
Abstract
Introduction
Chronic wounds remain a major clinical challenge. Human cryopreserved viable amniotic membrane (hCVAM) is among the most successful therapies, but the mechanisms of action remain loosely defined. Because proper regulation of macrophage behavior is critical for wound healing with biomaterial therapies, we hypothesized that hCVAM would positively regulate macrophage behavior in vitro, and that soluble factors released from the hCVAM would be important for this effect.
Materials and Methods
Primary human pro-inflammatory (M1) macrophages were seeded directly onto intact hCVAM or cultured in separation via transwell inserts (Soluble Factors) in the presence of pro-inflammatory stimuli (interferon-γ and lipopolysaccharide) to simulate the chronic wound environment. Macrophages were characterized after 1 and 6 days using multiplex gene expression analysis of 37 macrophage phenotype- and angiogenesis-related genes via NanoString™, and protein content from conditioned media collected at days 1, 3 and 6 was analyzed via enzyme linked immunosorbent assays.
Results and Discussion
Gene expression analysis showed that Soluble Factors promoted significant upregulation of pro-inflammatory marker IL1B on day 1 yet downregulation of TNF on day 6 compared to the M1 macrophage control. In contrast, intact hCVAM, which includes both extracellular matrix, viable cells, and soluble factors, promoted downregulation of pro-inflammatory markers TNF, CCL5 and CCR7 on day 1 and endothelial receptor TIE1 on day 6, and upregulation of the anti-inflammatory marker IL10 on day 6 compared to the M1 Control. Other genes related to inflammation and angiogenesis (MMP9, VEGF, SPP1, TGFB1, etc.) were differentially regulated between the Soluble Factors and intact hCVAM groups at both time points, though they were not expressed at significantly different levels compared to the M1 Control. Interestingly, Soluble Factors promoted increased secretion of the pro-inflammatory cytokine tumor necrosis factor-α (TNF-α), while direct contact with hCVAM inhibited secretion of TNF, relative to the M1 Control. Both Soluble Factors and intact hCVAM inhibited secretion of MMP9 and VEGF, pro-inflammatory proteins that are critical for angiogenesis and remodeling, compared to the M1 Control, with intact hCVAM having a stronger effect.
Conclusions
In a simulated pro-inflammatory environment, intact hCVAM has distinct anti-inflammatory effects on primary human macrophages, and direct macrophage contact with intact hCVAM is required for these effects. These findings are important for the design of next generation immunomodulatory biomaterials for wound repair and regenerative medicine that may include living cells, soluble factors, or a controlled drug delivery system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic wounds continue to be a major clinical problem affecting millions of patients and costing over $10.5 billion every year in the US alone.30,43 Human amniotic membrane (hAM), which is the innermost, avascular layer of the placenta, is composed of two main components: a matrix of extracellular matrix (ECM) proteins and a cellular layer consisting of epithelial cells, neonatal fibroblasts and mesenchymal stem cells (MSCs). hAM has been used as a treatment for chronic wounds for more than a century.11 More recently it has regained traction as a chronic wound graft and dressing due to its ease of availability, advancements in tissue preservation, its efficacy in treating burns and ophthalmic disorders,38 and association with a myriad of beneficial properties, including anti-inflammatory,16 anti-scarring,49 anti-bacterial,48 and pro-angiogenic37 behavior. Despite the appeal of hAM and positive clinical outcomes, there remains a limited understanding of its mechanisms of action in chronic wound healing.26
Currently, there are over 25 placental wound care products on the market14; one of the most successful is human cryopreserved, viable amniotic membrane (hCVAM), which retains cellular viability post-thaw while maintaining a shelf life comparable to other chronic wound treatments, and has shown efficacy compared to the standard of care in clinical trials.14,22,39 The mechanisms behind the pro-healing effects of hCVAM are poorly understood, and may derive from soluble factors released either from the ECM or from the cells within the membrane, the ECM itself, or synergistic effects of all three sources. In a recent study, a novel comparative effectiveness research technique was used to perform a retrospective non-randomized investigation to compare clinical trial data from chronic wound patients treated with hCVAM or a dehydrated-hAM.8,20 The results showed that wounds treated with hCVAM closed at a significantly higher rate (63%) than those treated with the dehydrated membrane (18.2%) over 12 weeks.20 Additionally, in vitro studies showed that hCVAM released lower levels of pro-fibrotic transforming growth factor-β1 (TGFB1), as well as decreased levels of pro-inflammatory factors including interferon-γ (IFNG), chemokine (C–C motif) ligand 5 (CCL5), epidermal growth factor (EGF), and thrombopoietin, compared to devitalized hAM.56 On the other hand, hAM-derived ECM itself has been shown to promote enhanced cell proliferation compared to intact hCVAM.53 While this study and others have shown positive effects of hAM-derived ECM or in combination with MCSs or other biomaterials,15,18,27,53,54 the processes of decellularization, devitalization, or dehydration have been shown to significantly alter the ECM architecture and change the composition of hAM.10,17,21 Thus, there remains a need to study the pro-healing effects of intact hCVAM in comparison to its released soluble factors.
The pro-healing effects of implanted or topically applied biomaterials are strongly associated with response of macrophages, the primary cell of the innate immune response,2,35 which play a major role in wound healing, vascularization, and integration or failure of biomaterials.2,25,40 Macrophages have been shown to exhibit different phenotypes in vitro and in vivo ranging from pro-inflammatory (M1) to anti-inflammatory (M2) as healing progresses.50,52,57 While both M1 and M2 macrophages are present throughout the normal healing process, M1 macrophages primarily accumulate at early stages, around 1–5 days post injury, while M2 macrophages accumulate later, from 4 to 14 days in vivo. 3 Conversely, in chronic wounds, this transition in macrophage phenotype is dysregulated. Recent studies in mice and humans have shown that chronic wounds are stalled in a low-grade pro-inflammatory state, where M1 macrophages are found in significantly higher numbers relative to the M2 phenotype.13,31,32,33,34, – 34,36,44 Additionally, two distinct subtypes of M2 phenotypes have been described, including M2a that are stimulated in vitro with interleukin-4 (IL4) and are associated with resolution of healing in vivo, and M2c, which are stimulated in vitro with IL10 and potentially involved in promoting angiogenesis and tissue remodeling at early stages of wound healing.28 However, it is now widely accepted that macrophages frequently exist on a diverse spectrum of phenotypes with complex functions.35,46,58
Because of the critical role of macrophages in wound healing, we hypothesized that hCVAM would directly modulate macrophage behavior. In this study, we co-cultured primary human macrophages directly in contact with hCVAM or with soluble factors secreted from hCVAM via separation with transwell inserts to probe the potential mechanisms. It is important to note that direct contact with hCVAM also contains any soluble factors from the tissue or cells within, and that the effects of these signals may be synergistic. Macrophages were pre-polarized into the M1 phenotype using IFNG and lipopolysaccharide (LPS) prior to initiating the experiment to simulate a more clinically relevant scenario, as macrophages in chronic wounds have been identified to be primarily of the M1 phenotype.32,34,44 The effects on macrophage behavior were analyzed over time using expression of a panel of 37 genes related to the M1, M2a, and M2c macrophage phenotypes (such as surface marker expression) and other aspects of inflammation and angiogenesis, as well as secretion of several key proteins. We chose to phenotype macrophages based primarily on gene expression, which has been proposed to be a more thorough method compared to analysis of surface markers46 because macrophages often considerably change behavior without detectable changes in surface marker expression.45,47
Methods
Cell Culture and Cell Seeding onto hCVAM
Primary human monocytes isolated peripheral blood from a single donor were purchased from University of Pennsylvania’s Human Immunology Core and differentiated into unactivated M0 macrophages over 5 days and polarized into M1 macrophages for an additional 48 h (Fig. 1a), as previously described.47 Briefly, monocytes were cultured in complete RPMI culture medium (cRPMI): RPMI 1640 with phenol red and l-glutamine supplemented with 10% heat-inactivated human serum, 1% penicillin streptomycin (Gibco™, Grand Island, NY), and 20 ng/mL of recombinant human macrophage colony stimulating factor (MCSF) (PeproTech®, Rocky Hill, NJ) for 5 days with a media change on the third day. On day 5, unactivated M0 macrophages were scraped, counted, and re-plated in cRPMI supplemented with 100 ng/mL of recombinant human IFNG (PeproTech®, Rocky Hill, NJ) and 100 ng/mL of LPS (Sigma Aldrich) (cRPMI-M1) for an additional two days to create M1 macrophages. After the full seven days, cells were scraped, counted, and used in subsequent experiments.

Schematic of experimental methods and design. (a) Primary human monocytes were cultured with macrophage colony stimulating factor (MCSF) for 5 days (including a media change on day 3) to differentiate the cells into unactivated (M0) macrophages. M0 macrophages were then cultured with MCSF supplemented with LPS and IFNG for 2 days according to previous methods47; (b) M1 macrophages were cultured in four different treatment groups each containing M1 macrophage culture medium (cRPMI-M1, detailed in the methods): M1 Control; M1 macrophages seeded onto intact hCVAM (hCVAM); M1 macrophages co-cultured with hCVAM separated by a semi-permeable transwell insert (Soluble Factors); and hCVAM without any seeded macrophages (hCVAM-only Control). All treatment groups containing hCVAM (hCVAM, Soluble Factors and hCVAM-only Control) were performed with n = 3 donors of tissue. All macrophages were obtained from one donor; (c) the experiment was initiated after 7 days of macrophage culture when M1 polarization was complete, which was considered day 0 for subsequent experiments. Samples for RNA extraction and gene expression analysis were collected on days 1 and 6 (denoted with ~), while conditioned media samples were collected on days 1, 3 (during media change), and 6 (denoted with +).
Preparation of hCVAM and Experimental Group Organization
Samples of hCVAM (Grafix®, Osiris Therapeutics, Inc.) were generously donated by Osiris Therapeutics, Inc. (Columbia, MD), and stored at −80 °C. In preparation for in vitro culture, hCVAMs (n = 3 donors) were thawed in their packaging in a 37 °C water bath for approximately 1 min. Thawed packaged hCVAMs were quickly moved into a biological safety cabinet, removed from packaging and washed in sterile phosphate buffered saline (PBS) (cellgro®, Manassas, VA) in Petri dishes, which is consistent with clinical practice. The membranes were biopsy-punched using a 10 mm punch before being organized into experimental groups that were designed to isolate the effects of soluble factors released from the matrix and/or from cells within the matrix (Soluble Factors) in comparison to the effects of direct contact with intact hCVAM on macrophage behavior over time (Fig. 1b). For the intact hCVAM group (n = 3 hCVAM donors; n = 3 experimental replicates per donor per time point), M1 macrophages (5.0 × 105 cells in 20 μL of cRPMI-M1) were seeded onto the stromal side of the membrane, which is the side placed onto chronic wounds clinically,22 and allowed to attach to the membrane for 30–45 min in a 37 °C, 5% CO2 cell culture incubator environment prior to the addition of 1 mL cRPMI-M1. For the Soluble Factors group (n = 3 hCVAM donors, n = 3 experimental replicates per donor per time point), M1 macrophages (5.0 × 105 cells) were plated in 1 mL of cRPMI-M1 in an ultra-low attachment tissue culture plate and allowed to sink to the bottom of the well before the adding a Millicell® cell culture insert (EMD Millipore Corporation, Billerica, MA) containing an hCVAM sample and 0.4 mL of cRPMI-M1. hCVAM controls and M1 Controls were also included. For the M1 Control group (n = 3 experimental replicates per time point), 5.0 × 105 M1 macrophages were plated in 1 mL of cRPMI-M1 in an ultra-low attachment tissue culture plate. For the hCVAM Control group (n = 3 donors, n = 3 experimental replicates per donor per time point), hCVAM was cultured in cRPMI-M1. All samples inclusive of any unadhered macrophages were collected for RNA extraction (hCVAM together with seeded macrophages and hCVAM alone in 1 mL of TRIzol® Reagent (Life Technologies, Carlsbad, CA), or macrophages alone in 350 μL of lysis buffer) on days 1 and 6 in Eppendorf tubes and stored at −80 °C, while conditioned media was collected on days 1, 3, and 6 and frozen at −80 °C until further analysis (Fig. 1c).
RNA Extraction and Multiplex Gene Expression Analysis
First, all samples were thawed on ice and homogenized in TRIzol® Reagent (Life Technologies, Carlsbad, CA). Sample homogenization for membrane-containing samples was performed using a Mini BeadBeater-16 (BioSpec Products, Bartlesville, OK) with three 2.3 mm stainless steel beads in each tube for 5–8 cycles of 10 s, placing samples on ice to maintain low temperature in between cycles. Next, 0.2 mL of chloroform was added to each sample and shaken rigorously by hand for 15 s, which were then incubated for 2–3 min at room temperature, and centrifuged for 15 min at 4 °C with a speed of 12,000×g. The aqueous layer was then removed into a fresh tube. Cell-only samples were lysed by repeatedly pipetting in lysis buffer up and down, as opposed to homogenization with the bead beater. Equal parts 70% ethanol was added to all samples to obtain a final concentration of 35% ethanol, which was then loaded onto the RNeasy Mini spin columns (Qiagen®, Hilden, Germany), where RNA was purified according to the manufacturers’ instructions. RNA from all samples was eluted in one volume of 30 μL and immediately frozen at −80 °C for future analysis. RNA was later thawed on ice and quantified using a NanoDrop 1000 (Thermo Scientific, Wilmington, DE) or a Bioanalyzer 2100 with RNA Nano 6000 kit (Agilent Technologies, Santa Clara, CA). Samples with 260/280 ratios between 1.7 and 2.2 or RIN numbers over 9.0 were considered pure and were utilized for multiplex gene expression analysis using NanoString (NanoString Technologies, Seattle, WA). Importantly, all hCVAM Control samples had extremely low concentrations of RNA (<4 ng/mL) confirmed with both Bioanalyzer and NanoDrop 1000; on the Bioanalyzer, samples had little to no visible bands in the RNA Nano chip above water-only controls. Therefore, hCVAM Controls were not included in the NanoString gene expression analysis, and gene expression data obtained from macrophage-seeded hCVAM samples are considered to derive primarily from the macrophages. NanoString gene expression analysis was performed according to the manufacturers’ instructions, where 100 ng of RNA per sample was hybridized with the capture and reporter probes of the custom codeset (Table 1), inclusive of 37 endogenous genes, 5 housekeeping genes, 8 External RNA Control Consortium (ERCC) negative controls, and 6 ERCC positive controls. Samples from the M1 Control, Direct Contact, and Soluble Factors treatment groups were divided between 9 different NanoString cartridges, which only held 12 samples each, so that 1–6 samples from each treatment and/or donor within a time point were included in each cartridge. Raw count data was extracted from nSolver™ Analysis Software 3.0 followed by quality control, normalization, and filtering, as described below.
Data Normalization and Analysis
Raw count data from NanoString was first normalized to the ERCC positive controls per NanoString’s recommendation. First, the geometric mean of all positive controls for all samples was calculated and then divided by the geometric mean of each individual samples’ ERCC positive controls, generating a ERCC positive control factor. All endogenous, housekeeping, ERCC negative and positive controls were multiplied by each samples’ ERCC positive control factor to account for NanoString individual lane assay efficiency. Positive control-normalized data was input into R Studio, where batch effects were removed for different lots of codeset and per NanoString cartridge, using the function removeBatchEffect within the limma package in R. The function to remove batch effects set the primary and secondary batch effects to two independent vectors and utilized a linear model to fit and correct the data. Next, data were normalized using the voomWithQualityWeights function within the limma package, which sets a weight factor to each sample treatment before performing a global normalization.24 Next, the maximum value from all of the ERCC negative controls from each lane was subtracted from each sample to determine which genes were not expressed above the negative controls. If any biological replicate (n = 3 donors of hCVAM) was missing two or more experimental replicates (of n = 3 replicates per donor), then that biological replicate was excluded, and if more than 50% of all replicates were missing per gene, then the gene was excluded, which included ALPL, ANGPT1, BGLAP, COL1A1, COL4A1, CTGF, and TEK. Next, fold change values were calculated for each individual replicate compared to the averaged M1 Control for each gene and used to generate a heatmap using the heatmap.2 function in R and a grouped column graph at each time point in GraphPad Prism 6. A dotted line at a fold change of 1.0 (or 0 on graphs of Log2-transformed data of values normalized to the M1 Control) on each individual gene represents no change vs. the M1 Control. Principal component analysis was performed on the Log2 voom-normalized data as implemented in the scikit-learn Decomposition package with missing values imputed using nearest neighbor estimation as implemented in the scikit-learn Preprocessing package,1 to holistically examine effects of treatment, donor, and time.
Protein Secretion
Protein secretion from conditioned media from each treatment group was analyzed using enzyme-linked immunosorbent assay (ELISA) according to the manufacturers’ instructions for human interleukin-10 (IL10), tumor necrosis factor-α (TNF-α), and vascular endothelial growth factor (VEGF; PeproTech®, Rocky Hill, NJ) and matrix metalloproteinase-9 (MMP9; R&D Systems, Inc., Minneapolis, MN).
Statistical Analysis
Normalized gene expression and protein secretion data were processed in GraphPad Prism 6.0. Statistical analysis of fold change data for individual gene expression was performed using a non-matched two-way analysis of variance (ANOVA) and Tukey’s post hoc multiple comparisons test with an adjusted p value of 0.01 to determine significant differences between treatment groups within a time point. Analysis of protein secretion was performed using a non-matched two-way ANOVA and a Tukey’s post hoc multiple comparisons test with an adjusted p-value of 0.01 to determine differences between treatment groups within a time point and over time. Statistical analysis of changes within a treatment group over time were performed utilizing a mixed effects model. Treatment and time were set as joint fixed effects while donor level variability was treated as a random effect. Regression was performed using the statsmodels MixedML package.42
Results
Multiplex Gene Expression Analysis
No RNA was detected in the hCVAM-only control group via Bioanalyzer 2100 and NanoDrop 1000; therefore, all detected gene expression is considered to derive solely from macrophages. Normalized NanoString gene expression data were first visualized using principal component analysis, which illustrated that there was no effect of hCVAM donor on global gene expression patterns (Fig. S1). A heatmap of all replicates organized by gene function revealed that the majority of genes were upregulated in the Soluble Factors group and downregulated in hCVAM group on day 1 compared to the M1 Control, while there appeared to be more subtle shifts in gene expression between treatment groups on day 6 (Fig. 2a). Plotting each gene individually at each time point (Figs. 2b and 2c) further confirmed these trends, and also showcased several genes with the most substantial changes. The genes with the most substantial differences between how direct contact with the hCVAM and Soluble Factors regulated macrophage behavior included CCL5, VEGF, CCL22, MRC1, PDGFB, MMP9, VCAN, ANGPT2, BMP2, NOTCH1, PECAM1, RUNX2, SMAD1, and TGFB1 on day 1 and CCL5, CD80, CD163, MMP7, PECAM1, SPP1, and VDR on day 6 (Figs. 2b and 2c).

(a) Heatmap of all gene replicates represented as Log2(value/M1 Control) generated in R using heatmap.2 function in gplots package; (b) all genes and samples from NanoString gene expression analysis represented as a grouped column graph of Log2(Value/M1 Control) for day 1 and (c) day 6. A dotted line at a fold change of 1.0 (or 0 on graphs of Log2-transformed data of values normalized to the M1 Control) represents no change vs. the M1 Control.
To explore individual gene expression differences, each gene was plotted over time and statistical analysis was conducted using a two-way ANOVA to assess differences between treatment groups within a time point, which identified 13 genes with significant differences (Fig. 3) and 17 genes without (Fig. S2); a mixed effects model was used to determine differences in expression over time (Fig. S3). Compared to the M1 Control, Soluble Factors promoted significant upregulation of the pro-inflammatory marker IL1B expression on day 1 (p < 0.01, indicated by # in Fig. 3a) but significant downregulation of the pro-inflammatory marker TNF expression on day 6 (Fig. 3b), while intact hCVAM promoted downregulation of pro-inflammatory markers TNF, CCL5 and CCR7 on day 1 (Figs. 3b–3d). Intact hCVAM also promoted upregulation of the anti-inflammatory marker IL10 (Fig. 3e) and downregulation of the gene encoding tyrosine kinase receptor TIE1 (Fig. 3f) compared to the M1 Control on day 6.

NanoString gene expression analysis of all differentially expressed genes (p < 0.01). hCVAM data are represented as Log2(Value/M1 Control) and as the mean of all experimental replicates (n = 4–9) ± standard error of the mean (SEM). A dotted line at a fold change of 1.0 (or 0 on graphs of Log2-transformed data of values normalized to the M1 Control) on each individual gene represents no change vs. the M1 Control; those samples significantly different from the M1 Control are shown with a # symbol (p < 0.01). Statistical significance between treatment groups (represented with black bars) was calculated using a two-way ANOVA with a Tukey’s post hoc multiple comparisons test, *p < 0.01, **p < 0.001, ***p < 0.0001.
There were additional differences observed solely between the hCVAM and Soluble Factors groups at both time points, though these differences were not always significant compared to the M1 Control. Expression of IL1B, TNF, CCL5, CCR7, IL10 (Figs. 3a–3e), MMP9, VEGF, NOTCH1, RUNX2, SPP1, and TGFB1 (Figs. 3g–3l) was higher by macrophages in the Soluble Factors group on day 1 compared to the hCVAM group. At the day 6 time point, CCL5 and TIE1 were also expressed at higher levels in the Soluble Factors group relative to the hCVAM group (Figs. 3c and 3f). Only IL10 and CD80 were expressed at lower levels in the Soluble Factors group relative to the hCVAM group, and these changes were observed at day 6 (Figs. 3e and 3m). Changes in gene expression were also explored over time within a given treatment group (Fig. S3); interestingly, the only gene that changed significantly over time was IL1B in the Soluble Factors group (Fig. 3a).
Protein Secretion Analysis
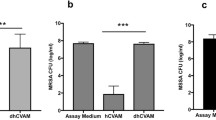
We next examined protein secretion of four proteins that are particularly important in inflammation, wound healing, and angiogenesis: TNF, IL10, MMP9 and VEGF (Fig. 4). Expression of these proteins on the gene level is shown in Figs. 3b, 3e, 3g, and 3h. Macrophages in the hCVAM group secreted significantly lower levels of TNF, VEGF, and MMP9 compared to the M1 Control and the Soluble Factors groups at all time points (Fig. 4). Additionally, macrophages in the Direct Contact group secreted significantly greater levels of IL10 compared to the Soluble Factors group at day 6 (Fig. 4b). Compared to the M1 Control at corresponding time points, macrophages in the Soluble Factors group secreted higher levels of TNF (all time points) and IL10 (day 3 only) and lower levels of MMP9 (day 1 only) and VEGF (day 3 only).

Protein content in the conditioned media generated from each treatment at days 1, 3, and 6. All data are represented as mean ± SEM of all experimental replicates within detection limits (n = 3–9 per treatment group per timepoint). Statistical analysis was performed using a two-way ANOVA with a Tukey’s post hoc multiple comparisons analysis to determine differences between treatments within a time point, *p < 0.01, **p < 0.001, ***p < 0.0001, + p < 0.01 compared to the hCVAM Control, and nd = no detection of protein.
Discussion
In this study, we explored the immunomodulatory actions of soluble factors derived from hCVAM in comparison to intact hCVAM on the response of M1 macrophages, which dominate the chronic wound environment in vivo. 32,44 Direct contact with intact hCVAM, which includes both ECM- and soluble factor-derived signals, promoted more changes in gene expression and protein secretion than Soluble Factors. These changes resulted in a shift toward an anti-inflammatory phenotype, even in the continued presence of pro-inflammatory stimuli. Collectively, these results suggest that the immunomodulatory effects of hCVAM require direct contact with the membrane, which have important implications for the design of next generation bioactive, immunomodulatory chronic wound treatments.
Macrophages are highly influenced by their microenvironment, which includes their interaction with structures, cytokines, signals, cells, or foreign materials.23 In keeping with the results shown in this study, recent studies have shown that ECM-derived signals are key modulators of macrophage behavior in vitro and in vivo. In one study, an acellular porcine bladder xenograft and rat body wall autograft promoted a more M2-like macrophage phenotype response (as measured via immunohistochemical staining with CD163) and were subsequently associated with a constructive remodeling healing outcomes in a rat abdominal wall repair model relative to their cellular counterparts.5,9 In addition, macrophages co-cultured with four commercially available ECM-derived wound matrices in vitro exhibited hybrid macrophage phenotypes that correlated with the products’ outcomes in vivo. 55,58 Collectively, these results illustrate the critical role of ECM-based biomaterials in modulating macrophage behavior for tissue repair.
The anti-inflammatory effects of hCVAM on macrophages that we report in this study are consistent with recent reports that have explored responses of other immune cells to hCVAM. For example, in one study peripheral blood mononuclear cells (PBMCs) were pretreated with LPS, activating the cells to secrete pro-inflammatory cytokines,19 and co-cultured with either hCVAM or devitalized hAM pre-treated with TNF. hCVAM pre-treated with TNF promoted significantly higher levels of secretion of the anti-inflammatory factor IL10 compared to devitalized hAM pre-treated with TNF.11 In another study, macrophages shifted to an anti-inflammatory phenotype when cultured in media conditioned by hAM-derived MSCs, even in the presence of pro-inflammatory stimuli.29 In a study of diabetic wound healing in vivo, authors micronized hCVAM via homogenization and filtration to obtain 300–600 μm particles, which were compared to particles derived from devitalized AM in a murine full-thickness diabetic wound model.59 The living micronized AM (LMAM), which retained cell viability, promoted significantly enhanced wound closure, increased recruitment of macrophages (F4/80+) that were predominately of an M2 phenotype (MRC1+), and reduced secretion of pro-inflammatory proteins [IL1B, interleukin-6 (IL6) and TNF] in vivo compared to amniotic membrane that was devitalized via multiple freeze/thaw cycles.59 Furthermore, conditioned media from LMAM caused macrophages to significantly downregulate expression of the M1 marker CCR7, upregulate expression of the M2a marker MRC1, and increase secretion of factors involved in chemotaxis, inflammation and angiogenesis, relative to devitalized hAM.59 Collectively, these studies and our results suggest that cell-hCVAM interactions promote an anti-inflammatory macrophage phenotype, even in the presence of pro-inflammatory stimuli.
The distinct changes in macrophage behavior, as measured by multiplex gene expression analysis and protein secretion, provide insight into the mechanisms of hAM-promoted chronic wound healing. Previous studies have shown that persistent, elevated TNF or blocked TNF have both been shown to be detrimental to angiogenesis and healing,6,51 while an early, yet transient, release of TNF, has been shown to enhance healing.4,41 These studies in combination with our work suggest that contact with hCVAM may play a unique role in modulating macrophage behavior via inhibition of TNF, which may minimize prolonged inflammation.41 Significant early downregulation of pro-inflammatory cytokines and markers TNF, CCL5, and CCR7 suggests that intact hCVAM may drive macrophages away from an M1 macrophage phenotype.47 While these effects may be beneficial for chronic wounds that are characterized by elevated inflammation, they may also have negative implications with respect to how macrophages handle bacterial infection.7
Lastly, there were limitations to this work. We attempted to simulate the chronic wound environment by conducting the experiments in the presence of the pro-inflammatory, M1-promoting stimuli IFNG and LPS, but even more anti-inflammatory effects might be observed in the absence of these signals. It would be interesting in future studies to investigate how macrophages of different phenotypes, including unactivated macrophages, differentially respond to hCVAM. Another limitation is that only a small number of genes and proteins was evaluated. Future work will explore a larger panel of genes related to inflammation and angiogenesis and include the use of functional assays, such as angiogenesis assays. Finally, there is always the possibility that in vitro results fail to accurately recapitulate interactions in vivo, where the microenvironment is considerably more complex. These limitations notwithstanding, this study illustrates that direct contact with hCVAM and soluble factors from hCVAM each have distinct roles in modulating macrophage behavior in vitro, with direct contact having more dominant effects in the continued presence of pro-inflammatory stimuli. These important, yet preliminary, findings lay the groundwork for more robust mechanistic studies of the immunomodulatory effects of hCVAM in vivo.
Abbreviations
- hAM:
-
Human amniotic membrane
- ANOVA:
-
Analysis of variance
- CCL5:
-
Chemokine (C–C motif) ligand 5
- cRPMI:
-
Complete RPMI culture medium
- cRPMI-M1:
-
Complete RPMI culture medium supplemented with M1-stimulating cytokines
- ECM:
-
Extracellular matrix
- EGF:
-
Epidermal growth factor
- ELISA:
-
Enzyme-linked immunosorbent assay
- ERCC:
-
External RNA Control Consortium
- hCVAM:
-
Human cryopreserved amniotic membrane
- IFNG:
-
Interferon-γ
- IL4:
-
Interleukin-4
- IL8:
-
Interleukin-8
- IL10:
-
Interleukin-10
- IL1A:
-
Interleukin-1α
- IL1B:
-
Interleukin-1β
- LPS:
-
Lipopolysaccharide
- LMAM:
-
Living micronized amniotic membrane
- MCSF:
-
Macrophage colony stimulating factor
- MMP9:
-
Matrix metalloproteinase-9
- MSCs:
-
Mesenchymal stem cells
- PBMCs:
-
Peripheral blood mononuclear cells
- PBS:
-
Phosphate buffered saline
- PDGFB:
-
Platelet derived growth factor
- PGE2:
-
Prostaglandin E2
- SEM :
-
Standard error of mean
- TGFB1:
-
Transforming growth factor-β1
- TNF-α :
-
Tumor necrosis factor-α
- VEGF:
-
Vascular endothelial growth factor
References
Abraham, A., et al. Machine learning for neuroimaging with scikit-learn. Front. Neuroinform. 8:14, 2014.
Anderson, J. M., A. Rodriguez, and D. T. Chang. Foreign body reaction to biomaterials. Semin. Immunol. 20:86–100, 2008.
Arnold, L., et al. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 204:1057–1069, 2007.
Ashcroft, G. S., et al. Tumor necrosis factor-alpha (TNF-α) is a therapeutic target for impaired cutaneous wound healing. Wound Repair Regen. 20:38–49, 2012.
Badylak, S. F., J. E. Valentin, A. K. Ravindra, G. P. McCabe, and A. N. Stewart-Akers. Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Eng. A 14:1835–1842, 2008.
Baluk, P., et al. TNF-α drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J. Clin. Investig. 119:2954–2964, 2009.
Benoit, M., B. Desnues, and J.-L. Mege. Macrophage polarization in bacterial infections. J. Immunol. 181:3733, 2008.
Berger, M. L., M. Mamdani, D. Atkins, and M. L. Johnson. Good research practices for comparative effectiveness research: defining, reporting and interpreting nonrandomized studies of treatment effects using secondary data sources: the ISPOR Good Research Practices for Retrospective Database Analysis Task Force Report—Part I. Value Health 12:1044–1052, 2009.
Brown, B. N., J. E. Valentin, A. M. Stewart-Akers, G. P. McCabe, and S. F. Badylak. Macrophage phenotype and remodeling outcomes in response to biologic scaffolds with and without a cellular component. Biomaterials 30:1482–1491, 2009.
Cooke, M., et al. Comparison of cryopreserved amniotic membrane and umbilical cord tissue with dehydrated amniotic membrane/chorion tissue. J. Wound Care 23:465–476, 2014.
Davis, J. S., et al. The use of skin grafts in the ambulatory treatment of ulcers: report of fifty cases. JAMA LXIV:558–560, 1915.
Duan-Arnold, Y., et al. Retention of endogenous viable cells enhances the anti-inflammatory activity of cryopreserved amnion. Adv. Wound Care (New Rochelle) 4:523–533, 2015.
Ferraro, N. M., W. Dampier, M. S. Weingarten, and K. L. Spiller. Deconvolution of heterogeneous wound tissue samples into relative macrophage phenotype composition via models based on gene expression. Integr. Biol. (Camb.) 9:328–338, 2017.
Gibbons, G. W. Grafix, a cryopreserved placental membrane, for the treatment of chronic/stalled wounds. Adv. Wound Care (New Rochelle) 4:534–544, 2015.
Guo, X., et al. Modulation of cell attachment, proliferation, and angiogenesis by decellularized, dehydrated human amniotic membrane in in vitro models. Wounds 29:28–38, 2017.
Hao, Y., D. H. Ma, D. G. Hwang, W. S. Kim, and F. Zhang. Identification of antiangiogenic and antiinflammatory proteins in human amniotic membrane. Cornea 19:348–352, 2000.
Hopkinson, A., et al. Optimization of amniotic membrane (AM) denuding for tissue engineering. Tissue Eng. C 14:371–381, 2008.
Huang, G., et al. Accelerated expansion of epidermal keratinocyte and improved dermal reconstruction achieved by engineered amniotic membrane. Cell Transplant. 22:1831–1844, 2013.
Jansky, L., P. Reymanova, and J. Kopecky. Dynamics of cytokine production in human peripheral blood mononuclear cells stimulated by LPS or infected by Borrelia. Physiol. Res. 52:593–598, 2003.
Johnson, E. L., J. T. Marshall, and G. M. Michael. A comparative outcomes analysis evaluating clinical effectiveness in two different human placental membrane products for wound management. Wound Repair Regen. 2017. doi:10.1111/wrr.12503.
Laurent, R., A. Nallet, L. Obert, L. Nicod, and F. Gindraux. Storage and qualification of viable intact human amniotic graft and technology transfer to a tissue bank. Cell Tissue Bank. 15:267–275, 2014.
Lavery, L. A., et al. The efficacy and safety of Grafix((R)) for the treatment of chronic diabetic foot ulcers: results of a multi-centre, controlled, randomised, blinded, clinical trial. Int. Wound J. 11:554–560, 2014.
Lavin, Y., et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159:1312–1326, 2014.
Law, C. W., Y. Chen, W. Shi, and G. K. Smyth. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15:R29, 2014.
Leibovich, S. J., and R. Ross. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am. J. Pathol. 78:71–100, 1975.
Litwiniuk, M., and T. Grzela. Amniotic membrane: new concepts for an old dressing. Wound Repair Regen. 22:451–456, 2014.
Liu, D., H. Xiong, P. Ning, J. Chen, and W. Lan. In: 2010 3rd International Conference on Biomedical Engineering and Informatics, Vol. 4, pp. 1633–1635, 2010.
Lurier, E. B., et al. Transcriptome analysis of IL-10-stimulated (M2c) macrophages by next-generation sequencing. Immunobiology 222(7):847–856, 2017.
Magatti, M., et al. Human amnion favours tissue repair by inducing the M1-to-M2 switch and enhancing M2 macrophage features. J. Tissue Eng. Regen. Med. 2016. doi:10.1002/term.2193.
Markova, A., and E. N. Mostow. US skin disease assessment: ulcer and wound care. Dermatol. Clin. 30:107–111, ix, 2012.
Mirza, R., L. A. DiPietro, and T. J. Koh. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 175:2454–2462, 2009.
Mirza, R. E., M. M. Fang, W. J. Ennis, and T. J. Koh. Blocking interleukin-1β induces a healing-associated wound macrophage phenotype and improves healing in Type 2 diabetes. Diabetes 62:2579–2587, 2013.
Mirza, R. E., M. M. Fang, E. M. Weinheimer-Haus, W. J. Ennis, and T. J. Koh. Sustained inflammasome activity in macrophages impairs wound healing in Type 2 diabetic humans and mice. Diabetes 63:1103–1114, 2014.
Mirza, R., and T. J. Koh. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 56:256–264, 2011.
Mosser, D. M., and J. P. Edwards. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8:958–969, 2008.
Nassiri, S., I. Zakeri, M. S. Weingarten, and K. L. Spiller. Relative expression of proinflammatory and antiinflammatory genes reveals differences between healing and nonhealing human chronic diabetic foot ulcers. J. Investig. Dermatol. 135:1700–1703, 2015.
Niknejad, H., G. Paeini-Vayghan, F. A. Tehrani, M. Khayat-Khoei, and H. Peirovi. Side dependent effects of the human amnion on angiogenesis. Placenta 34:340–345, 2013.
Parolini, O., and M. Caruso. Review: preclinical studies on placenta-derived cells and amniotic membrane: an update. Placenta 32(Suppl 2):S186–S195, 2011.
Regulski, M., et al. A retrospective analysis of a human cellular repair matrix for the treatment of chronic wounds. Ostomy Wound Manag. 59:38–43, 2013.
Roh, J. D., et al. Tissue-engineered vascular grafts transform into mature blood vessels via an inflammation-mediated process of vascular remodeling. Proc. Natl Acad. Sci. USA 107:4669–4674, 2010.
Sainson, R. C. A., et al. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood 111:4997–5007, 2008.
Seabold, S., and P. Josef. Statsmodels: econometric and statistical modeling with Python. In: Proceedings of the 9th Python in Science Conference, pp 57–61, 2010.
Sen, C. K., et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. Off. Publ. Wound Heal. Soc. Eur. Tissue Repair Soc. 17:763–771, 2009.
Sindrilaru, A., et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Investig. 121:985–997, 2011.
Singh, S., et al. Unbiased analysis of the impact of micropatterned biomaterials on macrophage behavior provides insights beyond predefined polarization states. ACS Biomater. Sci. Eng. 2017. doi:10.1021/acsbiomaterials.7b00104.
Spiller, K. L., and T. J. Koh. Macrophage-based therapeutic strategies in regenerative medicine. Adv. Drug Deliv. Rev. 2017. doi:10.1016/j.addr.2017.05.010.
Spiller, K. L., et al. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 35:4477–4488, 2014.
Talmi, Y. P., L. Sigler, E. Inge, Y. Finkelstein, and Y. Zohar. Antibacterial properties of human amniotic membranes. Placenta 12:285–288, 1991.
Tseng, S. C., D. Q. Li, and X. Ma. Suppression of transforming growth factor-beta isoforms, TGF-beta receptor type II, and myofibroblast differentiation in cultured human corneal and limbal fibroblasts by amniotic membrane matrix. J. Cell. Physiol. 179:325–335, 1999.
van Putten, S. M., D. T. A. Ploeger, E. R. Popa, and R. A. Bank. Macrophage phenotypes in the collagen-induced foreign body reaction in rats. Acta Biomater. 9:6502–6510, 2013.
Wetzler, C., H. Kämpfer, B. Stallmeyer, J. Pfeilschifter, and S. Frank. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Investig. Dermatol. 115:245–253, 2000.
Willenborg, S., et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 120:613–625, 2012.
Wilshaw, S. P., J. N. Kearney, J. Fisher, and E. Ingham. Production of an acellular amniotic membrane matrix for use in tissue engineering. Tissue Eng. 12:2117–2129, 2006.
Wilshaw, S. P., J. Kearney, J. Fisher, and E. Ingham. Biocompatibility and potential of acellular human amniotic membrane to support the attachment and proliferation of allogeneic cells. Tissue Eng. A 14:463–472, 2008.
Witherel, C. E., P. L. Graney, D. O. Freytes, M. S. Weingarten, and K. L. Spiller. Response of human macrophages to wound matrices in vitro. Wound Repair Regen. 24:514–524, 2016.
Wolbank, S., et al. Impact of human amniotic membrane preparation on release of angiogenic factors. J. Tissue Eng. Regen. Med. 3:651–654, 2009.
Wynn, T. A., and L. Barron. Macrophages: master regulators of inflammation and fibrosis. Semin. Liver Dis. 30:245–257, 2010.
Xue, J., et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40:274–288, 2014.
Zheng, Y., et al. Topical administration of cryopreserved living micronized amnion accelerates wound healing in diabetic mice by modulating local microenvironment. Biomaterials 113:56–67, 2017.
Acknowledgments
The authors would like to thank Yi Arnold-Duan and Matthew Moorman (Osiris Therapeutics, Inc.) for their helpful discussions and technical advice in handling hCVAM. This work was sponsored in part by Osiris Therapeutics, Inc., and by NHLBI Grant Number R01 HL130037 to KLS. CEW is grateful for the US Department of Education Graduate Assistance in Areas of National Need (GAANN) Interdisciplinary Collaboration and Research Enterprise (iCARE) Fellowship.
Conflict of interest
KLS discloses a potential conflict of interest: this study was funded in large part by Osiris Therapeutics, Inc. The study was designed by KLS and CEW, with some input from Osiris with respect to the potential impact of different experiments. Employees from Osiris had no part in interpretation of the study’s results. CEW, TY, MC, and WD declare that they have no conflicts of interest.
Ethical Approval
De-identified hCVAM samples were provided by Osiris Therapeutics as commercially available materials. De-identified human monocytes were purchased from the University of Pennsylvania Human Immunology Core. As such both human materials are exempt from review by the Institutional Review Board. No animal experiments were conducted for this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Associate Editor Alyssa Panitch oversaw the review of this article.
Kara Spiller is an Assistant Professor in Drexel University’s School of Biomedical Engineering, Science, and Health Systems. Dr. Spiller received Bachelor’s and Master’s Degrees in Biomedical Engineering from Drexel in 2007. As an NSF Graduate Research Fellow, she conducted her doctoral research in the design of semi-degradable hydrogels for the repair of articular cartilage in the Biomaterials and Drug Delivery Laboratory at Drexel and in the Shanghai Key Tissue Engineering Laboratory of Shanghai Jiao Tong University. After completing her Ph.D. in 2010, she conducted research in the design of scaffolds for bone tissue engineering on a Fulbright Fellowship in the Biomaterials, Biodegradables, and Biomimetics (the 3Bs) Research Group at the University of Minho in Guimarães, Portugal. She then conducted postdoctoral studies towards the development of immunomodulatory biomaterials for bone regeneration in the Laboratory for Stem Cells and Tissue Engineering at Columbia University, before joining the Faculty of Drexel in 2013. Her research is funded by grants from the National Science Foundation and the NIH, as well as grants from private foundations and industry. Her research interests include the role of inflammation in regenerative medicine, the design of immunomodulatory biomaterials, and international engineering education.

This article is part of the 2017 CMBE Young Innovators special issue.
Electronic supplementary material
Below is the link to the electronic supplementary material. Figure S1 Principal component analysis of principal component 1 (PC1) vs. principal component 2 (PC2), which account for 42.5 and 15% of the variance, respectively. Each treatment is represented by a color, M1 Control: grey, Direct Contact: blue, Soluble Factors: gold, while each donor is represented by a different symbol. Filled markers represent day 1 samples while outlined (no-fill) markers represent day 6 samples. Figure S2 NanoString gene expression analysis of all additional genes that were not differentially expressed. Data are represented as Log2(Value/M1 Control) and as the mean of all experimental replicates (n = 4–9) ± standard error of the mean (SEM). A dotted line at a fold change of 1.0 (or 0 on graphs of Log2-transformed data of values normalized to the M1 Control) on each individual gene represents no change vs. the M1 Control. Figure S3 Differentially expressed gene, IL1B, via mixed-effects regression with treatment and time held as fixed effects and donor as a random effect with ***p < 0.0001. Treatment group with a decreasing slope over time indicates a downregulation over time.
Rights and permissions
About this article

{kind=link}
Cite this article
Witherel, C.E., Yu, T., Concannon, M. et al. Immunomodulatory Effects of Human Cryopreserved Viable Amniotic Membrane in a Pro-Inflammatory Environment In Vitro . Cel. Mol. Bioeng. 10, 451–462 (2017). https://doi.org/10.1007/s12195-017-0494-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12195-017-0494-7