
Abstract
Nicotine has been known to play a pathogenic role in various cardiovascular disorders. However, the definite mechanism of nicotine-mediated endothelial dysfunction in vivo remains unclear because hemodynamic factor in most of in vitro studies was excluded. In this study, we investigated how nicotine affects human umbilical vein endothelial cells (HUVECs), from views of inflammatory and hemostatic responses of the cells, under a hemodynamic environment as occurred in vivo. Our results showed that both inflammation, reflected by production of reactive oxygen species and efficacy of monocytes adhesion, and hemostatic expression of HUVECs were abnormally enhanced after treated with 10−4 M nicotine and 12 dynes cm−2 laminar shear stress (LSS) simultaneously for 24 h, and that the protein expression levels of VCAM-1, ICAM-1, and PAI-1 were significantly enhanced 1.3-, 2- and 2-fold (p < 0.05 for each), respectively, as compared to the group with nicotine alone; 2.2-, 3- and 4.2-fold (p < 0.05 for each), respectively, as compared to the group with LSS alone. We reasoned that those irregular expressions were resulted from the reduction of endothelial nitric oxide synthase that was initially caused by nicotine exposure and exacerbated due to LSS treatment. Furthermore, all the impaired responses can be alleviated by use of 1 μg mL−1 recombinant tissue plasminogen activator, implicating that the irregular inflammation may be due to thrombosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nicotine is the major component among more than 4700 chemical constituents of cigarette smoking and has been recognized as a leading pathogenic factor for various cardiovascular diseases such as atherosclerosis,19 hypertension,26 thrombosis,10,20 and cardiomyopathy.42 Although the biological effects of nicotine have been extensively investigated in the past decades, the precise mechanisms of nicotine-induced endothelial dysfunction still remain uncertain in clinics because most of prior in vitro studies excluded hemodynamic factors that led to the experiments far from the real environment in vessels.
Knowingly vascular endothelial cells in vivo are continually subjected to hemodynamic forces particularly laminar shear stress (LSS) because they directly contact with flowing blood. LSS is a biomechanical force and its magnitude is determined by blood flow rate, vessel geometry and fluid viscosity. Since it enables to modulate endothelial structure and functions through the activities of local mechanotransduction mechanisms, LSS has long been recognized as a crucial regulator for the endothelial physiology and that the mostly known function is to provide endothelial protection such as anti-atherosclerosis.32 In general, atherosclerotic lesions usually initiate at bifurcations and/or sharp curves where the blood flow is interrupted and the mean LSS magnitude is low (<4 dynes cm−2).23,34 However, quite a few clinical studies showed that smoking-mediated atherosclerotic development does not follow this pattern. Instead, there is a strong association between smoking and large (proximal) vessel (e.g., aorta, artery) disease1,14,31 where the intensity of LSS is certainly higher than 4 dynes cm−2.28 In addition, it has been recognized that smoking is one of the major risk factors for peripheral arterial disease (PAD), and such smoking-mediated PAD is predominantly occurred at proximal arteries, especially aorta and iliac arteries.7,30 These substantial correlations lead to our hypothesis that high LSS may play a role in vascular injury while the endothelial cells are exposed to nicotine.
Inflammation is a protective response mediated by innate immune system following exposure to a range of harmful stimuli. Although inflammation is an essential mechanism in response to impairments such as tissue injury and/or microbial invasion, inappropriate or excessive inflammatory response may cause physiological disorder or even mortality for humans. Currently there is ongoing debate that whether nicotine functions as an inducer or inhibitor of endothelial inflammation under static condition. Although most studies point towards a more pro-inflammatory response, some efforts presented an opposite result (i.e., anti-inflammatory effect).15,33,41 This controversy may be explained by different modals or analytical methods used for the investigations. In terms of the correlation between LSS and endothelial inflammation, on the other hand, it has been widely demonstrated that LSS enables to reduce inflammatory response of endothelium through promoting the anti-inflammatory molecules such as shear stress responsive promoter elements (SSREs)25 and peroxisome proliferator-activated receptor gamma (PPARγ)-response elements,21 or reducing the cytokine stimulation.27 Recent studies have further showed that the LSS-mediated anti-inflammation may be contributed by regulation of MicroRNAs based on their effectiveness on the vascular homeostasis.3 Nonetheless, to the best of our knowledge, very little information is referred to the scenario of endothelial inflammation where the cells were treated with nicotine and LSS simultaneously.
Previously we have demonstrated that combined treatments of nicotine and LSS enabled to induce cytoskeleton collapse and exacerbate apoptosis in endothelial cells while those abnormal responses were not appeared in the group with either nicotine or LSS alone.18 In this study, we aimed to study how synergistic impact of nicotine and LSS leads to endothelial dysfunction, from views of inflammation and hemostasis, to further understand the influence of nicotine in vivo. Since the oxidative excess is often linked to a pro-inflammatory state of the endothelium,12 we first measured the degrees of oxidative stress suffered by the HUVECs with nicotine and/or LSS treatment through measurements of ROS expression, followed by detecting the levels of inflammatory and hemostatic responses in each group to evaluate the synergistic effect of nicotine and LSS on the endothelium as occurred in the vessels. Furthermore, the mechanism of abnormal cellular responses obtained and the efficacy of recombinant tissue plasminogen activator (rtPA), a protein responsible for clot breakdown, on alleviating the endothelial impairment were comprehensively investigated.
Materials and Methods
Cell Culture and Reagents
Human umbilical vein endothelial cells (HUVECs; Lonza, Walkersville, MD) were cultured by using Medium 199 supplemented with 20% fetal bovine serum (FBS; Biological Industries, Kibbutz Beit Haemek, Israel), endothelial cell growth supplement (ECGS; Millipore, Billerica, MA), 0.1% heparin sodium, 1% l-glutamine, and 1% penicillin/streptomycin (Biological Industries) and commonly maintained in tissue culture flasks at 37 °C with 5% CO2. HUVECs were transferred onto sterilized gelatin-coated glass slides 24 h prior to the experiment. THP-1 cells (human acute monocytic leukemia cell line) were cultivated using RPMI-1640 Medium supplemented with 0.05 mM of 2-mercaptoethanol, 1% penicillin/streptomycin, and 10% FBS (Biological Industries) in tissue culture flasks at 37 °C with 5% CO2. In this study, nicotine ((-)-nicotine; Sigma, St. Louis, MO) was used at concentration of 10−4 M to imitate physiologically relevant nicotine concentration in heavy/habitual smokers.39 rtPA (Cell Sciences, Canton, MA) was used at concentration of 1 μg mL−1. All reagents were used as received.
LSS Setup
To provide LSS on the cell surface, a laminar shearing system was established by which HUVECs were seeded on the glass slides and sheared in the parallel plate flow chamber as illustrated in Fig. 1. In this study, intensity of 12 dynes cm−2 of LSS (τ) was employed in all the shearing experiments to simulate the level of LSS in the proximal arteries,40 which can be obtained by setting appropriate flow rate of the medium (Q) according to Navier–Stokes equation:
where µ is the viscosity of the flowing solution (i.e., cell culture medium) with and without nicotine and/or rtPA (~0.01 dynes-s cm−2); b and h represent the width and height, respectively, of the space above the cell monolayer. Cells in the shearing experiments were stimulated by LSS, nicotine, and rtPA (if it was used) simultaneously.

Schematic diagram of the LSS shearing system and photographs of components of the parallel plate flow chamber. This LSS shearing system was partly consisted of a co-cylindrical medium tank, peristaltic pump, pulse damper, and a parallel plate flow chamber in a closed container where the environmental temperature was maintained at 37 °C by using a temperature sensor, a temperature controller, and a heater. The flushing medium in the co-cylindrical tank was injected with 5% CO2 mixture gas and maintained at 37 °C by water perfusion method as indicated by green and blue lines, respectively. The parallel plate flow chamber was composed of a plastic flow-leading block (B), rubber frame (C), glass slide (D), and rubber slice (E). All parts were encapsulated in a stainless steel cast (A and F) after sterilization. The HUVECs were seeded on the gelatin-coated glass slide and sheared with 12 dynes cm−2 for defined times. Red arrows indicate the flow direction.
Dichlorofluorescin Oxidation Assay
Twenty four hour prior to the experiment, HUVECs at passage 3–5 were seeded onto 16 gelatin-coated glass slides with 5 × 105 cells per each. For the shearing groups (six of 16 slides), cells were treated with 12 dynes cm−2 of LSS by using culture medium with and without 10−4 M nicotine for 6, 12, and 24 h. Cells in four of 16 slides were first sheared with 12 dynes cm−2 of LSS by using normal (two of four slides) and 10−4 M nicotine (two of four slides) medium for 24 h, followed by statically maintained in their shearing medium for additional 12 and 24 h. After washed twice with PBS, cells were recovered in 5 mL of culture medium containing 200 μM of 2′,7′ dichlorofluorescin diacetate (DCFH-DA; Sigma) for 40 min, followed by washing with PBS for three times. For the static groups (six of ten slides), cells were treated with 10−4 M nicotine for 0 (without nicotine), 6, 12, 24, 36, and 48 h, followed by addition of DCFH-DA as described above. The fluorescent images of DCFH-DA-treated HUVECs were first photographed by fluorescent microscopy. Afterward all of the cells on each slide were collected by trypsinization and the level of fluorescence expression of each group was detected using a spectrofluorometer with excitation wavelength of 485 nm and emission wavelength of 525 nm. In this study, the intensity of fluorescence was quantitatively represented by relative fluorescence units (RFUs) and the cell numbers of all groups were equalized before conduction of spectrofluorometry.
Monocyte Adhesion Assay
Twenty four hour prior to the experiment, HUVECs at passage 3–5 were inoculated onto 16 gelatin-coated glass slides with 5 × 105 cells per each. For the shearing groups (six of 16 slides), cells were treated with 12 dynes cm−2 of LSS by using culture medium with and without 10−4 M nicotine for 6, 12, and 24 h. Cells in four of 16 slides were first sheared with 12 dynes cm−2 of LSS by using normal (two of four slides) and 10−4 M nicotine (two of four slides) medium for 24 h, followed by statically maintained in their shearing medium for additional 12 and 24 h. After washed twice with PBS, 1 × 106 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein, acetoxymethyl ester (BCECF-AM; Sigma)-stained THP-1 cells were added to each group and co-cultured with the HUVECs at 37 °C for 40 min, followed by washing with PBS for three times. For the static groups (six of ten slides), cells were treated with 10−4 M nicotine for 0 (without nicotine), 6, 12, 24, 36, and 48 h, followed by the procedures of BCECF-AM-stained THP-1 adhesion as described above. After additional 1-h incubation at 37 °C post PBS wash, image of each group containing HUVECs and attached THP-1 cells were photographed by both phase-contrast and fluorescent microscopies. The fluorescence intensity of each group expressed from the adhered BCECF-AM-stained THP-1 cells was measured using a spectrofluorometer with excitation wavelength of 485 nm and emission wavelength of 525 nm, and quantified by RFU subsequently. The level of monocyte adhesion for each group was quantitatively represented by an R value which is the ratio of the detected fluorescence intensity to total HUVEC number counted by hemocytometry.
Immunocytochemistry and Western Blot
HUVECs post nicotine and/or LSS treatment were fixed and immunostained with monoclonal antibody (mAb) against intercellular adhesion molecule 1 (ICAM-1; 84H10, 10 μg mL−1; Cell signaling, Danvers, MA) followed by staining with fluorescently labeled secondary antibodies (10 μg mL−1) as described previously.35 Immunoblotting and preparation of sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE) were performed as previously described.38 Briefly, 30-μg whole protein extract of each sample was resolved by SDS-PAGE and transferred to 0.45-μm polyvinylidene fluoride membranes afterward. The membrane was then treated by blocking buffer (5% w/v skim dry milk in PBS-Tween-20 (0.05% v/v; PBS-T)) for 90 min at room temperature. The primary antibodies used in this study including ICAM-1, vascular cell adhesion molecule 1 (VCAM-1; Cell Signaling), plasminogen activator inhibitor1 (PAI-1; Cell Signaling), endothelial nitric oxide synthase (eNOS; Cell Signaling), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cell Signaling) were first diluted 1:1000 in blocking buffer, followed by incubation with membranes at 4 °C for 12 h. Membranes were then washed three times with PBS-T and incubated with HRP-conjugated goat anti-mouse/rabbit IgG secondary antibodies for 90 min at room temperature. After washed with PBS-T, the membranes were treated with chemiluminescent substrates and the level of each molecular band was analyzed by using the ChemiDoc-It 810 imaging system (UVP, Upland, CA). GAPDH was employed as the reference gene in all western blot analyzes.
Statistical Analysis
All data were obtained from triplicate experiments and presented as mean ± standard error (SE). Statistical analyzes were conducted by using MedCalc software in which comparisons for one condition between two groups were performed by Student’s t test followed by Dunnett’s post hoc test at a significance level of p < 0.05 throughout the study.
Results and Discussion
Synergistic Effect of Nicotine and LSS Enhanced ROS Production of HUVECs
Figure 2I exhibits photomicrographic images of DCFH-DA-stained HUVECs under various treatments in which the levels of green fluorescence represent the magnitudes of ROS produced from the cells. As compared to the group with neither nicotine nor LSS treatment (Fig. 2I(A); blank control), the ROS level in the 10−4 M nicotine-treated cells without LSS increased with time (Fig. 2I(B–F)) and was significantly enhanced 2.6 folds (p < 0.05) after 48 h (Fig. 2II). In terms of the shearing groups, the cells without nicotine exhibited mild ROS expression throughout the time course (Fig. 2I(G–I, X1, X2)), while that in the nicotine-treated group remarkably enhanced during the first 24-h shearing (Fig. 2I(J–L)) and reduced after turned to static incubation at 37 °C for 24 h (Fig. 2I(X4)). Based on the RFU analyzes (Fig. 2II), the ROS level in the cells with and without nicotine increased 4 folds (p < 0.05) and decreased 1.2 folds, respectively, after sheared with 12 dynes cm−2 LSS for 24 h, then reduced 28% and increase 15%, respectively, after incubated at 37 °C for additional 24 h in static.

Analyzes of ROS productions in HUVECs after different treatments. (I) The upper panel of cell images (A–F) represent photomicrographic images of DCFH-DA-stained HUVECs after stimulated with 10−4 M nicotine for 0 (A; without nicotine), 6 (B), 12 (C), 24 (D), 36 (E) and 48 (F) h in static. The bottom panel of cell images G–L represent photomicrographic images of DCFH-DA-stained HUVECs sheared with 12 dynes cm−2 LSS for 6 (G and J), 12 (H and K), and 24 (I and L) h using normal (G–I) or 10−4 M nicotine (J–L) medium. The bottom panel of cell images X1–X4 represent photomicrographic images of DCFH-DA-stained HUVECs first sheared with 12 dynes cm−2 LSS for 24 h using normal (X1 and X2) or 10−4 M nicotine (X3 and X4) medium following incubated in the normal (X1 and X2) or 10−4 M nicotine (X3 and X4) medium in static for additional 12 (X1 and X3) and 24 (X2 and X4) h. Arrows denote the flow direction. All images were photographed by fluorescent microscopy at 200× magnification. Scale bar 100 μm. (II) Quantitative analyzes of ROS levels of the HUVECs under different treatments within 48 h by using spectrofluorometry with excitation wavelength of 485 nm and emission wavelength of 525 nm. The RFU values were presented after normalized to the background signal. Values are mean ± SE (n = 3). *p < 0.05. †p < 0.05 as compared to the blank control.
Synergistic Impact of Nicotine and LSS Enhanced Efficacy of Monocyte Adhesion to HUVECS
Figure 3 exhibits the results of monocyte adhesion for the statically cultured (Fig. 3I) and 12 dynes cm−2 LSS-treated HUVECs using normal (Fig. 3II) or 10−4 M nicotine (Fig. 3III) medium for 48 h. For the static settings, our data showed that the number of adherent THP-1 cells increased with nicotine exposure time (Fig. 3I(a–f)) that the R value (normalized fluorescence intensity) significantly enhanced 2.8 folds (p < 0.05) after 48 h (Fig. 3IV). In terms of the shearing groups, it can be observed that the number of THP-1 cells on the HUVECs without nicotine slightly decreased in the first 24-h shearing (Fig. 3II(a–c)) and increased after maintained at 37 °C in static (Fig. 3II(d, e)), showing a 25% decrease and a 32% increase of R value in the first and second 24-h period, respectively (Fig. 3IV). Conversely, the amount of adherent THP-1 cells on the HUVECs with both nicotine and LSS treatments dramatically increased with shearing time (Fig. 3III(a–c)) and decreased after the LSS was terminated (Fig. 3III(d, e)), showing a 3.5-fold enhancement (p < 0.05) and a 20% decrease of R value in the first and second 24-h period, respectively (Fig. 3IV). Moreover, the efficacy of THP-1 adhesion on the cells was positively correlated with the expression level of surface ICAM-1 molecules of the HUVECs as illustrated in Fig. 3 (I–III; inset images in A–E).

Analyzes of monocyte adhesion efficacies of HUVECs after different treatments. (I) Micrographic images of HUVECs adhered with BCECF-AM-stained THP-1 cells after treated with 10−4 M nicotine for 0 (without nicotine stimulation; A/a), 6 (B/b), 12 (C/c), 24 (D/d), 36 (E/e), and 48 (F/f) h in static using phase-contrast (A–F) and fluorescent (a–f) microscopies. All images were photographed at 200× magnification. Scale bar 100 μm. (II and III) Micrographic images of HUVECs adhered with BCECF-AM-stained THP-1 cells after sheared with 12 dynes cm−2 LSS by normal (II) or 10−4 M nicotine (III) medium for 6 (A/a), 12 (B/b), and 24 (C/c) h. (D/d and E/e) Micrographic images of HUVECs adhered with BCECF-AM-stained THP-1 cells that first sheared with 12 dynes cm−2 LSS for 24 h by normal (II) or 10−4 M nicotine (III) medium following statically maintained in the normal (II) or 10−4 M nicotine (III) medium for additional 12 (D/d) and 24 (E/e) h. Images were photographed by phase-contrast (A–E) and fluorescent (a–e) microscopies. All images were photographed at 200× magnification. Scale bar 100 μm. Arrows denote the flow direction. The inset images in (I–III) represent the expression levels of ICAM-1 on the cell surface of each sample which were photographed by fluorescent microscopy at 400× magnification. (IV) Quantitative analyzes of the monocyte adhesion levels of HUVECs under different treatments within 48 h by using spectrofluorometry with excitation wavelength of 485 nm and emission wavelength of 525 nm. R the fluorescence intensity detected/total HUVEC number. Values are mean ± SE (n = 3). *p < 0.05. †p < 0.05 as compared to the blank control.
Through the DCFH-DA-staining (Fig. 2) and THP-1 adhesion (Fig. 3) analyzes, we found that the level of endothelial inflammation can be reduced by shearing with 12 dynes cm−2 LSS or enhanced by 10−4 M nicotine exposure for 24 h that represented an anti- and pro-inflammatory effect for each stimulus as reported previously.2,5 However, what is of interest is that the inflammatory response of the sheared HUVECs can be reversely enhanced while they were concurrently treated with nicotine, and the magnitude of induced inflammation was even higher than that obtained from the cells with nicotine alone. Moreover, such overexpressed inflammatory responses can be reduced after statically cultured in 37 °C for an additional 24 h (p = NS, Figs. 2II and 3IV), demonstrating that the irregular inflammation were mainly due to chronic shearing exposure. Overall these findings clearly showed that LSS played a promotive role in nicotine-induced endothelial inflammation.
Synergistic Impact of Nicotine and LSS on Protein Expressions of VCAM-1, ICAM-1, and PAI-1
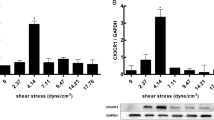
To fully address the expression levels of adhesion molecules in the HUVECs with different treatments, the total protein levels of VCAM-1 and ICAM-1 genes in all samples were further examined in addition to the surface ICAM-1 test as described above. Furthermore, in order to investigate how the excessive inflammatory response correlated with atherothrombosis in the vasculature, the levels of PAI-1, a protein which is encoded by serpin peptidase inhibitor clade E member 1 (SERPINE1) gene, in all settings were measured in parallel. Since the influence of LSS on the endothelial inflammation has been identified in the previous tests, the groups with set of turn-off-LSS were excluded in the following experiments. Figure 4a exhibits the western blot results of VCAM-1, ICAM-1, and PAI-1 molecules in the HUVECs under various conditions. After normalizing the intensity of each blot to GAPDH, we found that the expression levels of all three genes were up- and down-regulated in the groups with nicotine alone and LSS alone, respectively, within 24 h (Figs. 4b–4d), while those in the nicotine-treated cells can be further enhanced by 12 dynes cm−2 LSS that the levels of VCAM-1, ICAM-1, and PAI-1 significantly enhanced 1.3, 2, and 2 folds, respectively (Figs. 4b–4d; p < 0.05 for each), after 24 h.

Analyzes of inflammation- and hemostasis-related protein expressions in HUVECs under various treatments. (a) Western blots of VCAM-1, ICAM-1, PAI-1 and GAPDH proteins in (I) statically cultured and (II) 12 dynes cm−2 LSS-sheared HUVECs with and without 10−4 M nicotine (N) and/or 1 μg mL−1 rtPA for 0, 6, 12, and 24 h. The condition of each lane is indicated on the top of the blotting picture. The setting in static culture with neither nicotine nor rtPA was employed as the control. (b–d) Quantitative analyzes of the protein expressions of VCAM-1 (b), ICAM-1 (c), and PAI-1 (d) for the groups shown at left side of the dotted lines in the blot image. Each bar represents the level of relative optical intensity of the protein after normalized to GAPDH. Solid black: blank control group with neither nicotine nor LSS treatment; solid red: static group with nicotine; black stripe: shearing group without nicotine; red stripe: shearing group with nicotine. Values are mean ± SE (n = 3). *p < 0.05. †p < 0.05 as compared to the blank control. The quantitative analyzes of the protein expressions obtained from the groups with nicotine, LSS, and rtPA (subjects at right side of dotted line in blot image II) were presented in Fig. 7, whereas the blots images of the static groups with nicotine and rtPA (subjects at right side of dotted line in blot image I) were merely provided as references in this study.
Based on the western blot analysis, the outcomes of total protein expressions of adhesion molecules (VCAM-1 and ICAM-1) were consistent with the results of monocyte adhesion assay and expressions of surface ICAM-1 as shown in Fig. 3. Similarly, the expression level of PAI-1 in the HUVECs can be enhanced by nicotine treatment as reported previously,29,43 and such increased level can be further promoted by LSS as shown in Figs. 4a–4d. Since elevation of PAI-1 expression has been recognized as a risk factor for atherothrombosis36 and tumor invasion (i.e., cancer metastasis)8 due to its inhibitory effects on tissue/urokinase-type plasminogen activators and matrix metalloproteinases, our data implicated that the LSS may play an adverse role in nicotine-mediated vascular dysfunction and such impairment was likely induced through PAI-1 activation. Taken all together, these findings may potentially interpret the clinical consequence that nicotine (or cigarette smoking) is highly associated with proximal vessel disease since where the intensity of LSS is relatively high (≥12 dynes cm−2)28,40 and that may play a detrimental role in the lesion development.
Synergistic Impact of Nicotine and LSS Reduced eNOS Expression in the HUVECs
To investigate how the excessive inflammatory response and elevated atherothrombotic risk occurred in the cells with both nicotine and LSS stimulations, the expression levels of eNOS in the cells with different treatments were examined since NO has long been known as one of the inflammatory and hemostatic modulators.9 As shown in Fig. 5, the protein levels of eNOS in the cells with 10−4 M nicotine alone (Fig. 5I) and 12 dynes cm−2 LSS alone (Fig. 5II) significantly decreased 1.7-fold (p < 0.05; Fig. 5III) and increased 1.6-fold (p < 0.05; Fig. 5III), respectively, after 24-h treatment. In terms of the group treated with both nicotine and LSS, the expression level of eNOS decreased with operation time (Fig. 5II) and exhibited a 2.7-fold reduction (p < 0.05; Fig. 5III) after 24 h.

Analyzes of eNOS expressions in HUVECs treated by various conditions. Expressions of eNOS and GAPDH proteins in statically cultured (I) and 12 dynes cm−2 LSS-sheared (II) HUVECs treated with and without 10−4 M nicotine (N) and/or 1 μg mL−1 rtPA for 0, 6, 12, and 24 h were examined by western blot. The condition of each lane is indicated on the top of the blot image. The setting in static culture with neither nicotine nor rtPA was employed as the control. (III) Quantitative analyzes of the eNOS protein expressions for the groups without rtPA (subjects at left side of the dotted line in the blot image I and II). Each bar represents the level of relative optical intensity of the protein after normalized to GAPDH. Values are mean ± SE (n = 3). †p < 0.05 as compared to the blank control. *p < 0.05 as compared to the shearing group without nicotine under the same operation time. ‡p < 0.05 as compared to the static group with nicotine under the same operation time. The quantitative analyzes of the protein expressions for the groups with nicotine, LSS, and rtPA (subjects at right side of dotted line in the blot image II) were presented in Fig. 6, whereas the blot images of the static groups with nicotine and rtPA (subjects at right side of dotted line in the blot image I) were merely provided as references in this study.
Based on the results of eNOS expression analyzes, we reasoned that the irregular inflammatory and hemostatic responses were likely resulted from decrease of NO production in the cells. According to the previous study, synergistic impact of nicotine and LSS enabled to induce severe cytoskeleton collapse (interruption of mechanostasis) and thus triggered ROS production.18 These ROS may consequently generate peroxynitrite to degrade eNOS co-factor tetrahydrobiopterin and lead to eNOS dysfunction and NO reduction accordingly.17,24 However, NO is one of the central mediators in vascular physiology and has been known to regulate VCAM-1 and ICAM-1 expressions through activation of nuclear factor kappa B (NF-κB)16 and cytosolic phospholipase A2α (cPLA2α),13 respectively. Therefore, attenuation of NO production may contribute to endothelial inflammation/dysfunction and hence induce cardiovascular lesions such as atherosclerotic plaque formation.22 Similar with the adhesion molecules, the expression levels of PAI-1 in the cells with dual stimulations (Fig. 4d) were inversely correlated with eNOS (Fig. 5III), implicating that PAI-1 level may be regulated by eNOS activity/NO productivity. In fact, it has been reported that eNOS enables to protect the ischemic stroke in the clinic4 and that is explainable by this outcome. Overall, these efforts provided a rational interpretation for the correlation between smoking and thrombosis where the LSS was taken into the consideration.
rtPA Retrieved eNOS Expression in the HUVECs Treated with Nicotine and LSS Simultaneously
To circumvent the irregular inflammatory and hemostatic responses due to synergistic impact of nicotine and LSS, the efficacy of using rtPA, the most commonly used thrombolytic agent in the clinic, to retrieve the eNOS reduction was first explored. In this study, the rtPA was selected because it enables to alleviate the promoted fibrosis due to PAI-1 upregulation11 and which was occurred in the cells with both nicotine and LSS treatments. Through the analyzes of western blot results as shown in Fig. 5II (subjects at right side of the dotted line), our data showed that 1 μg mL−1 rtPA enabled to retrieve the expression level of eNOS in the cells with both nicotine and LSS treatments within 24 h and a level which was similar with that in the blank control (p = NS) can be obtained after ≥12 h rtPA administration (Fig. 6).

Efficacy of using rtPA to recover the impaired eNOS expressions of HUVECs caused by synergistic impact of nicotine and LSS. Protein expressions of eNOS in the samples sheared by 12 dynes cm−2 of LSS for 6, 12, and 24 h using normal or 10−4 M nicotine medium in the presence and absence of 1 μg mL−1 of rtPA were examined. The group with neither nicotine nor LSS treatment was employed as the blank control. The blot images of eNOS and GAPDH proteins in each group were presented in Fig. 5II (subjects at right side of the dotted line). The eNOS expression level of each setting was presented after normalized to GAPDH. Values are mean ± SE (n = 3). *p < 0.05. †p < 0.05 as compared to the blank control.
rtPA Reversed Irregular Expressions of Adhesion and Hemostasis Molecules in the HUVECs Treated by Nicotine and LSS
We subsequently examined how rtPA affected the protein expressions of VCAM-1, ICAM-1, and PAI-1 molecules while the cells were treated with both 10−4 M nicotine and 12 dynes cm−2 LSS simultaneously. Through the analyzes of western blot results as shown in Fig. 4a(II) (subjects at right side of the dotted line), our data showed that the overexpressions of all three genes in the cells with dual stimulations can be mitigated in the presence of 1 μg mL−1 rtPA, in which the levels of VCAM-1, ICAM-1 and PAI-1 after 24-h rtPA treatment significantly reduced 1.2 (Fig. 7a; p < 0.05), 2.2 (Fig. 7b; p < 0.05), and 2 (Fig. 7c; p < 0.05) folds, respectively, as compared to the values gained from the cells with identical treatments except no rtPA. Considering the correlations between eNOS and each of VCAM-1, ICAM-1, and PAI-1, as well as the effects of rtPA on recovering the expression levels of above four genes (Figs. 6 and 7), we reasoned that rtPA alleviated endothelial inflammation and atherothrombotic risk through upregulating eNOS level, and the excessive inflammatory response was likely resulted from the induced hemostasis. However, more experiments are definitely needed to ascertain these hypotheses. Nonetheless, these results have prompted a clinical interest since the rtPA administration may be helpful to mitigate cigarette smoking-induced vascular lesion.

Efficacy of using rtPA to reduce the enhanced expressions of adhesion and hemostatic molecules in HUVECs caused by synergistic impact of nicotine and LSS. Protein expressions of VCAM-1 (a), ICAM-1 (b), and PAI-1 (c) in samples sheared by 12 dynes cm−2 LSS for 6, 12, and 24 h using normal or 10−4 M nicotine medium in the presence and absence of rtPA (1 μg mL−1) were examined. The group with neither nicotine nor LSS treatment was employed as the blank control. The western blot images of VACM-1, ICAM-1, PAI-1, and GAPDH proteins in each setting were exhibited in Fig. 4a(I) (subjects at right side of the dotted line), and the expression level of each protein was presented after normalized to GAPDH. Values are mean ± SE (n = 3). *p < 0.05. †p < 0.05 as compared to the blank control.
In this study, we have demonstrated that, for the first time, the synergistic impact of nicotine and LSS may further enhance the nicotine-induced inflammatory and hemostatic responses in the endothelial cells. All the experiments were carried out in the parallel plate flow system that brought the study closer to an in vivo condition (i.e., hemodynamic environment). Of particular interesting is that we surprisingly found the LSS, which is usually thought to play a protective role in endothelial physiology, may conversely play a detrimental role in vasculature to exacerbate the endothelial injury while the cells are concurrently exposed to nicotine. Since the excessive inflammation and hemostasis are contributing factors for development of many cardiovascular diseases,6,37 the efforts presented in this study may provide an explanation for strong correlation between smoking and proximal vessel diseases in the clinic. Furthermore, the efficacies of rtPA on alleviating the impaired inflammatory and hemostatic responses were demonstrated in this study. To fully address the effectiveness of rtPA identified, further investigation is certainly required and efforts are currently in progress.
References
Aboyans, V., P. Lacroix, and M. H. Criqui. Large and small vessels atherosclerosis: similarities and differences. Prog. Cardiovasc. Dis. 50:112–125, 2007.
Albaugh, G., E. Bellavance, L. Strande, S. Heinburger, C. W. Hewitt, and J. B. Alexander. Nicotine induces mononuclear leukocyte adhesion and expression of adhesion molecules, VCAM and ICAM, in endothelial cells in vitro. Ann. Vasc. Surg. 18:302–307, 2004.
Boon, R. A., E. Hergenreider, and S. Dimmeler. Atheroprotective mechanisms of shear stress-regulated microRNAs. Thromb. Haemost. 108:616–620, 2012.
Casas, J. P., L. E. Bautista, S. E. Humphries, and A. D. Hingorani. Endothelial nitric oxide synthase genotype and ischemic heart disease: meta-analysis of 26 studies involving 23028 subjects. Circulation 109:1359–1365, 2004.
Cunningham, K. S., and A. I. Gotlieb. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Invest. 85:9–23, 2005.
Devaraj, S., D. Y. Xu, and I. Jialal. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation 107:398–404, 2003.
Diehm, N., A. Shang, A. Silvestro, D. D. Do, F. Dick, J. Schmidli, F. Mahler, and I. Baumgartner. Association of cardiovascular risk factors with pattern of lower limb atherosclerosis in 2659 patients undergoing angioplasty. Eur. J. Vasc. Endovasc. Surg. 31:59–63, 2006.
Duffy, M. J., P. M. McGowan, and W. M. Gallagher. Cancer invasion and metastasis: changing views. J. Pathol. 214:283–293, 2008.
Endemann, D. H., and E. L. Schiffrin. Endothelial dysfunction. J. Am. Soc. Nephrol. 15:1983–1992, 2004.
Fahim, M. A., A. Nemmar, S. Al-Salam, S. Dhanasekaran, M. Shafiullah, J. Yasin, and M. Y. Hassan. Thromboembolic injury and systemic toxicity induced by nicotine in mice. Gen. Physiol. Biophys. 33:345–355, 2014.
Ghosh, A. K., and D. E. Vaughan. PAI-1 in tissue fibrosis. J. Cell Physiol. 227:493–507, 2012.
Griendling, K. K., and G. A. FitzGerald. Oxidative stress and cardiovascular injury: part I: basic mechanisms and in vivo monitoring of ROS. Circulation 108:1912–1916, 2003.
Hadad, N., L. Tuval, V. Elgazar-Carmom, R. Levy, and R. Levy. Endothelial ICAM-1 protein induction is regulated by cytosolic phospholipase A2α via both NF-κB and CREB transcription factors. J. Immunol. 186:1816–1827, 2011.
Haltmayer, M., T. Mueller, W. Horvath, C. Luft, W. Poelz, and D. Haidinger. Impact of atherosclerotic risk factors on the anatomical distribution of peripheral arterial disease. Int. Angiol. 20:200–207, 2001.
Heeschen, C., J. J. Jang, M. Weis, A. Pathak, S. Kaji, R. S. Hu, P. S. Tsao, F. L. Johnson, and J. P. Cooke. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat. Med. 7:833–839, 2001.
Khan, B. V., D. G. Harrison, M. T. Olbrych, R. W. Alexander, and R. M. Medford. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc. Natl. Acad. Sci. USA 93:9114–9119, 1996.
Koppenol, W. H., J. J. Moreno, W. A. Pryor, H. Ischiropoulos, and J. S. Beckman. Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem. Res. Toxicol. 5:834–842, 1992.
Lee, Y. H., R. S. Chen, N. C. Chang, K. R. Lee, C. T. Huang, Y. C. Huang, and F. M. Ho. Synergistic impact of nicotine and shear stress induces cytoskeleton collapse and apoptosis in endothelial cells. Ann. Biomed. Eng. 43:2220–2230, 2015.
Lee, J., and J. P. Cooke. The role of nicotine in the pathogenesis of atherosclerosis. Atherosclerosis 215:281–283, 2011.
Lindenblatt, N., U. Platz, J. Hameister, E. Klar, M. D. Menger, and B. Vollmar. Distinct effects of acute and chronic nicotine application on microvascular thrombus formation and endothelial function in male and female mice. Langenbecks. Arch. Surg. 392:285–295, 2007.
Liu, Y., Y. Zhu, F. Rannou, T. S. Lee, K. Formentin, L. Zeng, X. Yuan, N. Wang, S. Chien, B. N. Forman, and J. Y. Shyy. Laminar flow activates peroxisome proliferator-activated receptor-gamma in vascular endothelial cells. Circulation 110:1128–1133, 2004.
Lum, H., and K. A. Roebuck. Oxidant stress and endothelial cell dysfunction. Am. J. Physiol. Cell. Physiol. 280:C719–741, 2001.
Malek, A. M., S. L. Alper, and S. Izumo. Hemodynamic shear stress and its role in atherosclerosis. J. Am. Med. Assoc. 282:2035–2042, 1999.
Milstien, S., and Z. Katusic. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 263:681–684, 1999.
Nagel, T., N. Resnick, C. F. Dewey, Jr, and M. A. Gimbrone, Jr. Vascular endothelial cells respond to spatial gradients in fluid shear stress by enhanced activation of transcription factors. Arterioscler. Thromb. Vasc. Biol. 19:1825–1834, 1999.
Narayanaswami, V., S. S. Somkuwar, D. B. Horton, L. A. Cassis, and L. P. Dwoskin. Angiotensin AT1 and AT2 receptor antagonists modulate nicotine-evoked [3H] dopamine and [3H] norepinephrine release. Biochem. Pharmacol. 86:656–665, 2013.
Ni, C. W., H. J. Hsieh, Y. J. Chao, and D. L. Wang. Shear flow attenuates serum-induced STAT3 activation in endothelial cells. J. Biol. Chem. 278:19702–19708, 2003.
Papaioannou, T. G., and C. Stefanadis. Vascular wall shear stress: basic principles and methods. Hellenic. J. Cardiol. 46:9–15, 2005.
Rodella, L. F., C. Rossini, G. Favero, E. Foglio, C. Loreto, and R. Rezzani. Nicotine-induced morphological changes in rat aorta: the protective role of melatonin. Cells Tissues Organs. 195:252–259, 2012.
Sackett, D. L., R. W. Gibson, I. D. Bross, and J. W. Pickren. Relation between aortic atherosclerosis and the use of cigarettes and alcohol. An autopsy study. N. Engl. J. Med. 279:1413–1420, 1968.
Smith, F. B., A. J. Lee, F. G. Fowkes, A. Rumley, and G. D. Lowe. Smoking, haemostatic factors and the severity of aorto-iliac and femoro-popliteal disease. Thromb. Haemost. 75:19–24, 1996.
Takabe, W., E. Warabi, and N. Noguchi. Anti-atherogenic effect of laminar shear stress via Nrf2 activation. Antioxid. Redox. Signal. 15:1415–1426, 2011.
Tonnessen, B. H., S. R. Severson, R. D. Hurt, and V. M. Miller. Modulation of nitric-oxide synthase by nicotine. J. Pharmacol. Exp. Ther. 295:601–606, 2000.
Traub, O., and B. Berk. Laminar shear stress: mechanisms by which endothelial cells transduce an atheroprotective force. Arterioscler. Thromb. 18:677–685, 1998.
van Buul, J. D., C. Voermans, V. van den Berg, E. C. Anthony, F. P. Mul, S. van Wetering, C. E. van der Schoot, and P. L. Hordijk. Migration of human hematopoietic progenitor cells across bone marrow endothelium is regulated by vascular endothelial cadherin. J. Immunol. 168:588–596, 2002.
Vaughan, D. E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 3:1879–1883, 2005.
Virdis, A., U. Dell’Agnello, and S. Taddei. Impact of inflammation on vascular disease in hypertension. Maturitas. 78:179–183, 2014.
Wang, W., C. H. Ha, B. S. Jhun, C. Wong, M. K. Jain, and Z. G. Jin. Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood. 115:2971–2979, 2010.
Xu, M., J. E. Scott, K. Z. Liu, H. R. Bishop, D. E. Renaud, R. M. Palmer, A. Soussi-Gounni, and D. A. Scott. The influence of nicotine on granulocytic differentiation—inhibition of the oxidative burst and bacterial killing and increased matrix metalloproteinase-9 release. BMC Cell Biol. 9:19, 2008.
Zarins, C. K., M. A. Zatina, D. P. Giddens, D. N. Ku, and S. Glagov. Shear stress regulation of artery lumen diameter in experimental atherogenesis. J. Vasc. Surg. 5:413–420, 1987.
Zhang, S., I. Day, and S. Ye. Nicotine induced changes in gene expression by human coronary artery endothelial cells. Atherosclerosis. 154:277–283, 2001.
Zhou, X., Y. Sheng, R. Yang, and X. Kong. Nicotine promotes cardiomyocyte apoptosis via oxidative stress and altered apoptosis-related gene expression. Cardiology. 115:243–250, 2010.
Zidovetzki, R., P. Chen, M. Fisher, F. M. Hofman, and F. M. Faraci. Nicotine increases plasminogen activator inhibitor-1 production by human brain endothelial cells via protein kinase C-associated pathway. Stroke 30:651–655, 1999.
Acknowledgements
This work was financially supported by Ministry of Science and Technology, R.O.C. (MOST 104-2221-E-008-095; Y.-H. Lee).
Conflict of interest
Yu-Hsiang Lee, Chi-Chung Lee, Chien-Hsun Huang, and Feng-Ming Ho declare no conflict of interest.
Ethical Standards
The authors for this article carried out no human studies. The authors for this article carried out no animal studies.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Associate Editor Joyce Wong oversaw the review of this article.
Rights and permissions
About this article

Cite this article
Lee, YH., Lee, CC., Huang, CH. et al. Laminar Shear Stress Promotes Nicotine-Induced Inflammation and Hemostatic Expression in Human Endothelial Cells. Cel. Mol. Bioeng. 9, 466–477 (2016). https://doi.org/10.1007/s12195-016-0434-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12195-016-0434-y