
Abstract
The bacterial diversity associated with eroding sponges belonging to the Cliona viridis species complex is scarcely known. Cliona thomasi described from the West Coast of India is a new introduction to the viridis species complex. In this study, we determined the bacterial diversity associated with C. thomasi using next-generation sequencing. The results revealed the dominance of Proteobacteria followed by Cyanobacteria, Actinobacteria and Firmicutes. Among Proteobacteria, the Alphaproteobacteria were found to be the most dominant class. Furthermore, at the genus level, Rhodothalassium were highly abundant followed by Endozoicomonas in sponge samples. The beta-diversity and species richness measures showed remarkably lower diversity in Cliona thomasi than the ambient environment. The determined lower bacterial diversity in C. thomasi than the environmental samples, thus, categorized it as a low microbial abundance (LMA). Functional annotation of the C. thomasi–associated bacterial community indicates their possible role in photo-autotrophy, aerobic nitrification, coupling of sulphate reduction and sulphide oxidization. The present study unveils the bacterial diversity in bioeroding C. thomasi, which is a crucial step to determine the functions of the sponge holobiont in coral reef ecosystem.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Marine sponges (phylum Porifera), one of the oldest multicellular animals (Metazoa), are an essential component of aquatic benthic communities (Wulff 2001; de Goeij et al. 2013). Sponges are also important members of coastal food webs and biogeochemical cycles and play a vital role in marine habitats by providing a significant number of important ecosystem services such as the establishment of a 3-dimensional structure that generates habitat for other organisms, water purification and nutrient cycling (Bell 2008). Interestingly, sponges are known to harbour the highest number of prokaryotic symbionts along with other invertebrates (Webster and Thomas 2016). Furthermore, the prokaryotic sponge symbionts were found to be stable, to a certain extent, at different physico-chemical environmental conditions such as variability in nutrient composition (Luter et al. 2014), temperature (Simister et al. 2012; Pita et al. 2013), pH (Ribes et al. 2012), light (Cárdenas et al. 2014) and at geographical (Steinert et al. 2016; Thomas et al. 2016) and temporal scales (Erwin et al. 2015). Such sponge-prokaryotic symbiotic association has recently been suggested to play roles in maintaining the fitness of sponges over other competitors in the same niche (Kiran et al. 2018; Pita et al. 2018). Sponges have also been considered as a reservoir of pharmacologically and biotechnologically important microbes (Villegas-Plazas et al. 2019). Growing evidence suggests that sponge microbial symbionts are the main producers of several documented sponge-derived bioactive compounds rather than the host itself (Pita et al. 2018). A number of sponge-holobiont publications reported microbial metabolic contributions to the host sponge, e.g. heterotrophy and autotrophy, and elemental recycling, along with the production of bioactive compounds (Webster and Thomas 2016; Bourne et al. 2016; Slaby et al. 2017).
Coral bioeroding sponges are the least studied for their microbiome association and functions. However, the role of sponge-holobiont in space competition and coral destruction is not clear. A few studies have shown that the sponges of Cliona viridis complex are associated with Symbiodiniaceae, which exert thermotolerance to them over their space competitor neighbouring corals (Ramsby et al. 2017; Achlatis et al. 2019). As a result, the invasion of sponge increased over the coral affected by the sudden increase in temperature, for instance, bleaching events (Carballo et al. 2013). The only reports available for microbial diversity analyses in Cliona viridis complex included C. viridis (Schmidt, 1862) from the Mediterranean (Blanquer et al. 2013; Soares 2016), C. varians (Duchassaing & Michelotti, 1864) from the Florida keys (Sacristán-Soriano et al. 2020) and C. orientalis Thiele, 1900 from the Great Barrier Reef (Pineda et al. 2016, 2017; Ramsby et al. 2018b). However, the prokaryotic associates of these bioeroding sponges were neglected for such sponge-holobiont interactions. Therefore, it is crucial to define the sponges and their associates as a holobiont unit rather than only the sponge as an autonomous entity (Roughgarden et al. 2017; Pita et al. 2018). Before gaining insights into the physiological plasticity induced in the host by a change in associated microbiome, it is necessary to investigate the communitity structure of this microbiome. Thus, the aim of the present study is to determine the bacterial community associated with coral excavating sponge Cliona thomasi Mote, Schönberg, Samaai, Gupta & Ingole, 2019. The same study also described C. thomasi as an aggressive benthic competitor in coral reef, which overgrows at a faster rate over the live coral colonies and outcompete live corals (Mote et al. 2019). This species belongs to the C. viridis species complex, which is considered to be among the most dominant and destructive macroborers on coral reefs (Schönberg et al. 2017). C. thomasi has been observed as a fast-growing, coral-infesting species with high ecological significance.
Cliona thomasi has also been listed as one of the WORMS top ten new marine species of 2019 (http://lifewatch.be/en/worms-top10-2018), thereby highlighting its high ecological importance. Interestingly, C. thomasi has shown an increase in its prevalence over coral after a mass bleaching event in the year 2015–2016. Our subsequent continuous monitoring of the site showed an increased abundance of C. thomasi. This observed increase in C. thomasi, at first instance, may be attributed to an increased SST induced coral bleaching providing favourable ground for the sponge to proliferate. The sea surface temperature (SST) data from the NOAA Coral Reef Watch (NOAA-CRW) platform revealed that the SST of the sponge habitat had exceeded the thermal bleaching threshold in the summer months (April–June) in the years 2015, 2016, 2017, 2018 and 2019 (Supplementary Fig. 1). During the monitoring of this site, an anomalous increase in SST was observed during summer months. In order to gain insights into the host associate–derived physiological benefits, this study defines the bacterial diversity associated with C. thomasi as a first step investigation.
Materials and methods
Sample collection
Samples of the bioeroding sponge, C. thomasi growing on colonies of the hard coral Turbinaria mesenterina (Lamarck, 1816), were collected from the Malvan Marine Sanctuary (16° 3′ 46.76″ N, 73° 27′ 17.18″ E) located on the central West Coast of India. Corals in the Malvan Marine Sanctuary are under stress due to elevated temperature-induced bleaching events (De et al. 2015), coral diseases (Hussain et al. 2016), sedimentation and physical damages from unregulated tourism (De et al. 2020). The samples were collected from a depth of 5–7 m, and care was taken while collecting the samples that each sample was distinct and separated from others by at least 5 m. The growth of C. thomasi over the coral species T. mesenterina is shown in Fig. 1. The sponge samples were collected in replicates of four. To compare the community with the ambient environment, surrounding seawater samples (n = 3) were collected from the same depth 1 m away from the sponge. Samples were immediately brought on board, fixed in liquid nitrogen and transported to the laboratory for further processing.

In-situ observation of sponge Cliona thomasi growing over Turbinaria mesenterina in Malvan Marine Sanctuary
DNA extraction and library preparation
The collected samples were homogenized in liquid nitrogen and processed for DNA extraction using a tissue DNA extraction kit according to the manufacturer’s protocol (Invitrogen, CA, USA). About 2 L of seawater was filtered using a 0.22-μm polycarbonate membrane filter (Whatman ®), and residue that remained on filter paper was used for DNA isolation. For bacterial community analysis, amplification of the 16S rRNA V3-V4 region was performed (Muyzer et al. 1993; Li et al. 2009). Illumina MiSeq 16S rRNA amplicon libraries were generated following a standard protocol (New England Biolabs, Frankfurt, Germany). The libraries were validated using 2100 Bioanalyzer (Agilent Technologies) for quality, and samples were sequenced using the 2 × 300 paired-end chemistry (MiSeq Reagent Kit).
Sequence assembly and prokaryotic community analysis
Illumina Miseq platform generated raw 16S rRNA amplicon reads for all the samples were processed for quality filtering and adaptor removal using FastQC V0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). After trimming, V3–V4 region sequences were assembled using the FLASH program 1.2.11 with a minimum overlap cutoff of 10 bp and maximum overlap cutoff of 240 bp. By default mismatches were allowed (default is 0) and as a result produced an average contig length 350–450 bp. The chimera sequences were then removed using the de novo based UCHIME method implemented in the USEARCH tool version 11.0.667. Assembled reads were subjected to QIIME version 2.0 pipeline for downstream bioinformatics analysis (Bolyen et al. 2019). First, assembled reads were clustered using UCLUST (Edgar, 2010) and operational taxonomic units (OTUs) were picked at a 97% similarity. The singleton OTUs (read abundance < 2) were discarded from the analysis (Caporaso et al. 2010). The most abundant reads for the remaining OTUs were selected as representatives and then mapped against rRNA database SILVA release (132 version using PyNAST) (Caporaso et al. 2010). Taxonomy for each mapped OTUs was assigned using the RDP classifier using a threshold value of 0.8 (Wang et al. 2007). The OTUs that did not match to any reference taxonomy were considered as unknown. We used METAGENassist (Arndt et al. 2012) for assessing putative functional analysis of sponge bacterial community.
Statistical analysis
The diversity indices such as species richness and Shannon index were determined for all the investigated samples in R, package vegan v.2.5-6 (Oksanen 2017). The rarefaction curves for the samples were derived from the genus summary using the same package. The significant differences among the diversity indices values were determined by the Kruskal-Wallis test (with Dunn’s post hoc test) in Past version 3. The differences in the beta-diversity of the bacterial community composition between the sponge and environmental samples were tested using PERMANOVA with the Bray Curtis test followed by 999 permutations in PRIMER v7 (Clarke and Gorley 2015). Similarity percentage (SIMPER) analysis was performed to calculate the contribution of each bacterial community to the dissimilarity within and between samples in the PRIMER v7 (Clarke and Gorley 2015). Bacterial community composition across samples was ordinated using non-metric multidimensional scaling (nMDS) and ANOSIM performed in R, package vegan v.2.5-6 (Oksanen 2017).
Results
Prokaryotic diversity associated with C. thomasi
In this study, high-throughput 16S rRNA gene (V3 and V4 regions) sequencing analysis was performed for the newly defined coral-eroding sponge C. thomasi to assess its associated bacterial diversity. The 16SrRNA analysis determined 3,17,333–5,51,000 quality reads assigned to 2531–9581 sponge OTUs while 3,64,284–4,79,774 reads were assigned to 18,672–34,773 environmental OTUs (Supplementary Table 1). The rarefaction curve showed high sequencing coverage and higher taxonomic assignments to the environmental samples than sponge samples (Supplementary Fig. 2). Alpha diversity indices obtained from the analysis showed higher diversity and richness in environment samples than the sponge samples. Shannon’s diversity index determined for the sponge and environment samples were 1.79 ± 0.12 and 3.16 ± 0.99, respectively (Supplementary Table 1). The determined diversity indices were found to be significantly different for sponge and environment samples based on the Kruskal-Wallis test, followed by Dunn’s posthoc test (p < 0.05).
The beta-diversity composition of sponge samples was found to be significantly different from the environment samples (PERMANOVA; p = 0.02, at permutation N: 999). The significant difference in the bacterial composition between sponge and environment samples were further confirmed by ANOSIM (Global R = 0.66, p < 0.023). The multivariate clustering from nMDS also confirmed the variation in the bacterial community composition among sponge and the surrounding environment. The clustering of samples based on bacterial taxonomic abundance variation is reflected in nMDS as well as in heatmap (Supplementary Figs. 3 and 4). SIMPER analysis determined the key bacterial genera that contributed to the differences in the sponge and environmental samples. The bacterial genera which contributed to 75.65% dissimilarity between the sponge and environmental samples were Rhodothalassium, Thalassomonas, Neptuniibacter, Vibrio, Endozoicomonas, Lutibacter and Synechococcus (Supplementary Table 2).
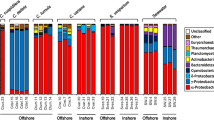
A total of 23 phyla were found to be associated with the sponge species C. thomasi while 33 phyla in environmental samples (ES). At the phylum level, all the samples included in this study were found to be dominated by Proteobacteria (sponge: 64.7.9 ± 4.90%, ES: 66.30 ± 20.0%), followed by Cyanobacteria (sponge: 12.69 ± 10.69%, ES: 1.20 ± 1.16%), Actinobacteria (sponge: 4.85 ± 3.94, ES: 3.07 ± 2.09%) and Bacteroidetes (sponge: 1.79 ± 3.94, ES: 14.53 ± 9.05%) (Fig. 2). Cyanobacteria was found significantly higher in sponge while Bacteroidetes in environmental samples (p value < 0.05).

Relative abundance of prokaryotic communities associated with the sponge Cliona thomasi and environmental samples at the phylum level
Within the Proteobacteria at class level, sponge samples were found to be dominated by Alphaproteobacteria (43.73 ± 11.11%) followed by Deltaproteobacteria (09.64 ± 6.89%), Gammaproteobacteria (2.20 ± 1.10%) and Betaproteobacteria (1.10 ± 0.01%) (Fig. 3a). The environmental samples, however, were found to be dominated with Gammaproteobacteria (50.33 ± 26.81%) followed by Alphaproteobacteria (8.05 ± 3.83%), Deltaproteobacteria (4.48 ± 3.21%) and Betaproteobacteria (0.06 ± 0.02%) (Fig. 3b). The significantly higher abundance of bacterial genera (p value < 0.05) found in sponge samples were Rhodothalassium (sponge: 24.17 ± 8.96%, ES: 0.007 ± 0.003%), Synechococcus (sponge: 5.82 ± 4.11%, ES: 0.24 ± 0.34%), Endozoicomonas (2.08 ± 1.47%, ES: 0.02 ± 0.01%), Prochlorococcus (sponge: 0.07 ± 0.11%, ES: 0.07 ± 0.01%), Shewanella (sponge: 0.55 ± 0.30%, ES: 0.02 ± 0.01%) and Corynebacterium (sponge: 0.10 ± 0.05%, ES: 0.01 ± 0.01%) (Fig. 4). The sponge-specific bacterial genera were dominated by Methylobacterium, Brevibacterium, Cupriavidus and Massilia, while the environmental samples were dominated by Vibrio, Neptuniibacter, Alteromonas, Thalassomonas, Photobacterium and Lutibacter (Fig. 4). Both the sponge and environmental samples showed significantly higher unidentified bacterial communities-58% and 62%, respectively. The unknown bacterial diversity was excluded from the study while analysing the data.

a, b Relative abundance variations within Proteobacteria associated with the sponge and environmental samples

Relative abundance of prokaryotic communities associated with the sponge Cliona thomasi and environmental samples at the genus level
Functional annotation
This study further determined the taxonomic based predictive functions of sponge-specific prokaryotic communities using the METAGENassist web portal (Supplementary Fig. 5). The sponge-associated bacterial population–based predicted metabolic processes mainly include the breakdown of complex organic molecules such as xylan, chitin and chlorophenol degraders, and sugar fermenters. Metabolism functions revealed that ammonia oxidizers, nitrite reducers, nitrogen fixers, sulphate reducers and sulphate oxidizers were enriched in the sponge. Other processes identified in sponge were Chlorophenol degradation, Dehalogenation, Lignin degradation and Naphthalene degradation.
Discussion
The present study describes the bacterial diversity associated with bioeroding sponge C. thomasi, belonging to the family Clionaidae. This species was known to harbour photosymbiont dinoflagellate Zooxanthallae (Mote et al. 2019), which may provide essential nutrients to the host as it does to other C. viridis complex species (Hill et al. 2011). Determining the bacterial diversity associated with C. thomasi in this study is the first step towards developing a baseline microbial diversity structure of C. thomasi.
Similar to earlier reports, C. thomasi investigated in this study was found to be dominated by Proteobacteria, together with the significant contributions by Cyanobacteria and Actinobacteria and frequent occurrence of Firmicutes, Bacteroidetes and Planctomycetes (Thomas et al. 2016). In the present study, C. thomasi showed the dominance of Alphaproteobacteria similar to the previous reports of sponge species from C. viridis complex and other Symbiodiniaceae-bearing sponges (Supplementary Table 3) (Blanquer et al. 2013; Soares 2016; Pineda et al. 2016, 2017; Ramsby et al. 2018b; Sacristán-Soriano et al. 2020). However, azooxanthellate sponges (without Symbiodiniaceae association) such as C. deltrix Pang, 1973 and C. celata Grant, 1826, were reported for the predominance of Gammaprotoeobacterial communities (Jeong et al. 2015; Thomas et al. 2016; Sacristán-Soriano et al. 2020).
The identification of cyanobacteria as sponge-specific photosynthetic associate of C. thomasi corroborates with the earlier reports of finding the Cyanobacteria (Synechococcus) as sponge-specific cluster and absent in seawater (Simister et al. 2012). The association of cyanobacteria in sponges benefits the host in various ways. For instance, compounds isolated from the cyanobacterial symbionts of the sponge Lamellodysidea herbacea deterred fish feeding (Sawhney and Mishra 2019; Schorn et al. 2019). Also, increased cyanobacteria abundance was shown to be an opportunistic proliferation in bleached C. orientalis, fulfilling the energy requirement of the host (Ramsby et al. 2018a). Morrow et al. (2015) showed the higher prevalence of Synechococcus in two different sponge species colonized in the acidified environment of a CO2 seep. Photosynthetic symbionts not only act as the energy source to the sponge but also provide photoprotective effects against the intermittent high light exposure to sponge (Steindler et al. 2002) and also known to produce cytotoxic secondary metabolites (Teruya et al. 2004; Matthew et al. 2010). In addition to photosynthetic symbionts, the other sponge-enriched microbes include Endozoicomonas, Williamsia, Shewanella and Vibrio. Endozoicomonas is a common bacterial genus widely reported from different sponges (Nishijima et al. 2013), a wide range of coral species and several other marine organisms (Jensen et al. 2010; Esteves et al. 2013). Endozoicomonas are known for multiple functions in sponges such as nutrient acquisition, structuring the sponge microbiome via signalling molecules or in host health stability (Nishijima et al. 2013; Gardères et al. 2015). The actinomycetes that belonged to the genera Williamsia are known for bioactive natural product synthesis in sponges (Audia et al. 2017). Shewanella and Vibrio are frequently encountered as sponge-associated microbes (Hentschel et al. 2012; Esteves et al. 2013).
The beta-diversity measures in this study showed a significant difference among bacterial diversity associated with the investigated sponge and environment samples (Supplementary Fig. 4). Recently, Steinert et al. (2019) also showed that the sponge’s prokaryotic community was significantly different from the surrounding water. Higher species richness and Shannon diversity indices for the environment samples attribute to their higher diversity than the sponge. The results highlighted less complexity of bacterial community in C. thomasi than its surrounding environment suggesting it as a low microbial abundance (LMA) sponge (Gloeckner et al. 2014). These results corroborate well with previous reports on other closely related Cliona species such as C. orientalis, C. varians and C. viridis (Blanquer et al. 2013; Poppell et al. 2014; Soares 2016; Pineda et al. 2016; Thomas et al. 2016; Sacristán-Soriano et al. 2020). The LMA characteristic of C. thomasi indicates a unique metabolic feature of higher availability of carbon in sponge choanosome than N and P through higher photosynthetic activity (Thacker and Freeman 2012). This fact is further supported by the identification of higher autotrophy through specifically determined Synechococcus together with sulphur metabolism–related bacteria in C. thomasi.
The sponge-enriched specific bacterial community is supportive of sponge holobiont with their distinct functions. The functions of sponge holobiont may broadly be classified into two categories (1) metabolism and (2) defence (Pita et al. 2018). The present study predicts the function of heterotrophic metabolism in C. thomasi through the degradation of complex polysaccharides such as xylan and chitin by the sponge-specific bacterial community. Heterotrophic carbon metabolism through hydrolysis of complex polysaccharide has widely been reported from the different sponge-associated bacterial community from different regions (Slaby et al. 2017). Heterotrophically metabolizing community association is further supported by the fact that the microbes are also dependent upon the sponge biomass itself or the components of the sponge extracellular matrix (Slaby et al. 2017; Tout et al. 2017). The enrichment of N metabolism–related functions identified in this study highlights the putative role of C. thomasi–associated microbes in the Nitrogen cycle, which is one of the limiting nutrients in the marine environment (Kiran et al. 2018; Pita et al. 2018). For instance, Rhodothalassium may play a role in nitrogen cycling in sponge (Parfrey et al. 2018; Ramsby et al. 2018b). Sponges are known to produce an excess of ammonia as a waste by-product, which is toxic to their photosymbionts if not being assimilated (Hentschel et al. 2012). The higher prevalence of functions related to ammonia oxidization in this study suggested the ammonia metabolism through either aerobic (nitrification, nitrogen fixation) or anaerobic (denitrification, anammox) manner (Hoffmann et al. 2009; Fiore et al. 2015; Ribes et al. 2015). Since nitrogen is the limiting factor for primary productivity in the marine ecosystem, an adaptation of microbial community capable of enriching N is essential in sponges for maintaining the symbiosis (Fan et al. 2012; Bayer et al. 2014; Moitinho-Silva et al. 2014). The determination of the higher abundance of nitrifying function in this study compared to denitrifying suggest that the sponge excrete nitrogen mainly in the form of ammonia (Southwell et al. 2008; Ribes et al. 2012; Morganti et al. 2017). The higher relative percentage of the function corresponding to nitrification further indicates that the C. thomasi holobiont act as a net source of bioavailable nitrogen (Fiore et al. 2013).
The other dominant function determined in C. thomasi based specific bacterial community was related to sulphur metabolism, mainly the sulphide oxidizer and sulphate reducer. Similar, endosymbiotic sulphur cycle through the association of sulphate-reducing and sulphate-oxidizing symbionts has been described in a marine oligochaete worm (Dubiller et al. 2001) and the cold water sponge Geodoa barrette (Hoffmann et al. 2003). These functions are mainly attributed to the Alphaproteobacteria and Gammaproteobacteria community (Jensen et al. 2017). The present study also determined the higher abundance of Rhodothalassium (Alphaproteobacteria) and Endozoicomonas (Gammaproteobacteria) community in C. thomasi. The coupling of sulphide oxidizer and sulphate reducer supports detoxification of sulphide produced by the metabolic activity of sponge or other associates under oxic or anoxic condition (Hoffmann et al. 2003). In the presence of oxygen, the sulphide is oxidized to sulphate by sulphide oxidizer. Nevertheless, under anoxic conditions in sponge tissue, the detoxification of sulphide was performed by sulphide reducers but necessitated the biologically available ferrous ions (Le Pennec et al. 2003; Orcutt et al. 2011). The presence of higher abundance of Endozoicomonas in Symbiodiniaceae-associated sponge C. thomasi indicates that it may play role in the sulphur cycle metabolizing of dimethylsulfoniopropionate (DMSP) to form dimethylsulphate (DMS) and dimethyl sulfoxide (DMSO) (Gardner et al. 2016; Osman et al. 2020). These molecules have potential roles in osmoregulation and antioxidant capacity of the host (Gardner et al. 2016).
In conclusion, the present study determined microbial community structure associated with a newly discovered coral-eroding sponge species. The results revealed that the sponges had microbial signature different from its surrounding environment. The microbial specificity associated with the C. thomasi highlights their putative roles benefiting the sponge holobiont. Investigating the sponge-holobiont is essential to realize a holistic regulatory, physiological and functional attributes to understand the space competitive mechanisms of sponge in the reef ecosystem.
Data availability
Accession numbers of nucleotide sequences submitted to NCBI are coded as follows: SRA submission SUB7245986, submission ID PRJNA623169 and Bio project PRJN623169.
References
Achlatis M, Schönberg CHL, van der Zande RM, LaJeunesse TC, Hoegh-Guldberg O, Dove S (2019) Photosynthesis by symbiotic sponges enhances their ability to erode calcium carbonate. J Exp Mar Bio Ecol 516:140–149. https://doi.org/10.1016/j.jembe.2019.04.010
Arndt D, Xia J, Liu Y, Zhou Y, Guo AC, Cruz JA, Sinelnikov I, Budwill K, Nesbo CL, Wishart DS (2012) METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res 40:88–W95. https://doi.org/10.1093/nar/gks497
Audia C, Afonso De Menezes B, Sanches Afonso R et al (2017) Williamsia spongiae sp. nov., an actinomycete isolated from the marine sponge Amphimedon viridis. Int J Syst Evol Microbiol 67:1260–1265. https://doi.org/10.1099/ijsem.0.001796
Bayer K, Moitinho-Silva L, Brümmer F, Cannistraci CV, Ravasi T, Hentschel U (2014) GeoChip-based insights into the microbial functional gene repertoire of marine sponges (high microbial abundance, low microbial abundance) and seawater. FEMS Microbiol Ecol 90:832–843. https://doi.org/10.1111/1574-6941.12441
Bell JJ (2008) The functional roles of marine sponges. Estuar Coast Shelf Sci 79:341–353. https://doi.org/10.1016/j.ecss.2008.05.002
Blanquer A, Uriz MJ, Galand PE (2013) Removing environmental sources of variation to gain insight on symbionts vs. transient microbes in high and low microbial abundance sponges. Environ Microbiol 15:3008–3019. https://doi.org/10.1111/1462-2920.12261
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS II, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857
Bourne DG, Morrow KM, Webster NS (2016) Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu Rev Microbiol 70:317–340. https://doi.org/10.1146/annurev-micro-102215-095440
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Carballo JL, Bautista E, Nava H et al (2013) Boring sponges, an increasing threat for coral reefs affected by bleaching events. Ecol Evol 3:872–886. https://doi.org/10.1002/ece3.452
Cárdenas CA, Bell JJ, Davy SK, Hoggard M, Taylor MW (2014) Influence of environmental variation on symbiotic bacterial communities of two temperate sponges. FEMS Microbiol Ecol 88:516–527. https://doi.org/10.1111/1574-6941.12317
Clarke KR, Gorley RN (2015) PRIMER v7 Plymouth Routines In Multivariate Ecological Research. www.primer-e.com. Accessed 15 Feb 2020
de Goeij JM, van Oevelen D, Vermeij MJA, Osinga R, Middelburg JJ, de Goeij AFPM, Admiraal W (2013) Surviving in a marine desert: the sponge loop retains resources within coral reefs. Science 342:108–110. https://doi.org/10.1126/science.1241981
De K, Sautya S, Mote S et al (2015) Is climate change triggering coral bleaching in tropical reef? Curr Sci 109:1379–1880
De K, Nanajkar M, Mote S, Ingole B (2020) Coral damage by recreational diving activities in a marine protected area of India: unaccountability leading to ‘tragedy of the not so commons. Mar Pollut Bull 155:111190. https://doi.org/10.1016/j.marpolbul.2020.111190
Dubiller N, Mülders C, Ferdelman T et al (2001) Endosymbiotic sulphate-reducing and sulphide-oxidizing bacteria in an oligochaete worm. Nature 411:298–302. https://doi.org/10.1038/35077067
Erwin PM, Coma R, López-Sendino P, Serrano E, Ribes M (2015) Stable symbionts across the HMA-LMA dichotomy: low seasonal and interannual variation in sponge-associated bacteria from taxonomically diverse hosts. FEMS Microbiol Ecol 91:1–11. https://doi.org/10.1093/femsec/fiv115
Esteves AIS, Hardoim CCP, Xavier JR, Gonçalves JMS, Costa R (2013) Molecular richness and biotechnological potential of bacteria cultured from Irciniidae sponges in the north-east Atlantic. FEMS Microbiol Ecol 85:519–536. https://doi.org/10.1111/1574-6941.12140
Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Webster NS, Thomas T (2012) Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci U S A 109:E1878–E1887. https://doi.org/10.1073/pnas.1203287109
Fiore CL, Baker DM, Lesser MP (2013) Nitrogen biogeochemistry in the Caribbean sponge, Xestospongia muta: a source or sink of dissolved inorganic nitrogen? PLoS One 8:e72961. https://doi.org/10.1371/journal.pone.0072961
Fiore CL, Labrie M, Jarett JK, Lesser MP (2015) Transcriptional activity of the giant barrel sponge, Xestospongia muta holobiont: molecular evidence for metabolic interchange. Front Microbiol 6(364):1–18. https://doi.org/10.3389/fmicb.2015.00364
Gardères J, Bedoux G, Koutsouveli V, Crequer S, Desriac F, Pennec G (2015) Lipopolysaccharides from commensal and opportunistic bacteria: characterization and response of the immune system of the host sponge Suberites domuncula. Mar Drugs 13:4985–5006. https://doi.org/10.3390/md13084985
Gardner SG, Nielsen DA, Laczka O, Shimmon R, Beltran VH, Ralph PJ, Petrou K (2016) Dimethylsulfoniopropionate, superoxide dismutase and glutathione as stress response indicators in three corals under short-term hyposalinity stress. Proc R Soc B Biol Sci 283:1–9. https://doi.org/10.1098/rspb.2015.2418
Gloeckner V, Wehrl M, Moitinho-Silva L, Gernert C, Schupp P, Pawlik JR, Lindquist NL, Erpenbeck D, Wörheide G, Hentschel U (2014) The HMA-LMA dichotomy revisited: an electron microscopical survey of 56 sponge species. Biol Bull 227:78–88. https://doi.org/10.1086/BBLv227n1p78
Hentschel U, Piel J, Degnan SM, Taylor MW (2012) Genomic insights into the marine sponge microbiome. Nat Rev Microbiol 10:641–654. https://doi.org/10.1038/nrmicro2839
Hill M, Allenby A, Ramsby B, Schönberg C, Hill A (2011) Molecular phylogenetics and evolution Symbiodinium diversity among host clionaid sponges from Caribbean and Pacific reefs: evidence of heteroplasmy and putative host-specific symbiont lineages. Mol Phylogenet Evol 59:81–88. https://doi.org/10.1016/j.ympev.2011.01.006
Hoffmann F, Rapp HT, Zöller T, Reitner J (2003) Growth and regeneration in cultivated fragments of the boreal deep water sponge Geodia barretti bowerbank, 1858 (Geodiidae, Tetractinellida, Demospongiae). J Biotechnol 100:109–118. https://doi.org/10.1016/S0168-1656(02)00258-4
Hoffmann F, Radax R, Woebken D, Holtappels M, Lavik G, Rapp HT, Schläppy ML, Schleper C, Kuypers MMM (2009) Complex nitrogen cycling in the sponge Geodia barretti. Environ Microbiol 11:2228–2243. https://doi.org/10.1111/j.1462-2920.2009.01944.x
Hussain A, De K, Thomas L et al (2016) Prevalence of skeletal tissue growth anomalies in a scleractinian coral: Turbinaria mesenterina of Malvan Marine Sanctuary, Eastern Arabian Sea. Dis Aquat Organ 121:79–83. https://doi.org/10.3354/dao03038
Jensen S, Duperron S, Birkeland N-K, Hovland M (2010) Intracellular Oceanospirillales bacteria inhabit gills of Acesta bivalves. FEMS Microbiol Ecol 74:523–533. https://doi.org/10.1111/j.1574-6941.2010.00981.x
Jensen S, Fortunato SAV, Hoffmann F, Hoem S, Rapp HT, Øvreås L, Torsvik VL (2017) The relative abundance and transcriptional activity of marine sponge-associated microorganisms emphasizing groups involved in sulfur cycle. Microb Ecol 73:668–676. https://doi.org/10.1007/s00248-016-0836-3
Jeong J-B, Kim K-H, Park J-S (2015) Sponge-specific unknown bacterial groups detected in marine sponges collected from Korea through barcoded pyrosequencing. J Microbiol Biotechnol 25:1–10
Kiran GS, Sekar S, Ramasamy P, Thinesh T, Hassan S, Lipton AN, Ninawe AS, Selvin J (2018) Marine sponge microbial association: towards disclosing unique symbiotic interactions. Mar Environ Res 140:169–179. https://doi.org/10.1016/j.marenvres.2018.04.017
Le Pennec G, Perovic S, Ammar MSA et al (2003) Cultivation of primmorphs from the marine sponge Suberites domuncula: morphogenetic potential of silicon and iron. J Biotechnol 100:93–108
Li H, Zhang Y, Li D, Xu H, Chen GX, Zhang CG (2009) Comparisons of different hypervariable regions of rrs genes for fingerprinting of microbial communities in paddy soils. Soil Biol Biochem 41:954–968. https://doi.org/10.1016/J.SOILBIO.2008.10.030
Luter HM, Gibb K, Webster NS (2014) Eutrophication has no short-term effect on the Cymbastela stipitata holobiont. Front Microbiol 5:1–10. https://doi.org/10.3389/fmicb.2014.00216
Matthew S, Salvador LA, Schupp PJ, Paul VJ, Luesch H (2010) Cytotoxic halogenated macrolides and modified peptides from the apratoxin-producing marine cyanobacterium Lyngbya bouillonii from Guam. J Nat Prod 73:1544–1552. https://doi.org/10.1021/np1004032
Moitinho-Silva L, Seridi L, Ryu T, Voolstra CR, Ravasi T, Hentschel U (2014) Revealing microbial functional activities in the Red Sea sponge Stylissa carteri by metatranscriptomics. Environ Microbiol 16:3683–3698. https://doi.org/10.1111/1462-2920.12533
Morganti T, Coma R, Yahel G, Ribes M (2017) Trophic niche separation that facilitates co-existence of high and low microbial abundance sponges is revealed by in situ study of carbon and nitrogen fluxes. Limnol Oceanogr 62:1963–1983. https://doi.org/10.1002/lno.10546
Morrow KM, Bourne DG, Humphrey C, Botté ES, Laffy P, Zaneveld J, Uthicke S, Fabricius KE, Webster NS (2015) Natural volcanic CO2 seeps reveal future trajectories for host-microbial associations in corals and sponges. ISME J 9:894–908. https://doi.org/10.1038/ismej.2014.188
Mote S, Schönberg CHL, Samaai T, Gupta V, Ingole B (2019) A new clionaid sponge infests live corals on the west coast of India (Porifera, Demospongiae, Clionaida). Syst Biodivers 17:190–206. https://doi.org/10.1080/14772000.2018.1513430
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Nishijima M, Adachi K, Katsuta A, Shizuri Y, Yamasato K (2013) Endozoicomonas numazuensis sp. nov., a gammaproteobacterium isolated from marine sponges, and emended description of the genus Endozoicomonas Kurahashi and Yokota 2007. Int J Syst Evol Microbiol 63:709–714. https://doi.org/10.1099/ijs.0.042077-0
Oksanen J (2017) Vegan: ecological diversity. R Packag. Version 2.4-4 11. https://cran.r-project.org/package=vegan
Orcutt BN, Sylvan JB, Knab NJ, Edwards KJ (2011) Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol Mol Biol Rev 75:361–422. https://doi.org/10.1128/MMBR.00039-10
Osman EO, Suggett DJ, Voolstra CR, Pettay DT, Clark DR, Pogoreutz C, Sampayo EM, Warner ME, Smith DJ (2020) Coral microbiome composition along the northern Red Sea suggests high plasticity of bacterial and specificity of endosymbiotic dinoflagellate communities. Microbiome 8:8. https://doi.org/10.1186/s40168-019-0776-5
Parfrey LW, Moreau CS, Russell JA (2018) Introduction: the host-associated microbiome: pattern, process and function. Mol Ecol 27:1749–1765
Pineda MC, Strehlow B, Duckworth A, Doyle J, Jones R, Webster NS (2016) Effects of light attenuation on the sponge holobiont-implications for dredging management. Sci Rep 6:39038. https://doi.org/10.1038/srep39038
Pineda M-C, Strehlow B, Sternel M, Duckworth A, Jones R, Webster NS (2017) Effects of suspended sediments on the sponge holobiont with implications for dredging management. Sci Rep 7:4925. https://doi.org/10.1038/s41598-017-05241-z
Pita L, Turon X, López-Legentil S, Erwin PM (2013) Host rules: spatial stability of bacterial communities associated with marine sponges (Ircinia spp.) in the Western Mediterranean Sea. FEMS Microbiol Ecol 86:268–276. https://doi.org/10.1111/1574-6941.12159
Pita L, Rix L, Slaby BM, Franke A, Hentschel U (2018) The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome 6:46. https://doi.org/10.1186/s40168-018-0428-1
Poppell E, Weisz J, Spicer L, Massaro A, Hill A, Hill M (2014) Sponge heterotrophic capacity and bacterial community structure in high- and low-microbial abundance sponges. Mar Ecol 35:414–424. https://doi.org/10.1111/maec.12098
Ramsby BD, Hill MS, Thornhill DJ, Steenhuizen SF, Achlatis M, Lewis AM, LaJeunesse TC (2017) Sibling species of mutualistic Symbiodinium clade G from bioeroding sponges in the western Pacific and western Atlantic oceans. J Phycol 53:951–960. https://doi.org/10.1111/jpy.12576
Ramsby BD, Hoogenboom MO, Smith HA, Whalan S, Webster NS (2018a) The bioeroding sponge Cliona orientalis will not tolerate future projected ocean warming. Sci Rep 8:1–13. https://doi.org/10.1038/s41598-018-26535-w
Ramsby BD, Hoogenboom MO, Whalan S, Webster NS (2018b) Elevated seawater temperature disrupts the microbiome of an ecologically important bioeroding sponge. Mol Ecol 27:2124–2137. https://doi.org/10.1111/mec.14544
Ribes M, Jiménez E, Yahel G, López-Sendino P, Diez B, Massana R, Sharp JH, Coma R (2012) Functional convergence of microbes associated with temperate marine sponges. Environ Microbiol 14:1224–1239. https://doi.org/10.1111/j.1462-2920.2012.02701.x
Ribes M, Dziallas C, Coma R, Riemann L (2015) Microbial diversity and putative diazotrophy in high- and low- microbial-abundance mediterranean sponges. Appl Environ Microbiol 81:5683–5693. https://doi.org/10.1128/AEM.01320-15
Roughgarden J, Scott GF et al (2017) Holobionts as units of selection and a model of their population dynamics and evolution. Biol Theory 0:3. https://doi.org/10.1007/s13752-017-0287-1
Sacristán-Soriano O, Turon X, Hill M (2020) Microbiome structure of ecologically important bioeroding sponges (family Clionaidae): the role of host phylogeny and environmental plasticity. Coral Reefs 39:1285–1298. https://doi.org/10.1007/s00338-020-01962-2
Sawhney S, Mishra JK (2019) Bioactive potential of bacterial endosymbionts isolated from Lamellodysidea herbacea, marine sponge from the coast of South Andaman, India, against human bacterial pathogens. J Appl Pharm Sci 9:1–8. https://doi.org/10.7324/JAPS.2019.90301
Schönberg CHL, Fang JKH, Carreiro-Silva M, Tribollet A, Wisshak M(2017) Bioerosion: the other ocean acidification problem. ICES J Mar Sci 74:895–925
Schorn MA, Jordan PA, Podell S et al (2019) Comparative genomics of cyanobacterial symbionts reveals distinct, specialized metabolism in tropical dysideidae sponges. MBio 10:e00821–e00819
Simister RL, Deines P, Botté ES, Webster NS, Taylor MW (2012) Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ Microbiol 14:517–524. https://doi.org/10.1111/j.1462-2920.2011.02664.x
Slaby BM, Hackl T, Horn H, Bayer K, Hentschel U (2017) Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J 11:2465–2478. https://doi.org/10.1038/ismej.2017.101
Soares AR (2016) Diversity and specificity of the marine sponge microbiome as inspected by next generation sequencing. Ph. D. dessertation. University of Algarve, Portugal. Accessed online https://core.ac.uk/download/pdf/61528073.pdf. Accessed 18 Jan 2020
Southwell MW, Weisz JB, Martens CS, Lindquist N (2008) In situ fluxes of dissolved inorganic nitrogen from the sponge community on Conch Reef, Key Largo, Florida. Limnol Oceanogr 53:986–996. https://doi.org/10.4319/lo.2008.53.3.0986
Steindler L, Beer S, Ilan M (2002) Photosymbiosis in intertidal and subtidal tropical sponges. Symbiosis 33:263–273
Steinert G, Taylor MW, Deines P, Simister RL, de Voogd NJ, Hoggard M, Schupp PJ (2016) In four shallow and mesophotic tropical reef sponges from Guam the microbial community largely depends on host identity. PeerJ 4:e1936. https://doi.org/10.7717/peerj.1936
Steinert G, Wemheuer B, Janussen D et al (2019) Prokaryotic diversity and community patterns in Antarctic continental shelf sponges. Front Mar Sci 6. https://doi.org/10.3389/fmars.2019.00297
Teruya T, Nakagawa S, Koyama T, Arimoto H, Kita M, Uemura D (2004) Nakiterpiosin and nakiterpiosinone, novel cytotoxic C-nor-D-homosteroids from the Okinawan sponge Terpios hoshinota. Tetrahedron 60:6989–6993. https://doi.org/10.1016/J.TET.2003.08.083
Thacker RW, Freeman CJ (2012) Sponge–microbe symbioses: recent advances and new directions. In: Becerro MA, Uriz MJ, Maldonado MTX (eds) Advances in sponge science: phylogeny, systematics, ecology, Advances in marine biology. Academic Press, Amsterdam, pp 57–111
Thomas T, Moitinho-Silva L, Lurgi M, Björk JR, Easson C, Astudillo-García C, Olson JB, Erwin PM, López-Legentil S, Luter H, Chaves-Fonnegra A, Costa R, Schupp PJ, Steindler L, Erpenbeck D, Gilbert J, Knight R, Ackermann G, Victor Lopez J, Taylor MW, Thacker RW, Montoya JM, Hentschel U, Webster NS (2016) Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun 7:11870. https://doi.org/10.1038/ncomms11870
Tout J, Astudillo-García C, Taylor MW, Tyson GW, Stocker R, Ralph PJ, Seymour JR, Webster NS (2017) Redefining the sponge-symbiont acquisition paradigm: sponge microbes exhibit chemotaxis towards host-derived compounds. Environ Microbiol Rep 9:750–755. https://doi.org/10.1111/1758-2229.12591
Villegas-Plazas M, Wos-Oxley ML, Sanchez JA, Pieper DH, Thomas OP, Junca H (2019) Variations in microbial diversity and metabolite profiles of the tropical marine sponge Xestospongia muta with season and depth. Microb Ecol 78:243–256. https://doi.org/10.1007/s00248-018-1285-y
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. https://doi.org/10.1128/AEM.00062-07
Webster NS, Thomas T (2016) The sponge hologenome. MBio 7:e00135–e00116. https://doi.org/10.1128/MBIO.00135-16
Wulff J (2001) Assessing and monitoring coral reef sponges: why and how? Bull Mar Sci 69:831–846
Acknowledgments
We are thankful to Dr. Christine H. L. Schönberg (University of Western Australia) for her valuable suggestions. Authors thank Dr. Kuldeep More for his kind support in nMDS and alpha diveristy indices analysis. This study forms part of the Ph.D. thesis of SM. SM and KD acknowledge the CSIR SRF and DST INSPIRE fellowships, respectively. All necessary permissions for sampling and field observation have been obtained by the authors from the competent authorities.
Funding
This study was financially support received by the Rajiv Gandhi Science and Technology Commission, Government of Maharashtra, India, under the Maharastra Gene Bank project (GAP2871).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethical approval
All applicable international, national and/or institutional guidelines for the care and use of animals were followed by the authors.
Sampling and field studies
All necessary permits for sampling and field observation have been obtained by the authors from the competent authorities.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 3733 kb)
Rights and permissions
About this article

Cite this article
Mote, S., Gupta, V., De, K. et al. Bacterial diversity associated with a newly described bioeroding sponge, Cliona thomasi, from the coral reefs on the West Coast of India. Folia Microbiol 66, 203–211 (2021). https://doi.org/10.1007/s12223-020-00830-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-020-00830-4