
Abstract
DNA extraction from environmental samples is a critical step for metagenomic analysis to study microbial communities, including those considered uncultivable. Nevertheless, obtaining good quality DNA in sufficient quantities for downstream methodologies is not always possible, and it depends on the complexity and stability of each ecosystem, which could be more problematic for samples from tropical regions because those ecosystems are less stable and more complex. Three laboratory methods for the extraction of nucleic acids from samples representing unstable (decaying coffee pulp and mangrove sediments) and relatively stable (compost and soil) environments were tested. The results were compared with those obtained using two commercial DNA extraction kits. The quality of the extracted DNA was evaluated by PCR amplification to verify the recovery of bacterial, archaeal, and fungal genetic material. The laboratory method that gave the best results used a lysis procedure combining physical, chemical, and enzymatic steps.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Obtaining DNA of adequate quality and in sufficient quantities from environmental samples is a critical stage of the analysis of microbial communities, as well as for searching for genes encoding novel enzymes with biotechnological applications. Several methodologies have been developed for nucleic-acid extraction and purification from various sample types, such as soil, compost, and sediment. To some degree, all of these methods are susceptible to contaminants such as humic acids, metals, and xenobiotics that interfere with the extraction process (Wintzingerode et al. 1997; He et al. 2009). In addition to these compounds, another factor that may influence DNA extraction is the ecosystem dynamics (physical and chemical changes) of the place where the sample was obtained. These dynamics can alter the structure of microbial communities and thus the sample’s composition (Wintzingerode et al. 1997).
While niche microenvironments like soils or marine sediments are highly dynamic in terms of microbial complexity, the presence of contaminants and the transformation of components, some degree of stability still exists even after environmental disturbances (Griffiths et al. 1999, 2004). This apparent stability has allowed the development of DNA extraction methodologies that have been marketed as kits that are suggested for use with this kind of samples.
In contrast to stable microenvironments, some ecosystems with great potential for interesting microbial activities have not achieved microenvironmental stability, which makes them highly variable and complex. Although DNA can be obtained from such environments, the yield and quality are often insufficient for downstream methodologies, and results from replicate extractions are often not reproducible. It is difficult to efficiently remove contaminants that inhibit the activity of enzymes used in subsequent DNA processing from these types of samples. These problems become worse because of the constantly changing microenvironmental factors in these ecosystems, such as decaying materials or zones with constant physical movement and the removal or the exchange of components (Alongi 1988). A clear example of this are mangrove sediments, which contain a very high amount of organic agents as well as contaminants, the concentration of which is constantly changing with time, which might be a determining factor influencing the success of DNA extraction.
In addition to the problems mentioned above, it has been found that DNA extraction from a particular microbial group (bacteria, fungi, or archaea) may be favored depending on the type of lysis used in different samples (Jiang et al. 2011). To overcome this bias, some authors have implemented extraction techniques using integrative lysis methods (Moré et al. 1994; Boon et al. 2000; Nelson et al. 2007; Jiang et al. 2011), which entail the combined use of chemical (SDS or phenol), physical (glass or zirconia beads, microwaves, frozen/thaw cycles), and enzymatic methods (lysozyme, proteinase K).
Lysis is followed by DNA extraction and in some cases, by an additional purification step. This final purification is done to remove impurities and inhibitors of enzymes that may affect subsequent DNA processing. Other reports recommend the use of columns or kits that substantially increase the cost of the purification procedure.
In this work, various methods of DNA extraction, including two commercial kits, were evaluated with samples from a tropical region. The following two samples representing unstable microenvironments were employed: decomposing coffee pulp and a mangrove sediment with a high organic matter content. Samples from both of these environments could be considered as complex, based on the difficulty of obtaining DNA of good quality and in sufficient quantities using routine extraction methods in our laboratory. For comparison, samples from two relatively stable environments (soil and compost) were also included.
Materials and methods
Sampling
Samples of five-day-old decomposing coffee pulp were obtained during the harvest season from the “Argovia” coffee farm (Tapachula-Nueva Alemania road, Chiapas, Mexico). These were collected from five sampling points in the pulp pile, at a depth of 50 cm and a surrounding temperature of 40–45 °C. Mangrove-sediment samples were obtained from the Pampa-Murillo lagoon system located at the municipality of Tapachula, Chiapas, Mexico. Seven 20-cm samples were taken with a 5.08-cm diameter (3-m long) PVC tube at different sampling points; five of these had a pasty appearance due to a high content of organic matter, and two had a gritty appearance. A composite sample was prepared using an equal mass of all samples. All samples were stored at −20 °C until processing.
Sawdust and coffee-waste compost samples were donated by the “La Concepción del Soconusco” farm, located in the parcel Felipe Carrillo Puerto, in Tapachula, Chiapas. A representative 2-kg compost sample was obtained by mixing samples from different points in a mature compost pile with a mean temperature of approximately 25 to 30 °C. The soil sample was obtained from the central garden at El Colegio de la Frontera Sur in Tapachula Chiapas, Mexico. This sample was taken at a 5-cm depth and processed immediately.
DNA extraction methods
Phenol-chloroform (PC)
The phenol-chloroform method used here was modified from Sambrook and Russel (2001). Briefly, a cell concentrate was made from 5 g of the sample by adding 5 mL of 0.1 mol/L sodium phosphate buffer (pH 8.0), vortexing for 1 min and allowing the mixture to stand for 1 min. The supernatant was concentrated by centrifugation at 2500g for 1 min. The pellet was lysed by adding 400 μL of deionized water, 0.2 g of sterile glass beads (450–600 μm diameter, Sigma catalogue G9268), 10 μL of 20 % SDS, and 500 μL of equilibrated phenol (pH 8.0). The sample was vortexed for 2 min and incubated at 60 °C for 10 min. The aqueous phase was extracted twice with chloroform, vortexed for 2 min at maximum speed, and centrifuged at 13,000g for 5 min. DNA was precipitated by adding 50 μL of 3 mol/L CH3COONa (pH 5.2) and two volumes of chilled isopropanol, incubating for 1 h at −20 °C and centrifuging for 20 min at 13,000g at 10 °C. The pellet was washed with 70 % ethanol, air-dried, and resuspended in 50 μL of TE buffer (10 mmol/L Tris-HCl; 1 mol/L EDTA, pH 8).
Enzymatic lysis (EL)
This method is based on the chemical-enzymatic lysis protocol for compost DNA extraction of Yang et al. (2007) with some modifications. Samples (0.25 g) were washed twice with 1 mL of 0.12 mol/L sodium phosphate (pH 8), incubated at 25 °C with shaking (150 rpm for 5 min), and centrifuged at 6000g for 10 min at 4 °C. The pellet was resuspended in 375 μL of extraction buffer (0.1 mol/L Tris-HCl, 0.1 mol/L EDTA, 0.1 mol/L NaH2PO4, 1.5 mol/L NaCl, and 1 % CTAB (pH 8.0)) and 2.5 μL of proteinase K (10 mg/mL). The sample was incubated for 30 min at 37 °C with shaking (300 rpm), then 50 μL of 10 % SDS was added followed by incubation for 1 h at 65 °C with shaking (300 rpm). The sample was mixed by inversion every 15 min. After centrifugation at 6000g for 5 min, the supernatants were transferred to pre-chilled tubes kept on ice. Two consecutive extractions of the pellet were made with 12.5 μL of 10 % SDS and 125 μL of extraction buffer, which were incubated for 10 min at 65 °C and 300 rpm. The supernatants from both extractions were combined. The crude extract was purified twice with one volume of phenol:chloroform:isoamyl alcohol (25:24:1), followed by two extractions with chloroform:isoamyl alcohol (24:1). The aqueous phase was transferred to a fresh tube, and DNA was precipitated with two volumes of chilled absolute ethanol and 0.1 volume of 3 mol/L sodium acetate (pH 5.2), incubated at −20 °C for 1 h and centrifuged at 13,000g for 20 min. The pellet was washed with 70 % ethanol, and the DNA was resuspended in 50 μL of TE buffer.
Lysozyme method (L)
Samples (0.5 g) were washed twice in 1.0 mL of 0.1 mol/L phosphate buffer (pH 8.0), vortexed for 15 min and centrifuged for 10 min at 6000g. The pellet was resuspended in 1 mL of lysis solution (150 mmol/L NaCl, 0.1 mol/L EDTA, 5 mg/mL lysozyme) and incubated at 37 °C for 2 h. Two-hundred μL of SDS solution (0.1 mol/L NaCl, 0.5 mol/L Tris-HCl, 10 % SDS) were added, and the sample was vortexed at room temperature for 5 min. Samples were subjected to three freeze (−70 °C) and thaw (65 °C) cycles. Proteinase K (2.5 mg/mL) was then added; the samples were incubated at 50 °C for 15 min and then vortexed and centrifuged immediately for 5 min at 13,000g. The supernatant was transferred to a fresh tube, and one volume of chloroform:isoamyl alcohol (24:1) was added. The sample was vortexed and then centrifuged at 13,000g for 5 min. DNA was precipitated with 0.1 volumes of 3.0 mol/L sodium acetate (pH 5.2) and two volumes of chilled absolute ethanol, with incubation at −20 °C overnight. The sample was centrifuged for 20 min at 13,000g and the pellet washed with chilled 70 % ethanol. Once dry, it was resuspended in 50 μL of sterile deionized water.
DNA extraction with commercial kits (KQ and KZ)
Samples of 0.2 g were used with the following two DNA extraction kits: QIAamp DNA Stool Mini KitTM (Qiagen), referred to as KQ; and the ZR Soil Microbe DNA MiniprepTM (Zymo Research), referred as to KZ. Extraction was done according to the manufacturer’s recommendations, and the DNA was eluted in 50 μL of the elution buffer provided in the kit.
Improved method for DNA extraction from decaying coffee pulp (IM-Z)
Based on the enzymatic lysis (EL) method described above, this extraction was modified to include a cellular concentration in which the sample (5 g) was washed with 50 mL of 20 mmol/L EDTA, homogenized for 5 min, vortexed at medium speed, and allowed to stand for sedimentation, thus obtaining a cellular concentrate (CC) from the supernatant. This was centrifuged at 2370g for 10 min. Each CC was processed following the EL method to obtain a crude extract, followed by purification with the ZR Soil Microbe DNA MiniprepTM kit, following the manufacturer’s recommendations but omitting the lysis steps. DNA was resuspended in 30 μL of the elution buffer provided in the kit.
DNA quantitation
To quantify DNA sample concentrations, we used Kodak Molecular Imaging software with the region of interest (ROI) method, which consists of manually selecting the area of interest to determine the quantity of pixels proportional to a known DNA quantity, using a DNA standard (O’GeneRuler 1 Kb Plus DNA ladder).
Samples that could not be quantified this way (e. g., smeared DNA) were analyzed by Paint Shop Pro 8 version 8.10, using the histogram tool to determine the quantity of pixels in the selected channel (Greyscale). By delimiting a specific area, we subtracted the background value from the mean value displayed and compared this value with the mean value obtained from the histogram of the reference-DNA sample (ladder) of a known concentration. A similar strategy was used to compare the proportion of impurities observed in some samples.
DNA quality assessment
To assess the quality of DNA and detect a broad range of microorganisms from the samples extracted with different methods, we performed PCR amplification on molecular markers from bacteria, fungi, and archaea, as described by Das et al. (2007), simulating a diversity study using the DGGE technique. The primers used are shown in Table 1, and triplicate PCRs were done for all assays. For bacterial analysis, oligonucleotides directed to a 16S rDNA region at positions 968 to 1401 were used; for fungi, an 18S rDNA region from position 20 to 368 was amplified (Das et al. 2007). Archaea identification was performed using a nested amplification as described by Vissers et al. (2009), which was specific for the 16S rDNA region from 519–915 nt. Positive controls for PCR amplification of bacteria, fungi, or archaea were DNA from Escherichia coli, Trichoderma versicolor, or anaerobic sludge from a wastewater treatment plant, respectively.
Reactions contained 1 × buffer, 0.2 ρmol of each primer, 0.2 mmol/L dNTPs, 1.5 mmol/L MgCl2, 0.4 U Taq DNA polymerase (Fermentas), and 7.0 ng/μL of DNA crude extract (or 10−1 and 10−2 dilutions) in a final volume of 20 μL. For bacteria and fungi, a second PCR using 0.2, 1.0 or 3.0 μL of the primary PCR product as the template was also done. PCR protocols shown in Table 1 all included an activation step of 5 min at 94 °C and a final extension for 5 min at 72 °C.
Results
Comparison of different DNA extraction methods
All of the methods evaluated reproducibly extracted DNA from the samples; because many of the extracts had impurities visible to the naked eye (brown or black pellets) or apparent in agarose gels (Fig. 1), these could not be quantified by spectrophotometric methods. The samples that could be quantified by image analysis were taken as a reference to estimate the amounts of DNA recovered using gel-band intensity measurements. Mangrove sediment had the greatest amount of impurities when extracted by PC and was used as a reference (100 %) to estimate the proportion of impurities for the other extracts. Thus, both the relative amounts of DNA extracted and the impurities visible in the gels were established.

Representative sample of DNA obtained showing visible impurities
To compare the efficiency of DNA recovered from the different samples, the yield (μg of DNA per g sample) was estimated for each extraction method (Table 2). Decomposing coffee pulp and mangrove-sediment samples yielded the lowest amounts of DNA (Fig. 2). The highest yields were obtained from compost (up to 21.9 μg DNA per g sample) and soil (15.6 μg DNA per g sample) samples extracted by KZ. However, compost samples contained more impurities when extracted with most of the methods, compared with the soil samples which had fewer impurities.

Quality of DNA obtained from environmental samples using different extraction methods, where black bars show the yield of DNA obtained; white bars indicate the relative percentage of impurities observed in the gel related to the maximum amount of impurities observed in Sd extracted by PC (set at 100 %). C compost, S soil, Sd mangrove sediment, P decomposing coffee pulp
Among the conventional extraction methods, the EL method gave a higher DNA yield (except for coffee pulp), whereas PC method had the lowest yield in all cases. Of the commercial kits, KZ gave a higher yield of DNA from compost, soil, and decomposing coffee pulp but not from mangrove-sediment samples, compared with KQ, which yielded a visibly clean pellet for all of the samples, including compost. For complex mangrove-sediment samples, the most efficient methods were KQ and EL, whereas for coffee pulp, the L and KZ methods performed best.
Assessment of DNA quality
The quality of DNA obtained with the different extraction methods was assessed by PCR amplification, using primers designed to detect different microbial groups. Unfortunately, the amounts of DNA from coffee pulp and mangrove-sediment samples obtained were not sufficient to perform all KZ PCR reactions, and it was impossible to get equivalent samples to complete the assays. With bacteria-targeted oligos, amplicons were observed for all samples and methods except for soil and compost obtained by EL (Table 3). It is noteworthy that in most cases, the amplification was obtained from secondary PCRs and with template dilutions.
For fungi, most of the amplifications also gave products in secondary PCRs. The DNA obtained by the L method allowed the detection of fungi in compost, sediment, and pulp samples. Fungi were detected in PCRs of compost-sample DNA extracted by the KZ, PC, and L methods, in pulp samples prepared by PC, L, and KQ extraction and in mangrove sediment extracted by L and KQ. In contrast, fungal amplification products were not detected in the DNA of soil samples extracted by any of the methods, even using template dilutions.
For Archaea, a higher proportion of successful amplification was observed in samples extracted by the KZ method, followed by the KQ and L methods. DNA samples obtained with the PC and EL methods showed no archaeal amplification, but pulp samples yielded some products when extracted by PC as did mangrove-sediment samples when extracted by EL.
The least successful method for PCR amplification of different microbial groups was EL, which yielded very few bacterial amplicons from pulp and mangrove-sediment samples. The best results with non-commercial methods were obtained with L, where all samples except soil were PCR-positive with the selected primers.
Improvement of DNA extraction from decaying pulp
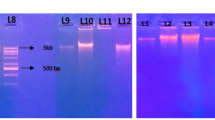
Because decomposing coffee pulp proved to be the most difficult material from which to obtain DNA, a method for this sample was optimized. To achieve this, the modified method of Yang et al. (2007) was employed with a preliminary step of cell concentration and an extra purification step using Zymo kit columns to remove impurities and inhibitors. With this optimization, we obtained four times the amount of DNA (approximately 100 ng/μL) as the highest yield obtained with the best-unmodified method (L) for pulp samples. No impurities were detected despite the smeared bands observed in gels (Fig. 3a). This method gave a lower average yield than the other protocols (Table 2), but the DNA pellets obtained were more concentrated and free from impurities than with other extraction methods and proved suitable for PCR using bacterial primers (Fig. 3b), even in the primary PCR with crude DNA extract (not shown).

a DNA extracted with improved method for decaying coffee pulp samples. b Secondary PCR amplification with primers for bacteria. 1 kb ladder; (+, −) positive and negative controls for PCR. C compost, P decomposing coffee pulp, Sd mangrove sediment, S soil
This method was not appropriate for obtaining DNA from the other samples tested, nevertheless, a little DNA was recovered from the compost and soil samples (Fig. 3a) (12.5 and 7.5 ng/μL, respectively). In bacterial PCR assays, only the compost sample showed a slight product band from secondary PCR (Fig. 3b).
Discussion
Environmental samples from tropical regions are very appealing to investigate for biological resources for biotechnological applications; however, such a search could be very complex due to the highly-dynamic characteristics of this ecosystem. Obtaining DNA from these kinds of samples could be difficult; we had previously tried to extract nucleic acids from mangrove sediment and decaying coffee pulp samples, testing various methods and modifications based on protocols suggested in the literature, including the use of 1.2 % CTAB in the buffer used prior to cell lysis or the addition of a solution of 12 % chelex-100 after lysis, or a combination of these techniques, with little success or with results that were not reproducible (unpublished data).
For this work, we selected those laboratory methods that provided the best extractions from the minimum quantity of sample from which we could obtain consistent results to evaluate and compare the performance of these methods using commercial kits. The DNA quality and efficiency of recovery was different in each case. The presence of impurities was evident in the compost, sediment, and soil samples extracted by the PC method. For compost and soil samples, most of the impurities were probably due to the presence of humic acids. For mangrove-sediment samples, impurities might be complex organic compounds from plants or other organic wastes. It is important to mention that the streams that feed the lagoon system where mangrove sediments were collected are adjacent to cattle grazing and banana cultivation areas, so they might contain agrochemicals such as pesticides (Castro et al. 2005; Hernández-Romero et al. 2004), which may have affected the DNA-recovery efficiency from these samples.
In general, decaying coffee pulp was the sample that had the lowest DNA yield for all of the tested methods, in spite of having microscopic evidence of have contained apparently higher richness of microorganisms compared with the soil and compost samples (not shown). This could be due not only to the high content of aromatic compounds, tannins, and polyphenols, along with polysaccharides and waxes from the coffee fruit pericarp (Esquivel and Jiménez 2012) but also because the decay process makes the pulp less homogeneous, giving it a highly variable composition and physicochemical characteristics.
Of all the extraction methods used, the L method had the most consistent performance, although its efficiency was no better than that of the other methods (Fig. 2, Table 2). Among the conventional methods, L gave DNA with the highest quality and allowed PCR amplification of markers for all microbial groups (bacteria, archaea, and fungi) in all samples, except for fungi in the soil sample (for which the other methods failed as well). The improved performance of this method in terms of the microbial diversity detected could stem from the integration of the different cell-lysis procedures used; physical (temperature shock), chemical (detergents), and enzymatic (proteinase K and lysozyme). Thus, the L method appears to be more robust than the other methods due to the efficiency of its lysis procedure, which allows the recovery of DNA from different types of organisms. A similar result was reported by Jiang et al. (2011) using mangrove sediment; and, along with our results, indicates that this strategy is suitable for other types of complex samples such as decomposing coffee pulp.
We cannot explain why soil fungi were not detected with any of the tested methods. This does not appear to be due to the methods used because fungal amplicons were obtained from the other sample types. It is possible that the primers used were not matched to the diversity of fungi in the soil sample, or there was little fungal biomass present at the time the samples were taken (consistent with microscopic observation, not shown). It is known that the type and condition of the plants (grass, in this case) and the soil influence the communities of fungi present (Johnson et al. 1992; Callaway et al. 2003).
Finally, for very complex samples, conventional extraction methods can be optimized by combining different purification procedures. In this study, the modifications to the EL method consisted of a cellular pre-concentration stage and the use of a commercial column for purification that yielded DNA of sufficient quantity and quality for PCR amplification. We did not evaluate DNA recovery for microbial groups other than bacteria for decaying coffee pulp, but the DNA quality yielded by the extraction method used for this sample was suitable for amplification of bacterial communities.
References
Alongi DM (1988) Bacterial productivity and microbial biomass in tropical mangrove sediments. Microb Ecol 15(1):59–79. doi:10.1007/BF02012952
Boon N, Marlé C, Top EM, Verstraete W (2000) Comparison of the spatial homogeneity of physico-chemical parameters and bacterial 16S rRNA genes in sediment samples from a dumping site for dredging sludge. Appl Microbiol Biot 53:742–747. doi:10.1007/s002530000325
Callaway RM, Mahall BE, Wicks C, Pankey J, Zabinski C (2003) Soil fungi and the effects of an invasive forb on grasses: neighbor identity matters. Ecology 84(1):129–135. doi:10.1890/0012-9658(2003)084[0129:SFATEO]2.0.CO;2
Castro V, Siu-Rodas Y, González-Huerta LV, Sokolov MY (2005) Contaminación por plaguicidas organoclorados en la Laguna Pampa El Cabildo, Chiapas, y su efecto tóxico en postlarvas de camarón blanco (Litopenaeus vannamei) (Decapoda:Penaeidae) de Chiapas, México. Rev Biol Trop 53(1):141–151. doi:10.15517/rbt.v53i1-2.14407
Coolen M, Hopmans E, Rijpstra IC, Muyzer G, Schouten S, Volkman JK, Sinninghe-Damsté J (2004) Evolution of the methane cycle in Ace Lake (Antarctica) during the Holocene: response of methanogens and methanotrophs to environmental change. Org Geochem 35:1151–116. doi:10.1016/j.orggeochem.2004.06.009
Das M, Todd R, Leff L (2007) Diversity of fungi, bacteria, and actinomycetes on leaves decomposing in a stream. Appl Environ Microb 73(3):756–767. doi:10.1128/AEM.01170-06
DeLong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89(12):5685–5689. doi:10.1073/pnas.89.12.5685
Esquivel P, Jiménez VM (2012) Functional properties of coffee and coffee by-products. Food Res Int 46(2):488–495. doi:10.1016/j.foodres.2011.05.028
Griffiths BS, Ritz K, Ebblewhite N, Dobson G (1999) Soil microbial community structure: effects of substrate loading rates. Soil Biol Biochem 31:145–153. doi:10.1016/S0038-0717(98)00117-5
Griffiths BS, Kuan HL, Ritz K, Glover LA, McCaig AE, Fenwick C (2004) The relationship between microbial community structure and functional stability, tested experimentally in an upland pasture soil. Microb Ecol 47(1):104–113. doi:10.1007/s00248-002-2043-7
He Y, Zhao Y, Zhou G, Huang M (2009) Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from aged refuse for microbial community analysis. Word J Microb Biotechnol 25(11):2043–2051. doi:10.1007/s11274-009-0106-3
Hernández-Romero H, Tovilla-Hernández C, Malo E, Bello-Mendoza R (2004) Water quality and presence of pesticides in a tropical coastal wetland in southern Mexico. Mar Pollut Bull 48:1130–1141. doi:10.1016/j.marpolbul.2004.01.003
Heuer H, Krsek M, Baker P, Smalla K, Wellington E (1997) Analysis of actinomycete communities by specific amplification of genes encoding 16S rRNA and gel-electrophoretic separation in denaturing gradients. Appl Environ Microbiol 63(8):3233–3241
Jiang YX, Wu JG, Yu KQ, Ai CX, Zou F, Zhou HW (2011) Integrated lysis procedures reduces extraction biases of microbial DNA from mangrove sediments. J Biosci Bioeng 111(2):153–157. doi:10.1016/j.jbiosc.2010.10.006
Johnson NC, Tilman D, Wedin D (1992) Plant and soil controls on mycorrhizal fungal communities. Ecology 73(6):2034–2042. doi:10.2307/1941453
May LA, Smiley B, Schmidt MG (2001) Comparative denaturing gradient gel electrophoresis of fungal communities associated with whole plant corn silage. Can J Microbiol 47(9):829–841. doi:10.1139/w01-086
Moré MI, Herrick JB, Silva MC, Ghiorse WC, Madsen EL (1994) Quantitative cell lysis of indigenous microorganisms and rapid extraction of microbial DNA from sediment. Appl Environ Microbiol 60(5):1572–1580
Nelson DM, Ohene-Adjei S, Hu FS, Cann IKO, Mackie RI (2007) Bacterial diversity and distribution in the holocene sediments of a northern temperate lake. Microbial Ecol 54(2):252–263. doi:10.1007/s00248-006-9195-9
Nübel U, Engelen B, Felske A, Snaidr J, Weishuber A, Amann RI, Ludwig W, Backhaus H (1996) Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J Bacteriol 178(19):5636–5643
Sambrook J, Russell D (2001) Molecular cloning: A laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, NY
Vissers E, Bodelier P, Muyzer G, Laanbroek H (2009) A nested PCR approach for improved recovery of archaeal 16S rRNA gene fragments from freshwater samples. FEMS Microbiol Lett 298(2):193–198. doi:10.1111/j.1574-6968.2009.01718.x
White T, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. A guide to methods and applications. Academic Press, Inc., New York, pp 315–322
Wintzingerode FV, Göbel UB, Stackebrandt E (1997) Determination of microbial diversity in environmental samples: pitfalls of PCR-based rRNA analysis. FEMS Microbiol Rev 21(3):213–229. doi:10.1111/j.1574-6976.1997.tb00351.x
Yang ZH, Xiao Y, Zeng GM, Xu ZY, Liu YS (2007) Comparison of methods for total community DNA extraction and purification from compost. Appl Microbiol Biot 74(4):918–925. doi:10.1007/s00253-006-0704-z
Acknowledgements
This work was funded by the National Council of Science and Technology project CB-2008-01-101389. MYL-C thanks scholarship number 14877 from the National Council of Science and Technology. We thank Guillermo Vázquez-Martínez for technical assistance with PCR reactions. We thank Michael F. Dunn for reviewing the manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Guillén-Navarro, K., Herrera-López, D., López-Chávez, M.Y. et al. Assessment of methods to recover DNA from bacteria, fungi and archaea in complex environmental samples. Folia Microbiol 60, 551–558 (2015). https://doi.org/10.1007/s12223-015-0403-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-015-0403-1