region sequence variation (isomiRs) and high conservation among miRNA family members it is challenging to design specific probes that all affect the specificity. PCR based arrays are also broadly used for determining miRNA expression profile. It has a very wide detection range but also requires probe design similarly to hybridization methods. A broadly accepted “gold standard” method in miRNA expression study is a two-step approach using looped miRNA specific reverse transcription primers and TaqMan probes for quantification. Next-generation-sequencing based approaches are becoming more and more popular in various molecular biological studies. Related to miRNA expression, compared to microarrays and PCR based methods, deep sequencing does not require predesigned probes hence it is able to identify novel miRNAs and can distinguish isomiRs. Although some NGS library preparation methods and the sequencing technology are not developed for short (<35 bp) sequences.
Our recent data showed that the inter-platform correlation was the weakest between NGS and the other two methods. We tested whether the bioinformatics settings i.e. the minimum expression level (min. read number) would affect the results. Our results showed that changing this cut-off number did not have a major effect on inter-platform correlation. On the other hand, the unidentified miRNAs by SOLiD System but detected by the other two platforms were in the low detection range suggesting that that this early version of the SOLID System technology had a less sensitivity compared to PCR or hybridization based platforms. It has also to be mentioned that these undetected miRNAs are usually excluded from the statistical analysis because either of their short lengths or because of not unique alignment. We also observed that these reads contains more nucleotide repeats compared to those miRNAs that were detected by SOLiD and in 75% the first nucleotide of the “undetected reads” were A or T suggesting that the discrepancy may arise from improper adapter ligation. This observation is in line with others showing that the library preparation techniques can introduce sequence-bias by amplifying some miRNAs while reducing others due to the sequence preferences of the ligation enzymes or to the differences in the secondary structures of RNAs [24].
According to technical validation we found an acceptable percentage for all platforms. Although TLDA and individual TaqMan RT-qPCR are similar methodologically the percentage of validation (76.2%) could be expected higher. The differences between TLDA and RT-qPCR protocols (pooled reverse transcription in TLDA vs. individual RT and qPCR; preamplification used in TLDA measurement vs. without preamplification; differences in the reaction volume 1 μl in TLDA vs. 15 μl in individual RT-qPCR) can influence the efficiency of both the RT and qPCR leading to this discrepancy that was found between the two approaches. Also, high variance of the replicates at TLDA system (median: 8.3%; min-max: 0.3-19.1%) has been also described in literature [25].
Altogether, the small overlap among different platforms found by us can explain why it is difficult to compare different miRNA studies showing very different results. Until now three miRNA profiling studies have been presented. We checked the expression of the 21 miRNAs identified with concordant expression in these three datasets and it was found that 8 of 21 were common in Bottoni and our previous study, 13 of 21 were common in Liang and our previous publication but we could not find any common miRNA between the significant miRNA lists of Bottoni and Liang studies [26, 27] (Table 3). It is noteworthy that the adenoma groups were varied among the three studies which may additionally increase the weak overlap.
Going further in the understanding of the biological role of miRNAs in NFPA tumorigenesis and keeping in mind the redundant effects of miRNAs it seems that the whole miRNome would characterize better one state than only a limited sets of miRNAs. Therefore, in order to decipher the biological relevance of miRNAs a pathway and network analysis were carried out. Network analysis is a relatively novel tool for analyzing high-throughput data. Comparing it to pathway analysis it is considered to be less biased because while pathway analysis basically performs a gene set enrichment analysis on pre-defined gene sets (groups of molecules belonging to certain pathways) network analysis visualize and analyze single interactions among molecules. Several common pathways were recognized for individual miRNA lists but, interestingly, we could not identify a common signaling pathway showing the same activation score (activation score indicates the activation state of the pathways based on the expression level and direction of miRNAs). This finding is a probable consequence of the poor overlap of differentially expressed miRNAs in NFPA vs. NPs among the various platforms used. The same result was observed in the network analysis as well. However, the miRNA HUBs in the networks were different but there were a remarkable overlap among the miRNAs targeted gene HUBs. These data may underline again the redundancy of target prediction and may be related to the miRNAs’ divergent and convergent function.
After having in hands these disappointing results we focused on the 25 miRNAs which were significantly differentially expressed between NFPA and normal pituitary samples measured by all three platforms. The Gene ontology and pathway analysis showed that these miRNAs are involved in regulation of developmental processes through targeting Wnt and TGF signaling. Wnt signaling is described to be activated in both pituitary organogenesis and its mature function [28]. In pituitary similarly to other tissue types Wnt signaling pathways control cell activity and may stimulate cell proliferation [28]. Indeed, Elston et al. reported that Wnt pathway inhibitors are strongly down-regulated in pituitary tumors (both non-functioning and clinically functioning pituitary tumors) compared with normal pituitary controls in all pituitary subtypes [29]. They suggested that WIF1 may be a tumor suppressor, specifically in NFPAs, and the Wnt pathway is important in pituitary tumorigenesis [29]. However, this finding was not validated by other reports showing no activation of canonical and non-canonical Wnt pathway activation in pituitary adenoma [30, 31]. Based on these discrepant results further studies are warranted for clarification of the role of miRNAs targeting molecules involved in Wnt signaling in pituitary adenomagenesis.
The second pathway for platform independent miRNAs was the TGF-β pathway. Several earlier studies, including ours, reported the possible involvement of this pathway in pituitary adenomagenesis [20, 32,33,34]. We showed the downregulation of the TGFβ pathway through miRNAs targeting Smad3 in NFPAs compared to NP, while Zhenye [34] reported that the activity of TGF-β signaling might be restrained in NFPAs and this result correlated with the development and invasion of NFPAs. The Smad3 and Phospho-Smad3 protein levels were found to be gradually decreased from normal anterior pituitaries, to non-invasive NFPAs and to invasive NFPAs [34]. All these data may confirm that TGFβ signaling seems to be important in development of NFPA.
Conclusion
Our study demonstrated that miRNA expression profiling has several limitations and the platform dependent effects may cause significant bias. However, it is also true that individual miRNA expression data obtained from high throughput techniques were replicable in an acceptable percentage by qRT-PCR suggesting these tools are useful in identification of miRNAs with potential biological function. On the other hand the redundancy observed in target prediction (and therefore in pathway and network analysis) may weaken or mask the differences of individual miRNAs showing a better overlap among pathways than significant miRNA lists obtained among different platforms. In summary it is highly warranted to validate the miRNA expression obtained by any high throughput method using another platform and an extended sample set.
Pathway and network analysis of platform-independent miRNAs and their potential targets demonstrated that differentially expressed miRNAs were likely involved in the tumorigenesis of NFPA through regulating developmental pathways.
References
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T (2001) Identification of novel genes coding for small expressed RNAs. Science 294:853–858. https://doi.org/10.1126/science.1064921
Place RF, Li L-C, Pookot D et al (2008) MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 105:1608–1613. https://doi.org/10.1073/pnas.0707594105
Ørom UA, Nielsen FC, Lund AH (2008) MicroRNA-10a binds the 5’UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell 30:460–471. https://doi.org/10.1016/j.molcel.2008.05.001
Zhang Z, Florez S, Gutierrez-Hartmann A et al (2010) MicroRNAs regulate pituitary development, and microRNA 26b specifically targets lymphoid enhancer factor 1 (Lef-1), which modulates pituitary transcription factor 1 (Pit-1) expression. J Biol Chem 285:34718–34728. https://doi.org/10.1074/jbc.M110.126441
Chen K, Rajewsky N (2006) Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet 38:1452–1456. https://doi.org/10.1038/ng1910
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. https://doi.org/10.1016/j.cell.2004.12.035
Dolecek TA, Propp JM, Stroup NE, Kruchko C (2012) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol 14(Suppl 5):v1–49. https://doi.org/10.1093/neuonc/nos218
Dworakowska D, Grossman AB (2009) The pathophysiology of pituitary adenomas. Best Pract Res Clin Endocrinol Metab 23:525–541. https://doi.org/10.1016/j.beem.2009.05.004
Mayson SE, Snyder PJ (2014) Silent (clinically nonfunctioning) pituitary adenomas. J Neuro-Oncol 117:429–436. https://doi.org/10.1007/s11060-014-1425-2
Sivapragasam M, Rotondo F, Lloyd RV et al (2011) MicroRNAs in the human pituitary. Endocr Pathol 22:134–143. https://doi.org/10.1007/s12022-011-9167-6
Li X-H, Wang EL, Zhou H-M et al (2014) MicroRNAs in Human Pituitary Adenomas. Int J Endocrinol 2014:435171. https://doi.org/10.1155/2014/435171
Pritchard CC, Cheng HH, Tewari M (2012) MicroRNA profiling: approaches and considerations. Nat Rev Genet 13:358–369. https://doi.org/10.1038/nrg3198
Mestdagh P, Hartmann N, Baeriswyl L et al (2014) Evaluation of quantitative miRNA expression platforms in the microRNA quality control (miRQC) study. Nat Methods 11:809–815. https://doi.org/10.1038/nmeth.3014
Git A, Dvinge H, Salmon-Divon M et al (2010) Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA N Y N 16:991–1006. https://doi.org/10.1261/rna.1947110
Butz H, Likó I, Czirják S et al (2010) Down-regulation of Wee1 kinase by a specific subset of microRNA in human sporadic pituitary adenomas. J Clin Endocrinol Metab 95:E181–E191. https://doi.org/10.1210/jc.2010-0581
Butz H, Németh K, Czenke D et al (2016) Systematic Investigation of Expression of G2/M Transition Genes Reveals CDC25 Alteration in Nonfunctioning Pituitary Adenomas. Pathol Oncol Res POR. https://doi.org/10.1007/s12253-016-0163-5
Thompson IR, Chand AN, King PJ et al (2012) Expression of guanylyl cyclase-B (GC-B/NPR2) receptors in normal human fetal pituitaries and human pituitary adenomas implicates a role for C-type natriuretic peptide. Endocr Relat Cancer 19:497–508. https://doi.org/10.1530/ERC-12-0129
Trivellin G, Butz H, Delhove J et al (2012) MicroRNA miR-107 is overexpressed in pituitary adenomas and inhibits the expression of aryl hydrocarbon receptor-interacting protein in vitro. Am J Physiol Endocrinol Metab 303:E708–E719. https://doi.org/10.1152/ajpendo.00546.2011
Harmati M, Tarnai Z, Decsi G et al (2016) Stressors alter intercellular communication and exosome profile of nasopharyngeal carcinoma cells. J Oral Pathol Med Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol. https://doi.org/10.1111/jop.12486
Butz H, Likó I, Czirják S et al (2011) MicroRNA profile indicates downregulation of the TGFβ pathway in sporadic non-functioning pituitary adenomas. Pituitary 14:112–124. https://doi.org/10.1007/s11102-010-0268-x
Butz H, Szabó PM, Nofech-Mozes R et al (2014) Integrative bioinformatics analysis reveals new prognostic biomarkers of clear cell renal cell carcinoma. Clin Chem 60:1314–1326. https://doi.org/10.1373/clinchem.2014.225854
Butz H, Szabó PM, Khella HWZ et al (2015) miRNA-target network reveals miR-124as a key miRNA contributing to clear cell renal cell carcinoma aggressive behaviour by targeting CAV1 and FLOT1. Oncotarget 6:12543–12557. 10.18632/oncotarget.3815
Wang B, Howel P, Bruheim S et al (2011) Systematic evaluation of three microRNA profiling platforms: microarray, beads array, and quantitative real-time PCR array. PLoS One 6:e17167. https://doi.org/10.1371/journal.pone.0017167
Chevillet JR, Lee I, Briggs HA et al (2014) Issues and prospects of microRNA-based biomarkers in blood and other body fluids. Mol Basel Switz 19:6080–6105. https://doi.org/10.3390/molecules19056080
Farr RJ, Januszewski AS, Joglekar MV et al (2015) A comparative analysis of high-throughput platforms for validation of a circulating microRNA signature in diabetic retinopathy. Sci Rep 5:10375. https://doi.org/10.1038/srep10375
Bottoni A, Zatelli MC, Ferracin M et al (2007) Identification of differentially expressed microRNAs by microarray: a possible role for microRNA genes in pituitary adenomas. J Cell Physiol 210:370–377. https://doi.org/10.1002/jcp.20832
Liang S, Chen L, Huang H, Zhi D (2013) The experimental study of miRNA in pituitary adenomas. Turk Neurosurg 23:721–727. https://doi.org/10.5137/1019-5149.JTN.7425-12.1
Chambers TJG, Giles A, Brabant G, Davis JRE (2013) Wnt signalling in pituitary development and tumorigenesis. Endocr Relat Cancer 20:R101–R111. https://doi.org/10.1530/ERC-13-0005
Elston MS, Gill AJ, Conaglen JV et al (2008) Wnt pathway inhibitors are strongly down-regulated in pituitary tumors. Endocrinology 149:1235–1242. https://doi.org/10.1210/en.2007-0542
Colli LM, Saggioro F, Serafini LN et al (2013) Components of the canonical and non-canonical Wnt pathways are not mis-expressed in pituitary tumors. PLoS One 8:e62424. https://doi.org/10.1371/journal.pone.0062424
Formosa R, Gruppetta M, Falzon S et al (2012) Expression and clinical significance of Wnt players and survivin in pituitary tumours. Endocr Pathol 23:123–131. https://doi.org/10.1007/s12022-012-9197-8
Lebrun J-J (2009) Activin, TGF-beta and menin in pituitary tumorigenesis. Adv Exp Med Biol 668:69–78
Ruebel KH, Leontovich AA, Tanizaki Y et al (2008) Effects of TGFbeta1 on gene expression in the HP75 human pituitary tumor cell line identified by gene expression profiling. Endocrine 33:62–76. https://doi.org/10.1007/s12020-008-9060-3
Zhenye L, Chuzhong L, Youtu W et al (2014) The expression of TGF-β1, Smad3, phospho-Smad3 and Smad7 is correlated with the development and invasion of nonfunctioning pituitary adenomas. J Transl Med 12:71. https://doi.org/10.1186/1479-5876-12-71
Acknowledgements
This work has been funded by National Research, Development and Innovation Office – NKFIH PD116093 to Henriett Butz. Attila Patocs is a recipient of “Lendulet” grant from Hungarian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The authors declare no potential conflicts of interest.
Electronic supplementary material
Supplemental Table 1
(DOCX 12 kb)
Supplemental Table 2
(DOCX 13 kb)
Supplemental Table 3
(DOCX 14 kb)
Supplemental Table 4
(DOCX 19 kb)
Supplementary Figure 1
Numbers of differentially expressed miRNAs in the same direction with a fold change cut-off 2 identified by different platforms (GIF 182 kb)
Supplementary Figure 2
a Pathway analysis of experimentally validated targets of differentially expressed miRNA lists obtained by different platforms b Comparison of most significant pathway activation z-scores of different platforms. Z-scores of pathways are presented where the scores showed the same direction at least two studies. Z-score infers the activation states of predicted pathway by investigating the activator or inhibitor function of the enriched genes in the particular pathway (GIF 43 kb)
Supplementary Figure 3
Pathway analysis of “platform independent” miRNAs (GIF 75 kb)
Supplementary Figure 4
Gene ontology analysis of “platform independent” miRNAs (GIF 183 kb)
Rights and permissions
About this article

Cite this article
Darvasi, O., Szabo, P.M., Nemeth, K. et al. Limitations of high throughput methods for miRNA expression profiles in non-functioning pituitary adenomas. Pathol. Oncol. Res. 25, 169–182 (2019). https://doi.org/10.1007/s12253-017-0330-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-017-0330-3