
Abstract
Osteosarcoma is the most common type of bone cancer in dogs and humans, with significant numbers of patients experiencing treatment failure and disease progression. In our search for new approaches to treat osteosarcoma, we previously detected multiple chaperone proteins in the surface-exposed proteome of canine osteosarcoma cells. In the present study, we characterized expression of representative chaperones and find evidence for stress adaptation in canine osteosarcoma cells relative to osteogenic progenitors from normal bone. We compared the cytotoxic potential of direct (HA15) and putative (OSU-03012) inhibitors of Grp78 function and found canine POS and HMPOS osteosarcoma cells to be more sensitive to both compounds than normal cells. HA15 and OSU-03012 increased the thermal stability of Grp78 in intact POS cells at low micromolar concentrations, but each induced distinct patterns in Grp78 expression without significant change in Grp94. Both inhibitors were as effective alone as carboplatin and showed little evidence of synergy in combination treatment. However, HMPOS cells with acquired resistance to carboplatin were sensitive to inhibition of Grp78 (by HA15; OSU-03012), Hsp70 (by VER-155008), and Hsp90 (by 17-AAG) function. These results suggest that multiple nodes within the osteosarcoma chaperome may be relevant chemotherapeutic targets against platinum resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The molecular chaperones are a large group of proteins that are critical for maintaining cellular proteostasis under normal conditions. They include multiple family members that can be upregulated in rapid response to internal or external stress signals to preserve normal cell function (Balch et al. 2008; Saibil 2013; Shemesh et al. 2021; Walter and Ron 2011). These proteins are located in all the major compartments of the cell where they act to fold, unfold, and stabilize client proteins, and thereby protect against protein misfolding and accumulation of cytotoxic aggregations (Hartl et al. 2011; Saibil 2013; Walter and Ron 2011). Molecular chaperones, together with co-chaperones and other accessory proteins, make up a functional signaling network known as the chaperome (Finka and Goloubinoff 2013; Wang et al. 2006). Studies of cultured cancer cells and patient samples indicate that the chaperome of some human tumors changes relative to normal cells and can be transformed into an epichaperome characterized by an enhanced signal transduction network with greater functional efficiency to match the increased physiological demands of the cancer cell (Finka and Goloubinoff 2013; Joshi et al. 2018; Rodina et al. 2016). The discovery that some cancers utilize the epichaperome for a survival advantage also raises the possibility that an understanding of crosstalk in the chaperome network, as well as the individual components, is a critical consideration for future design and development of effective therapeutics to target this kind of tumor dependence (Joshi et al. 2018; Kourtis et al. 2018; Rodina et al. 2016).
Chaperone proteins are organized into subfamilies based on their molecular mass and structural homology, but also show tissue-specific distribution patterns that can be further subdivided into the core chaperones and those that show variable expression (Shemesh et al. 2021). This level of normal physiological complexity presents an even greater challenge in that changes in chaperone proteins can underlie, or occur as a secondary consequence of, human disease (Macario and Conway de Macario 2002). With respect to cancer, molecular chaperones belonging to the stress-inducible heat shock protein (Hsp) family display increased expression in some tumors, along with changes in the function of specific proteins and in the context of the chaperome network (Calderwood 2018; Finka and Goloubinoff 2013; Hadizadeh Esfahani et al. 2018; Rodina et al. 2016). Additionally, Hsp family members also show aberrant expression patterns in cancer cells. For example, Hsp90 and Hsp70 typically function as intracellular cytosolic proteins, yet these proteins can also be (1) secreted outside of the cell via a nonconventional route, (2) localized at the plasma membrane without specific membrane localization domains, and (3) associated with the cell surface proteome (Calderwood 2018; Mambula and Calderwood 2006; Mambula et al. 2007; Nolan et al. 2015, 2017; Shin et al. 2003; Vega et al. 2008).
In our previous analysis of intact canine osteosarcoma cells, we identified Hsp and glucose-regulated protein (Grp) family members as components of the surface-exposed proteome (Milovancev et al. 2013). After sequential biotin-streptavidin labeling and purification of surface proteins from two different canine osteosarcoma cell types, multiple unique peptides corresponding to Hsp90 (alpha and beta isoforms), Grp78, Grp94 (with AMP-PNP bound), Hsp60, and a constitutively expressed core chaperone (Hsc70) were detected using liquid chromatography tandem mass spectrometry (LC–MS/MS). Peptides corresponding to these molecular chaperones were not, however, identified as components of the surface-associated proteome of normal canine osteogenic progenitor cells (Supplementary Material (Milovancev et al. 2013)). Grps are a subfamily of the variable, stress-inducible Hsp family discovered for their ability to respond to depletion in intracellular glucose (Lee 2014; Pouyssegur et al. 1977; Shiu et al. 1977). In normal cells, Grps are primarily located within the lumen of the endoplasmic reticulum (ER) and have specific functions within the ER microenvironment as protein-folding chaperones for new polypeptides entering the ER secretory pathway (Eletto et al. 2010; Marzec et al. 2012; Zimmermann et al. 2011). With the possibility that chaperone proteins could hold prognostic and diagnostic value in canine osteosarcoma (Milovancev et al. 2013), the present study sought to understand the relative expression of major molecular chaperones detected in canine osteosarcoma cells relative to nonmalignant osteogenic progenitor cells. We also explored the feasibility of inhibiting specific molecular chaperones in canine osteosarcoma cells with a focus on Grp78. We compared the mechanism of action and cytotoxic potential of a direct inhibitor of Grp78, termed HA15 (Cerezo et al. 2016), to the atypical Grp78 inhibitor OSU-03012 (also known as AR-12) (Booth et al. 2012; Park et al. 2008). Our studies of canine osteosarcoma cells with acquired resistance to the chemotherapy agent carboplatin revealed broad sensitivity of these cells to inhibitors of Grp78 and small molecule inhibitors of Hsp90 and Hsp70. These findings reveal cross signaling and connectivity within the canine osteosarcoma chaperome with implications for future drug development aiming to improve salvage options for patients failing established treatments.
Materials and methods
Chemicals, reagents, and antibodies
Thapsigargin (#T9033), HA15 (#SML2118), and 17-(allylamino)-17-demthoxygeldanamycin 17 (AAG; #100,068) were purchased from Millipore Sigma (Darmstadt, Germany). OSU-03012 (AR-12; #A2846-5), Z-VAD-FMK (#A1902), and VER-155008 (#A4387) were purchased from APExBIO Technology (Houston, TX, USA). Apratoxin A was isolated and purified from a laboratory culture of the marine cyanobacterium Moorea producens as described previously (Thornburg et al. 2013). Dry compounds were reconstituted in 100% cell culture grade dimethyl sulfoxide (DMSO), aliquoted, and stored at − 20 °C. Working stock solutions were prepared in DMSO on the day of the experiment; final concentrations of DMSO never exceeded 0.1%. Carboplatin (National Drug Code: 61,703–339-50) was purchased as an aqueous solution (10 mg/mL) from Hospira, Inc. (Lake Forest, IL, USA). Anti-HDAC2 (sc-9959) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA), anti-calreticulin (EPR3942; ab92516) from Abcam (USA), and anti-integrin β-1 (AF1778) from R&D Systems (Minneapolis, MN, USA). Cy3-conjugated anti-goat (111–165-003) and Cy2-conjugated anti-rabbit (611–225-215) secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PN, USA). All other primary and secondary antibodies were from Cell Signaling Technology, Inc. (Danvers, MA, USA), with specific codes for each product as follows: GRP78/BiP (#3177), phospho-Akt-Thr308 (#13,018), phospho-Akt-Ser473 (#4060), Akt (#4691), HSP90 (#4877), HSP70 (#4873), HSP40 (#4871), α-tubulin (#2125), CHOP (#5554), GAPDH (#5174), and EGFR (#4267). General laboratory reagents were from VWR International (Radnor, PA, USA).
Mammalian cell culture
All cells were maintained as adherent cultures under standard laboratory conditions at 37 °C in an atmosphere of 5% CO2. Human DU145 prostate, U251-MG and U87-MG glioblastoma, and MCF-7 breast and canine D17 osteosarcoma cells were cultured in Minimum Essential Media (Corning Cellgro, Corning, NY, USA) supplemented with l-glutamine (2 mM). Human MDA-MB-231 breast cancer cells were cultured in Dulbecco’s modified Eagle’s medium (Corning Cellgro). Human SAOS-2 osteosarcoma, SK-ES-1 Ewing’s sarcoma, HCT116 colon, and SKOV-3 ovarian cancer cells were cultured in McCoy’s 5A medium (Millipore Sigma). Human SF295 and canine osteosarcoma cell lines COS, POS, and highly metastatic POS (HMPOS) were cultured in RPMI 1640 (Corning Cellgro). All medium was supplemented with 10% fetal bovine serum (FBS; VWR International) and 1% penicillin and streptomycin (100 IU/mL penicillin/100 µg/mL streptomycin; Mediatech, Inc., Manassas, VA, USA). Canine osteogenic progenitor cells, isolated from normal canine bone (# Cn406K-05), were purchased from Cell Applications, Inc. (San Diego, CA, USA) and maintained in canine osteoblast growth medium (# Cn417K-500).
Generation of carboplatin-resistant cells
The generation and characterization of chemoresistant canine osteosarcoma HMPOS has been described previously (Weinman et al. 2021). Briefly, cells were grown in complete medium and treated sequentially, and continuously, with increasing concentrations of carboplatin starting with 0.5 µM for 72 h. With each concentration, the surviving cell population was allowed to grow, and if more than 30% of cell death was observed after 72 h of treatment, the same concentration of carboplatin was repeated for another 72 h. If less than 30% cell death was observed, then the cells were exposed to the next concentration of carboplatin. Subsequent to the starting concentration of 0.5 µM carboplatin, the progressive concentration steps were 1 µM, 2.5 µM, 4 µM, 6 µM, 8 µM, and 10 µM. This procedure was followed until 2.5 µM and 10 µM carboplatin-resistant cells were expanded and named HMPOS-2.5R and HMPOS-10R, respectively.
Immunocytochemistry
Cells were seeded and grown on glass coverslips in complete medium. Once attached, cells were washed twice in phosphate-buffered saline (PBS) and fixed with 4% formaldehyde for 20 min at room temperature. Cells were again washed twice in PBS, and the formaldehyde was quenched with complete medium for 10 min at room temperature. Following two additional washes in PBS, permeabilized cells were incubated with 10% Triton X-100. Cells were then washed twice with PBS and blocked with 1% bovine serum albumin (BSA) in PBS (BSA/PBS) for 15 min. Cells were incubated for 1 h with anti-GRP78 and anti-integrin β1 diluted in 1:500 in BSA/PBS. The coverslips were washed in BSA/PBS for three washes with 10 min each and then incubated with an Alexa Fluor–conjugated secondary antibody, diluted 1:500 BSA/PBS for 30 min at room temperature. Coverslips were washed again three times, for 10 min each, in BSA/PBS then mounted onto glass microscope slides with ProLong™ Gold Antifade with DAPI for permeabilized cells and without DAPI for cell surface expression analysis.
Images were taken using a Zeiss microscope using AxioVision software. In the survey of Grp78 on the cell surface and in permeabilized cells, the intensity was determined by auto-optimization through the software. When comparing the Grp78 and integrin beta 1 expression on the POS cell surface, the exposure was held to the integrin stain as a cell surface control. Cells were marked yes or no for determination of the percentage of integrin-positive cells that are also Grp78 positive. Individual cell fluorescence of integrin-positive cells for Grp78 and integrin beta 1 was quantified using ImageJ software. The raw integrated density was corrected by calculating the corrected total cell fluorescence (CTCF) using the following equation: integrated density − (area of selected cell × mean fluorescence of background readings).
Cell lysis, fractionation, and melting curve analysis
Whole cell lysates were prepared using fresh ice-cold lysis buffer containing Tris–HCl (50 mM; pH 7.5), EDTA (1 mM), EGTA (1 mM), 1% Triton X-100, sucrose (0.27 M), sodium fluoride (50 mM), sodium orthovanadate (1 mM), sodium pyrophosphate (5 mM), PMSF (1 mM), and benzamidine (1 mM). All cell lysates were cleared by centrifugation at 16,000 × g for 20 min at 4 °C. A subcellular protein fractionation kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to sequentially separate and enrich protein extracts to yield nuclear, cytoplasmic, and membrane fractions using the manufacturer’s protocol. The protein concentration in all extracts was determined by the bicinchoninic acid (BCA) method (Thermo Fisher Scientific, Inc.) and normalized to the same concentration for subsequent analysis. Melting curves were determined for Grp78 and α-tubulin according to the method detailed in Jafari et al. (2014). Briefly, 4 × 107 POS cells were seeded into T175 flasks and allowed to adhere for 18 h under standard growth conditions. The next day, cells were treated for 1 h with 0.1% DMSO (vehicle), HA15, or OSU-03012 (at concentrations indicated), dissociated from each flask with trypsin and transferred to 15-mL tubes for collection by centrifugation (300 × g for 3 min). After careful removal of the supernatant, cell pellets were washed and resuspended in PBS with protease inhibitors. The cell suspension was distributed into four tubes (per treatment) to a volume of 100 µL per tube and subjected to heat treatment (3 min) at the temperatures 45 °C, 48 °C, 51 °C, and 54 °C using a Veriti 96-well thermal cycler (Applied Biosystems # 4,375,786). Samples were cooled to room temperature and cells subsequently lysed through two successive rounds of freeze–thaw cycles. All samples were immediately processed for immunoblot analysis under standard denaturing conditions.
Western blot analysis
Samples were prepared in 4 × SDS sample loading buffer and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred to polyvinylidene fluoride (PVDF) membranes (Thermo Fisher Scientific), the membranes blocked in 5% (w/v) nonfat dry milk in Tris-buffered saline (TBS): Tris–HCl (50 mM; pH 7.4) and NaCl (150 mM) containing 0.05% Tween-20 (TBS-T). Membranes were then incubated for 16 h at 4 °C with gentle rotation in the appropriate primary antibody diluted in 5% (w/v) BSA in TBS-T. On the following day, membranes were washed with TBS-T 2 × for 5 min, then incubated in appropriate horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. Membranes were finally washed in TBS-T 3 × for 5 min, and target proteins detected by chemiluminescence using detection reagents (Amersham ECL Chemiluminescent Reagents, GE Healthcare Bio-Sciences, Pittsburg, PA, USA) and visualized using a myECL™ Imager system (Thermo Fisher Scientific).
Analysis of caspase activity and cell viability
For caspase activity assays, cells were seeded at a density of 3000 cells/well in 96-well white-walled, clear-bottom plates (Greiner Bio-One, Monroe, NC, USA) and treated with OSU-03012 (100 nM–3 µM) or vehicle (0.1% DMSO). At the end of treatment caspase-3,7 activity was measured using a luminescence-based caspase-Glo 3/7 assay (Promega, WI, USA). The caspase-Glo reagent, which also serves to lyse the cells, was added directly to each well and the resulting luminescence was measured every 30 min for 2 h using a Synergy HT microplate reader (BioTek Instruments, Vermont, USA). For viability assays, cells were seeded at a density of 3000 cells/well in 100 µL of complete medium in 96-well plates. After 18 h, cells were treated with increasing concentrations of OSU-03012, HA15, apratoxin A, carboplatin, or vehicle (0.1% DMSO). General cell viability was assessed using a colorimetric 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay in which viable cells generate a formazan product detected at 490 nm using a microplate reader (BioTek Instruments). For caspase inhibitor assays, cells were treated with Z-VAD-FMK (50 µM) 5 min before the addition of OSU-03012 or vehicle (0.1% DMSO). For these studies, viability was assessed after 24 h using a luminescence-based assay designed to detect cellular ATP in viable cells (CellTiter Glo®, Promega). In all assays, the viability of vehicle-treated cells at the end of the study was used to define 100% cell viability.
Data analysis
Concentration–response relationships were analyzed using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA, USA), and EC50 values were determined by nonlinear regression analysis fit to a logistic equation. Analysis of potential synergy between carboplatin and OSU-03012 or HA15 was determined using the Chou-Talalay combination index method with CompuSyn (CompuSyn, Inc., Paramus, NJ, USA) software (Chou 2006; Chou and Talalay 1984). Fraction-affected (Fa) values were calculated from mean cell viability of technical replicates (subtracted from 1 for each condition). For immunoblot analysis, signals were normalized to the intensity of control proteins (α-tubulin or GAPDH) and quantified relative to control using ImageJ software (rsbweb.nih.gov/ij). Statistical significance of data derived from cell viability assays and quantification of immunoblot assay were performed using a one-way analysis of variance (ANOVA) followed by a Student’s t test comparing untreated controls and treatment groups. P values less than 0.05 were considered significant.
Results
Canine osteosarcoma cells show increased basal expression of glucose-regulated and heat shock proteins
As a follow-up to our earlier study (Milovancev et al. 2013), we compared the expression pattern of representative molecular chaperones in whole cell lysates prepared from POS osteosarcoma cells, HMPOS osteosarcoma cells, and nonmalignant osteogenic progenitor cells from normal canine bone. Immunoblot analysis of Grp78, Grp94, Hsp90, Hsp70, and Hsp40 showed increased expression of all five variable chaperones in POS and HMPOS cancer cells relative to primary osteoblasts (Fig. 1A). We also compared the general subcellular expression pattern of Grp78 and Hsp90 in POS and HMPOS osteosarcoma cells relative to other control proteins enriched in membrane (EGFR), cytoplasmic (GAPDH), and nuclear (HDAC-2) compartments. Membrane fractions derived from POS or HMPOS osteosarcoma cells were strongly immunoreactive for Grp78 relative to cytoplasmic fractions, whereas nuclear fractions showed weak to no detectable immunoreactivity (Fig. 1B). In contrast, Hsp90 was most abundant in the cytoplasmic fraction of both cell types and absent from nuclear fractions (Fig. 1B). With a significant fraction of total Grp78 protein associated with cell membranes, we focused on the potential for surface Grp78 expression in these cells as observed in our proteomic study (Milovancev et al. 2013). Immunocytochemistry of POS osteosarcoma cells under nonpermeabilized conditions revealed a diffuse pattern of cell surface Grp78 associated with most cells in each visual field, whereas permeabilized cells showed a distinct punctate pattern of Grp78 immunoreactivity (Fig. 1C). Further analysis of Grp78 immunoreactivity, relative to that of the integrin β-1 receptor, showed wide surface expression of integrin β-1 in POS cells under nonpermeabilized conditions, and in co-stained cells, 82.6% of all integrin β-1-positive cells also scored positive for Grp78 (Fig. 1C).

Localization of Grp78 in canine osteosarcoma cells. A Immunoblot analysis of Grp78, Grp94, Hsp90, Hsp70, Hsp40, and α-tubulin expression in whole cell lysates prepared from normal osteogenic progenitor cells (OPCs) from normal bone, POS, and HMPOS canine osteosarcoma cancer cells. B Immunoblot analysis of Grp78 and representative control proteins in cytoplasmic (C), membrane (M), and nuclear (N) fractions from canine POS and HMPOS osteosarcoma cells. Samples were probed with anti-Grp78 and primary antibodies for detection of cytosolic (Hsp90, GAPDH), membrane (EGFR), and nuclear (HDAC2) proteins as indicated. Whole cell lysates (panel A) or cell fractions (panel B) were prepared at the same time; images are representative of three independent comparisons of all cell lines. C Immunocytochemical analysis of Grp78 distribution in canine POS osteosarcoma cells. Top row: nonpermeabilized or permeabilized cells were stained with anti-Grp78 (green) or anti-Grp78 plus DAPI to visualize nuclei stained (blue), respectively. Bottom row: nonpermeabilized cells were stained with anti-integrin β-1 (red) or anti-integrin β-1 plus anti-Grp78 (merged). D Quantification of corrected total cell fluorescence (CTCF) in cells co-stained for integrin β-1 plus Grp78 measured from three independent comparisons (n = 29 cells)
Next, we compared total Grp78 expression in untreated, canine osteosarcoma cells and several human cancer cell lines representing different histological subtypes. Grp78 expression in three canine osteosarcoma cell lines (D17, COS; POS) was comparable, or higher, to that detected in human SK-ES-1 Ewing sarcoma and SAOS-2 osteosarcoma cells, respectively (Fig. 2A). Other human cancer cell types showed high Grp78 expression including glioblastoma (U87-MG, SF295, U251-MG), a triple-negative breast cancer (MDA-MB-231), a prostate (DU-145), and colon (HCT116) cancer cell line (Fig. 2A). In contrast, human breast (MCF-7), ovarian (SKOV-3), and osteosarcoma (SAOS-2) cells showed low basal expression of Grp78. Taken together, these data support and extend the results of our earlier proteomic study and indicate that canine osteosarcoma cells show cell surface expression of the resident ER chaperone Grp78 and high basal accumulation of multiple molecular chaperones.

Comparative analysis of basal and stress-induced Grp78 expression in canine and human cancer cells. A Immunoblot analysis of Grp78 and α-tubulin expression in (from left to right) human SAOS-2 osteosarcoma, human SK-ES-1 Ewing’s sarcoma, canine osteosarcoma (D17, COS, POS), human glioblastoma (SF-295, U87-MG, U251-MG), human breast (MDA-MB-231, MCF-7), human DU145 prostate, human SAOS-2 osteosarcoma, human HCT116 colon, and human SKOV-3 ovarian cancer cells. Whole cell lysates, for each panel, were prepared at the same time and processed for Western blot analysis with the antibodies indicated. Lysates were analyzed on separate gels with human SAOS-2 osteosarcoma cells common to the 5- and 9-panel comparison; images are representative of three independent comparisons of each panel. B Immunoblot analysis of Grp78, CHOP, and α-tubulin expression in the absence (0.1% DMSO) or presence of thapsigargin (3 µM) in human SAOS-2 osteosarcoma, canine D17 or COS osteosarcoma cells, and human SK-ES-1 Ewing’s sarcoma, canine POS, or HMPOS osteosarcoma cells. Cells were grown under standard conditions and treated with, or without, thapsigargin for 24 h. Whole cell lysates were probed with antibodies against Grp78, CHOP, and α-tubulin (loading control) as indicated. Images are representative of a comparison that was repeated three times
To determine if Grp78 expression could be further induced in canine osteosarcoma cells, D17, COS, POS, and HMPOS cells were treated with a fixed concentration of thapsigargin (3 µM), a potent inducer of ER stress (Lytton et al. 1991; Wong et al. 1993). After 24 h, all four canine osteosarcoma cell types showed large upregulation of Grp78 expression relative to vehicle-treated cells in a manner that was qualitatively indistinguishable from human SK-ES-1 Ewing sarcoma cells or SAOS-2 osteosarcoma cells treated under the same conditions (Fig. 2B). Increases in Grp78 were accompanied by induction of the ER stress–associated transcription factor CCAAT-enhancer-binding protein homologous protein (CHOP), which was detected in all thapsigargin-treated canine and human cells (Fig. 2B) and consistent with a typical pattern of ER stress signaling.
Grp78 inhibitors are cytotoxic to canine osteosarcoma cells
To explore the feasibility of perturbing Grp78 function in canine osteosarcoma cells, we compared the action of two different pharmacological inhibitors of Grp78: (1) HA15, a specific inhibitor of Grp78 (Cerezo et al. 2016), and (2) OSU-03012, a derivative of the nonsteroidal anti-inflammatory agent celecoxib that has been reported to decrease Grp78 expression (Booth et al. 2012; Park et al. 2008). The cyanobacterial natural product apratoxin A was also included as a reference inhibitor of nascent Grp78 biosynthesis for comparison with OSU-03012 (Liu et al. 2009; Paatero et al. 2016). For these studies, POS osteosarcoma cells were treated with or without OSU-03012, HA15, or apratoxin A, and whole cell lysates collected and analyzed for expression of Grp78, Grp94, and calreticulin relative to vehicle (0.1% DMSO)-treated cells. Immunoblot analysis of cell lysates harvested 8 h or 24 h after treatment revealed Grp78 protein expression to be sensitive to all three compounds; however, each compound induced a different pattern of change in the three resident ER proteins surveyed (Fig. 3). OSU-03012 induced a biphasic response that was unlike the effect of apratoxin A or HA15; Grp78 expression was enhanced at 8 h, and decreased significantly by 24 h in the presence of OSU-03012 without statistically significant changes in expression Grp94 or calreticulin. Apratoxin A induced statistically significant decreases in Grp78 immunoreactivity at 8 h and 24 h, without significant changes in Grp94 expression over this timeframe, and was the only compound to inhibit calreticulin expression. HA15 treatment induced statistically significant increases in Grp78 immunoreactivity, relative to vehicle-treated POS cells, without statistically significant changes in Grp94 or calreticulin expression (Fig. 3).

Analysis of Grp78 and Grp94 expression in response to treatment with OSU-03012, apratoxin A, or HA15. A Expression of Grp78 and Grp94, relative to calreticulin and α-tubulin, in canine POS osteosarcoma cells in the absence (0.1% DMSO) or presence of OSU-03012 (3 µM), apratoxin A (300 nM), and HA15 (10 µM) after 8 h or 24 h. At the end of treatment, whole cell lysates were processed for immunoblot analysis and probed with antibodies against Grp78, Grp94, calreticulin, and α-tubulin as indicated. B Histograms show quantification of Grp78 and Grp94 expression from three independent experiments. Signals were normalized to α-tubulin and statistical significance in treated versus untreated cells indicated as *p < 0.05, **p < 0.01, ***p < 0.001, or ****p < 0.0001
To assess the feasibility of targeting canine Grp78 with these molecules, we compared the viability of normal canine cells with canine and human osteosarcoma cells after exposure to each of the three compounds. Apratoxin A showed low nanomolar cytotoxicity to nonmalignant canine osteogenic progenitors (Fig. 4A) and was potently cytotoxic to SAOS-2 (Fig. 4B), POS (Fig. 4C), and HMPOS (Fig. 4D) osteosarcoma cells (Table 1). In contrast, distinct differences were observed in the effects of OSU-03012 and HA15 on osteogenic progenitor cells from normal bone. OSU-03012 showed micromolar cytotoxicity (EC50 = 5.8 µM), whereas HA15 was relatively nontoxic to nonmalignant cells which remained over 75% viable after a 72-h exposure to a relatively high (30 µM) concentration of HA15 (Fig. 4A). HA15 was also relatively nontoxic to human SAOS-2 osteosarcoma cells (Fig. 4B) but was efficacious against canine POS (Fig. 4C) and HMPOS (Fig. 4D) cells at low micromolar concentrations (Table 1). OSU-03012 showed similar cytotoxic potency to HA15 against canine POS and HMPOS cells and was a slightly more efficacious cytotoxin; OSU-03012 was characterized by a steep concentration–response curve and induced a greater cell kill than HA15 in both POS and HMPOS osteosarcoma cells at the highest concentration (30 µM) tested (Fig. 4C, D). These data indicate that both HA15 and OSU-03012 are more toxic to canine osteosarcoma cells than canine osteogenic progenitors from normal bone and, of the two compounds, HA15 would be predicted to have a more favorable therapeutic index.

Concentration–response analysis of OSU-03012-, apratoxin A-, and HA15-induced changes in cell viability in normal canine osteoblasts relative to representative human and canine osteosarcoma cells. Cell viability of A canine osteogenic progenitor cells (OPCs), B human SAOS-2 osteosarcoma, C canine POS osteosarcoma, and D canine HMPOS osteosarcoma cells exposed at the same time to increasing concentrations of OSU-03012, apratoxin A, HA15, or vehicle (0.1% DMSO) for 72 h. Cell viability was determined by the MTT assay. The viability of vehicle-treated cells was defined as 100%. Graphs show a single comparison of three compounds across four cell types that was repeated three times. Points refer to average viability (n = 3 wells) and curves represent the fit of data points by nonlinear regression analysis to a logistic equation
OSU-03012 and HA15 bind Grp78 and induce apoptosis in canine osteosarcoma cells
The growth inhibitory effects of OSU-03012 at low (3 to 5 µM) micromolar concentrations were first attributed to direct inhibition of PDK1 in cancer cells (Zhu et al. 2004). However, molecular docking studies later predicted that OSU-03012 binds directly to the ATPase domain of Grp78 (Bhattacharjee et al. 2015). To understand if OSU-03012 can engage Grp78 in living canine osteosarcoma cells, we utilized a cellular thermal shift technique in which the action of OSU-03012 was compared with the specific Grp78 inhibitor HA15 (Cerezo et al. 2016). For these studies, POS cells were incubated under normal growth conditions for 1 h at 37 °C with either OSU-03012, HA15, or vehicle (DMSO 0.1%), and intact cells collected and resuspended in PBS. Cells were then heated for 3 min at fixed temperatures to denature all unbound proteins, cooled, and subjected to freeze–thaw samples to lyse the cells. Immunoblot analysis of samples prepared from cells treated with OSU-03012 (3 µM) or HA15 (3 µM and 10 µM) resulted in detection of an immunoreactive band corresponding to Grp78 at 45 °C and 48 °C (Fig. 5A). In samples subjected to 51 °C and higher temperatures, this band lacked specificity or was lost (Fig. 5A). Stabilization of Grp78 protein, by OSU-03012 (3 µM) or HA15 (10 µM), was statistically significant at 48 °C (Fig. 5B); however, we were unable to titrate OSU-03012 to lower concentrations and see a thermally stable band at 48 °C or at 45 °C. Taken together, these data suggest that OSU-03012 can bind Grp78 in intact cells at a concentration that is physiologically relevant based on the calculated EC50 (Table 1), but that this interaction is unstable at lower concentrations.

OSU-03012 and HA15 stabilize Grp78 in intact canine osteosarcoma cells and induce cell death. A Analysis of Grp78 thermal stability in the presence of 0.1% DMSO (V), 3 µM OSU-03012 (OSU), or 3 µM and 10 µM HA15. Melting curves were generated by exposing intact POS cells (4 × 107) to each treatment as indicated for 1 h. Cells were collected by gentle centrifugation, divided into equal aliquots, and subjected to a single fixed temperature (ranging from 45 to 54 °C). Immunoblot analysis of Grp78 and α-tubulin expression is representative of a single melting curve that was repeated three times with similar results. B Quantification of melting curve data (at highest concentration tested) from three independent studies. C OSU-03012 and D HA15 induce concentration-dependent increases in caspase-3,7 activity in POS and HMPOS osteosarcoma cells. Bars represent mean luminescence values (n = 3 wells per treatment) determined after the 24-h exposure to each compound, as indicated, and are expressed as relative light units ± SE from three independent determinations. E Comparison of the ability of a pan-caspase inhibitor to rescue POS or HMPOS cells from OSU-03012- or HA15-induced cytotoxicity. Cells were exposed to vehicle (0.1% DMSO), OSU-03012 (1 µM, 3 µM, or 10 µM), and HA15 (1 µM, 3 µM, or 10 µM), with or without V-ZAD-fmk (50 µM), and cell viability was assessed using the MTT assay after 24 h. The viability of vehicle-treated cells was defined as 100%. Data points show mean viability ± SE (n = 3 wells per treatment) from a representative comparison that was repeated in three independent experiments. Statistical significance of change in treated, relative to vehicle-treated (0.1% DMSO), cells is indicated as *p < 0.05, **p < 0.01, and ***p < 0.001
Having established that both OSU-03012 and HA15 are cytotoxic to canine osteosarcoma cells, we analyzed POS and HMPOS cells for additional markers of cell death. Treatment with OSU-03012 (Fig. 5C) or HA15 (Fig. 5D) induced statistically significant increases in caspase-3,7 activity in both cell types at low micromolar concentrations at 24 h, relative to vehicle-treated cells. Further, when cells were co-treated with the pan caspase inhibitor Z-VAD-fmk, in combination with OSU-03012 or HA15, the viability of POS and HMPOS cells was enhanced to varying levels corresponding to statistically significant rescue of cell viability (Fig. 5E). HMPOS cells were generally more sensitive to the presence of Z-VAD-fmk than POS cells, and HA15-induced cell death more sensitive to Z-VAD-fmk than OSU-03012 (Fig. 5E). These combined results provide biochemical evidence for apoptosis as a likely cell death mechanism in response to OSU-03012 or HA15 exposure in these osteosarcoma cell types.
Carboplatin-resistant osteosarcoma cells are sensitive to inhibitors of protein chaperone function
Carboplatin is widely used as adjuvant chemotherapy in canine osteosarcoma as it produces a significant increase in median survival time, relative to dogs that receive surgery alone (Skorupski et al. 2016). Using the same experimental conditions used to assess the toxicity of Grp78 inhibitors to normal osteoblasts (Fig. 4), we tested the potency and efficacy of carboplatin treatment alone and in combination with either OSU-03012 or HA15. Carboplatin alone (0.003 nM to 100 µM) induced a concentration-dependent reduction in the viability of both POS (Fig. 6A) and HMPOS (Fig. 6B) cells with EC50 values of 3.7 µM and 4.0 µM for POS and HMPOS cells, respectively (Table 2). When cells were exposed to carboplatin (3 nM to 100 µM) in the presence of a fixed, sublethal concentration of OSU-03012 (1 µM), the concentration–response curves were shifted to the left for POS (Fig. 6A) and HMPOS (Fig. 6B) cells. Nonlinear regression analysis of these data for carboplatin in the presence of OSU-03012 revealed EC50 values of 2.1 µM and 1.8 µM for POS and HMPOS cells, respectively (Table 2). Treatment with carboplatin and HA15 (1 µM) tended to potentiate the cytotoxicity of carboplatin at low concentrations (< 0.03 nM) and also resulted in a moderate increase in cytotoxic potency (Table 2). Further analysis of these cell viability data using the Chou-Talalay combination index method (Chou 2006; Chou and Talalay 1984) indicated the presence of mild synergy for the combination of carboplatin and OSU-03012 at lower concentrations of carboplatin (Fig. 6C, D), while the combination of carboplatin and HA15 failed to show synergy using this prediction (Fig. 6C, D).

Analysis of the cytotoxic efficacy of carboplatin in combination with OSU-03012 or HA15 treatment. A POS and B HMPOS osteosarcoma cell viability in response to carboplatin alone (3 nM to 100 µM) and in combination with a fixed concentration of OSU-03012 (1 µM) or HA15 (1 µM). Cells were exposed to single or combination treatments, as indicated, and cell viability was determined at 72 h with the viability of vehicle-treated (0.1% DMSO) cells defined as 100% viability. Data points represent mean viability ± SE (n = 3 wells per treatment), and curves represent the fit of data points by nonlinear regression analysis to a logistic equation. Cell viability curves are representative of a comparison that was repeated three times. Combination index plots for C POS and D HMPOS osteosarcoma cells for fixed combination treatments of carboplatin plus HA15 or OSU-03012 using the Chou-Talalay combination index method. Points below 1 (indicated by the dashed line) indicate synergy
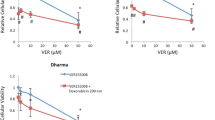
Treatment resistance and failure is a major complication in the treatment of high-grade osteosarcoma in both human and veterinary medicine (Simpson et al. 2017). To assess the extent to which carboplatin-resistant cells are sensitive to Grp78 inhibitors we tested OSU-03012 and HA15 against canine osteosarcoma cells with two different levels of carboplatin resistance. These sublines were created by exposure of parental canine HMPOS cells to progressively increasing concentrations of carboplatin (0.5 to 10 µM) and subsequent selection of surviving cells at each concentration (Weinman et al. 2021). Parental HMPOS, HMPOS-2.5R, and HMPOS-10R, corresponding to drug-resistant sublines with acquired resistance to 2.5 µM or 10 µM carboplatin, respectively, were validated for resistance to carboplatin (Fig. 7A) and incubated in parallel with increasing concentrations of OSU-03012 (Fig. 7B); HA15 (Fig. 7C), a control Hsp70 inhibitor; VER-155008 (Fig. 7D), a control Hsp90 inhibitor; 17-AAG (Fig. 7E); or vehicle (DMSO 0.1%). The viability of all cells was then assessed at 72 h relative to vehicle-treated control cells. While HMPOS-10R and HMPOS-2.5R remained fully or moderately resistant to carboplatin, respectively (Fig. 7A), both drug-resistant sublines were sensitive to all four small molecule inhibitors tested (Fig. 7B–E). Each compound reduced cell viability in a concentration-dependent manner with low micromolar potency and reasonable cytotoxic efficacy (Table 3). The efficacy of each inhibitor against HMPOS-2.5R cells was comparable to their effectiveness against parental HMPOS cells at the highest concentrations tested; HA15 and 17-AAG induced nearly 100% cell kill, whereas OSU-03012 and VER-155008 reduced viability by approximately 75% (Fig. 7B–E). HMPOS-10R cells were slightly more resistant than HMPOS-2.5R cells, but all inhibitors reduced viability by greater than 50% at the highest concentrations tested (Fig. 7B–E). These findings indicate that carboplatin resistance in HMPOS osteosarcoma does not confer cross-resistance to any of the small molecule inhibitors tested and raises the possibility of targeting glucose-regulated or heat shock protein family members in canine osteosarcoma.

Carboplatin-resistant canine osteosarcoma cells remain sensitive to Grp78 inhibitors and small molecular inhibitors of other molecular chaperones. A Characterization of chemoresistant HMPOS canine osteosarcoma cell lines. Parental canine HMPOS osteosarcoma and two carboplatin-resistant variants of HMPOS cell lines (resistant 2.5 CB HMPOS and resistant 10 CB HMPOS) were exposed to increasing concentrations of carboplatin for 72 h. B–E Concentration–response analysis of parental and chemoresistant canine osteosarcoma cells in the presence of B OSU-03012, C HA15, D Hsp70 inhibitor VER-155008, and E Hsp90 inhibitor 17-AAG. Cells were treated at the same time, and cell viability was assessed using a MTT assay. Data points represent mean viability ± SE (n = 3 wells per treatment) relative to vehicle control (0.1% DMSO). Each panel is representative of a comparison that was repeated three times in independent determinations
Discussion
Primary osteosarcoma is one of several spontaneous cancers affecting humans and dogs where a comparative oncology approach has the potential to accelerate our understanding of the disease and the development of new treatments to benefit both species (Geller and Gorlick 2010; Schiffman and Breen 2015; Simpson et al. 2017; Vail and MacEwen 2000). Current standard of care proceeds with a combination of surgical resection of the primary disease followed by chemotherapy treatment. However, lung metastases are a significant complication leading to extremely low survival rates in patients with progressive disease (Aljubran et al. 2009; Geller and Gorlick 2010; Simpson et al. 2017; Skorupski et al. 2016). In the dog, low survival rates are attributed to the fact that most patients have micrometastases that are clinically undetectable at the time of diagnosis and progress with time, despite the use of systemic therapy (MacEwen and Kurzman 1996; Szewczyk et al. 2015). In our search for better treatments to improve the prognosis of osteosarcoma, we previously identified multiple molecular chaperones as surface-exposed proteins in canine osteosarcoma cells (Milovancev et al. 2013). In the present study, we confirmed cell surface expression of Grp78 in canine osteosarcoma cells by immunocytochemistry and found general upregulation of glucose-regulated and heat shock protein family members in osteosarcoma cells, relative to osteogenic progenitor cells. These data are consistent with the pattern of changes that have been reported in several aggressive types of human cancers and are thought to represent a functional change in the chaperome of malignant versus nonmalignant cells (Finka and Goloubinoff 2013; Rodina et al. 2016; Roy et al. 2017; Shin et al. 2003). Differences between normal cells and tumor cells have reinforced the idea that druggable targets lie within the tumor chaperome and may eventually lead to new or adjuvant treatments with an acceptable margin of safety (Calderwood 2018; Joshi et al. 2018). Towards this goal, our biochemical studies provide insight into the ability of the investigational drug OSU-03012 to bind Grp78 in cells. However, while Grp78 inhibitors alone showed promising cytotoxic efficacy against POS, HMPOS, and carboplatin-resistant HMPOS cells, these data indicate that this response was not specific to Grp78; highly metastatic and carboplatin-resistant HMPOS canine osteosarcoma cells were also sensitive to inhibition of Grp78, Hsp90, and Hsp70 function.
Although chaperones are some of the most abundant proteins in human and canine cancer cells, a relatively small number of these are currently under consideration as viable therapeutic targets (Finka and Goloubinoff 2013; Roy et al. 2017; Shin et al. 2003; Yang et al. 2021). To date, drug development in this area has concentrated on the generation of compounds to disrupt the function or expression of specific chaperones and biologics to block signaling mediated via cell surface chaperones (Yang et al. 2021). A full understanding of the proposed site of action of some of these molecules is, however, complicated by the knowledge that the same protein can exist in multiple cellular compartments and also be secreted to the extracellular space (Calderwood 2018). Estimates of the total number of chaperone proteins in representative human cancer cells previously revealed that the ER compartment has significantly fewer chaperone family members than the cytosolic compartment (Finka and Goloubinoff 2013), and thus, we targeted ER proteostasis in canine cells with two pharmacological inhibitors of Grp78. Although HA15 and OSU-03012 had significant cytotoxic potential against canine osteosarcoma cells, HA15 was relatively nontoxic to nonmalignant, osteogenic progenitors from normal bone at the maximum concentration tested (10 µM), consistent with previous reports where HA15 had a promising safety profile against normal human melanocytes under standard culture conditions (Cerezo et al. 2016; Szasz et al. 2021). Our studies also showed canine POS, HMPOS, and carboplatin-resistant HMPOS cells to be more sensitive to HA15 than human SAOS-2 osteosarcoma cells. As we did not exceed 10 µM HA15 in our analysis or include other human osteosarcoma cell lines, it would be premature to conclude that this compound has unique efficacy for canine osteosarcoma cells without further follow-up. Rather, as HA15 is known to be a strong inducer of autophagy (Cerezo et al. 2016; Han et al. 2021), it is more likely that the pro-survival and pro-death functions of autophagy in response to HA15 were incompletely defined in our concentration–response analysis of SAOS-2 cells (Levine and Kroemer 2008).
HA15 inhibits the ATPase function of Grp78 and induces cell death through a lethal ER stress response with compensatory upregulation of Grp78 (Cerezo et al. 2016). As melanoma cells with high levels of Grp78 were found to be most sensitive to HA15, this mechanism is thought to underlie the observed selectivity of HA15 for cancer cells versus normal melanocytes or normal fibroblasts (Cerezo et al. 2016). Our analysis of canine POS osteosarcoma cells in response to HA15 treatment appears consistent with this mechanism; Grp78 expression was increased by HA15 without statistically significant changes in Grp94 or calreticulin expression. When the action of HA15 and OSU-03012 was compared in side-by-side studies, OSU-03012 shared some of the same characteristics. Both ligands stabilized Grp78 protein in POS cells at low micromolar concentrations and were qualitatively indistinguishable over a narrow range of temperatures in thermal stability studies. However, in straightforward Western blot analyses, short-term increases in Grp78 immunoreactivity at 8 h were not sustained and were, instead, significantly decreased by 24 h. These results are consistent with at least two lines of thought regarding the mechanism of action of OSU-03012: (1) in silico molecular docking studies have predicted that OSU-03012 binds directly to the ATPase domain of Grp78 (Bhattacharjee et al. 2015), and (2) cycloheximide chase experiments have shown that OSU-03012 accelerates Grp78 degradation resulting in a shorter half-life of the mature protein (Booth et al. 2012). With low micromolar activity, it is likely that OSU-03012 has mixed, or off target, actions in cancer cells that are ultimately difficult to separate. Inhibition of PDK-1 is typically cited as the primary mechanism of OSU-03012 action which occurs at low micromolar concentrations (IC50 = 5.0 µM (Zhu et al. 2004)). In addition, other studies have concluded that OSU-03012 lacks specificity and binds other heat shock protein family members resulting in broad-spectrum chaperone inactivation (Booth et al. 2016a, b). In the present study, OSU-03012 induced changes in Grp78 expression whereas Grp94 showed remarkably consistent expression in response to all three compounds tested including a broad-spectrum inhibitor of ER secretory pathway protein biosynthesis. Decreases in Grp78 and calreticulin expression observed in canine osteosarcoma cells were in agreement with previous analyses of these proteins in human U2OS osteosarcoma cells treated with apratoxin A (Liu et al. 2009), whereas Grp94 was relatively insensitive to apratoxin A at 24 h in canine POS cell lysates. Grp94, Grp78, and calreticulin all contain an N-terminal signal peptide that directs each nascent protein to the co-translocation machinery for processing at the entrance to the conventional secretory pathway and site of apratoxin A binding (Huang et al. 2016; Liu et al. 2009; Paatero et al. 2016). Although all three resident ER chaperones are Sec61 substrates, the present study revealed unequal sensitivity of these proteins to apratoxin with a ranked order of sensitivity (greatest to least) where calreticulin > Grp78 > > Grp94. Differences in the sensitivity of client proteins to broad inhibition of the Sec61 translocon channel have previously been observed in human breast cancer cells, where human epidermal growth factor receptor 3 (HER3) and HER1 were considerably more sensitive to apratoxin A than HER2 (Kazemi et al. 2021; Weijing et al. 2021).Taken together, these findings suggest that Grp94 may also be less vulnerable to an acute block in Sec61-dependent import of nascent Grp94 protein.
Carboplatin is one of three commonly used chemotherapy agents, along with cisplatin and doxorubicin, that prolong median survival time in canine osteosarcoma, relative to dogs that receive surgery alone (Mauldin et al. 1988; Skorupski et al. 2016). Despite the widespread use of these agents, acquired and intrinsic chemoresistance remains a major complication both in canine osteosarcoma and in human patients with recurrent disease (Hattinger et al. 2021; Szewczyk et al. 2015). In side-by-side comparisons, we found pharmacological inhibition of Grp78 function to be as effective as carboplatin in decreasing the viability of cultured POS and HMPOS cells. Perhaps more significant, however, was the finding that 17-AAG, VER-155008, OSU-03012, and HA15 were efficacious cytotoxins against carboplatin-resistant HMPOS cells. The geldanamycin analogue, 17-AAG, was the most potent of the four compounds tested, while other compounds induced between a 50% (VER-155008) and 75% (OSU-03012) cell kill against the most resistant HMPOS-10R derivative at 30 µM, suggesting that cellular dependence on the function of at least three chaperones (Hsp90, Hsp70, and Grp78) was independent of acquired resistance to carboplatin. The mechanistic basis of platinum resistance has been studied extensively in different histological cancer types and is attributed to several major adaptations, although a tumor supportive role for stress-inducible heat shock proteins has been noted (Galluzzi et al. 2014; Mandic et al. 2003; Shen et al. 2012; Yun et al. 2019). In an independent characterization of the same carboplatin-resistant cells, we previously detected Hsp27 in both lysates and exosomes and Hsp105 in the exosomal cargo of HMPOS-10R cells (Weinman et al. 2021). Further, the proteomic signature of the carboplatin-resistant HMPOS derivatives was found to be relevant to the study of spontaneous osteosarcoma in dogs (Weinman et al. 2021). Although Hsp27 and Hsp105 were not targeted in the present study, these observations raise important practical considerations in that 17-AAG, VER-155008, OSU-03012, and HA15 may not necessarily act exclusively at intracellular sites. For example, although Hsp90 was abundant in whole cell lysates and on the cell surface of POS and HMPOS osteosarcoma cells relative to normal osteoblasts, this chaperone was also previously detected as an exosomal cargo protein in blood samples from dogs with and without osteosarcoma (Brady et al. 2018; Milovancev et al. 2013). Taken together, these findings suggest that canine osteosarcoma is an example of a cancer type with a stress-adapted chaperome to support pro-survival signaling.
In summary, we have extended our analysis of protein chaperones in canine osteosarcoma cells and compared the action of pharmacological inhibitors of Grp78, Hsp70, and Hsp90 function against established cell lines and carboplatin-resistant derivatives. Canine osteosarcoma cells shared a similar adaptive response to ER stress induction by thapsigargin and were highly sensitive to the loss of secretory pathway proteostasis in response to the Sec61 inhibitor apratoxin A. Our results provide insight into the mixed action of the celecoxib analogue OSU-03012 and support the recent designation of this compound as a direct modulator of Grp78 that, in contrast to HA15, acts to downregulate Grp78 protein expression (Bailly and Waring 2019). The sensitivity of carboplatin-resistant osteosarcoma cells to several prototypical Hsp-targeting tool compounds, coupled with concentration–response analyses in normal canine osteogenic progenitor cells, suggests that the cancer chaperome may represent a viable drug target for canine osteosarcoma.
References
Aljubran AH, Griffin A, Pintilie M, Blackstein M (2009) Osteosarcoma in adolescents and adults: survival analysis with and without lung metastases. Ann Oncol 20(6):1136–1141
Bailly C, Waring MJ (2019) Pharmacological effectors of GRP78 chaperone in cancers. Biochem Pharmacol 163:269–278
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319(5865):916–919
Bhattacharjee R, Devi A, Mishra S (2015) Molecular docking and molecular dynamics studies reveal structural basis of inhibition and selectivity of inhibitors EGCG and OSU-03012 toward glucose regulated protein-78 (GRP78) overexpressed in glioblastoma. J Mol Model 21(10):272
Booth L, Cazanave SC, Hamed HA, Yacoub A, Ogretmen B, Chen CS, Grant S, Dent P (2012) OSU-03012 suppresses GRP78/BiP expression that causes perk-dependent increases in tumor cell killing. Cancer Biol Ther 13(4):224–236
Booth L, Roberts JL, Ecroyd H, Reid SP, Proniuk S, Zukiwski A, Jacob A, Damonte E, Tunon MJ, Dent P (2016a) AR-12 inhibits chaperone proteins preventing virus replication and the accumulation of toxic misfolded proteins. J Clin Cell Immunol 7(5)
Booth L, Shuch B, Albers T, Roberts JL, Tavallai M, Proniuk S, Zukiwski A, Wang D, Chen CS, Bottaro D et al (2016b) Multi-kinase inhibitors can associate with heat shock proteins through their NH2-termini by which they suppress chaperone function. Oncotarget 7(11):12975–12996
Brady JV, Troyer RM, Ramsey SA, Leeper H, Yang L, Maier CS, Goodall CP, Ruby CE, Albarqi HAM, Taratula O et al (2018) A preliminary proteomic investigation of circulating exosomes and discovery of biomarkers associated with the progression of osteosarcoma in a clinical model of spontaneous disease. Transl Oncol 11(5):1137–1146
Calderwood SK (2018) Heat shock proteins and cancer: intracellular chaperones or extracellular signalling ligands? Philos Trans R Soc Lond Ser B, Biol Sci 373(1738)
Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E, Botton T, Ronco C, Abbe P, Amdouni H et al (2016) Compounds triggering er stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell 29(6):805–819
Chou TC (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58(3):621–681
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22:27–55
Eletto D, Dersh D, Argon Y (2010) Grp94 in er quality control and stress responses. Semin Cell Dev Biol 21(5):479–485
Finka A, Goloubinoff P (2013) Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 18(5):591–605
Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G (2014) Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 5:e1257
Geller DS, Gorlick R (2010) Osteosarcoma: a review of diagnosis, management, and treatment strategies. Clin Adv Hematol Oncol 8(10):705–718
Hadizadeh Esfahani A, Sverchkova A, Saez-Rodriguez J, Schuppert AA, Brehme M (2018) A systematic atlas of chaperome deregulation topologies across the human cancer landscape. PLoS Comput Biol. 14(1):e1005890
Han C, Xie K, Yang C, Zhang F, Liang Q, Lan C, Chen J, Huang K, Liu J, Li K et al (2021) HA15 alleviates bone loss in ovariectomy-induced osteoporosis by targeting HSPA5. Exp Cell Res. 406(2):112781
Hartl FU, Bracher A, Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475(7356):324–332
Hattinger CM, Patrizio MP, Fantoni L, Casotti C, Riganti C, Serra M (2021) Drug resistance in osteosarcoma: emerging biomarkers, therapeutic targets and treatment strategies. Cancers (Basel) 13(12)
Huang KC, Chen Z, Jiang Y, Akare S, Kolber-Simonds D, Condon K, Agoulnik S, Tendyke K, Shen Y, Wu KM et al (2016) Apratoxin A shows novel pancreas-targeting activity through the binding of Sec 61. Mol Cancer Ther 15(6):1208–1216
Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Martinez MD (2014) The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9(9):2100–2122
Joshi S, Wang T, Araujo TLS, Sharma S, Brodsky JL, Chiosis G (2018) Adapting to stress - chaperome networks in cancer. Nat Rev Cancer 18(9):562–575
Kazemi S, Kawaguchi S, Badr CE, Mattos DR, Ruiz-Saenz A, Serrill JD, Moasser MM, Dolan BP, Paavilainen VO, Oishi S et al (2021) Targeting of HER/ErbB family proteins using broad spectrum Sec61 inhibitors coibamide A and apratoxin A. Biochem Pharmacol. 183:114317
Kourtis N, Lazaris C, Hockemeyer K, Balandran JC, Jimenez AR, Mullenders J, Gong Y, Trimarchi T, Bhatt K, Hu H et al (2018) Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. Nat Med 24(8):1157–1166
Lee AS (2014) Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 14(4):263–276
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132(1):27–42
Liu Y, Law BK, Luesch H (2009) Apratoxin A reversibly inhibits the secretory pathway by preventing cotranslational translocation. Mol Pharmacol 76(1):91–104
Lytton J, Westlin M, Hanley MR (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem 266(26):17067–17071
Macario AJ, Conway de Macario E (2002) Sick chaperones and ageing: a perspective. Ageing Res Rev 1(2):295–311
MacEwen EG, Kurzman ID (1996) Canine osteosarcoma: amputation and chemoimmunotherapy. Vet Clin North Am Small Anim Pract 26(1):123–133
Mambula SS, Calderwood SK (2006) Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol 177(11):7849–7857
Mambula SS, Stevenson MA, Ogawa K, Calderwood SK (2007) Mechanisms for Hsp70 secretion: crossing membranes without a leader. Methods 43(3):168–175
Mandic A, Hansson J, Linder S, Shoshan MC (2003) Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem 278(11):9100–9106
Marzec M, Eletto D, Argon Y (2012) GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta 1823(3):774–787
Mauldin GN, Matus RE, Withrow SJ, Patnaik AK (1988) Canine osteosarcoma. Treatment by amputation versus amputation and adjuvant chemotherapy using doxorubicin and cisplatin. J Vet Intern Med. 2(4):177–180
Milovancev M, Hilgart-Martiszus I, McNamara MJ, Goodall CP, Seguin B, Bracha S, Wickramasekara SI (2013) Comparative analysis of the surface exposed proteome of two canine osteosarcoma cell lines and normal canine osteoblasts. BMC Vet Res 9:116
Nolan KD, Franco OE, Hance MW, Hayward SW, Isaacs JS (2015) Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J Biol Chem 290(13):8271–8282
Nolan KD, Kaur J, Isaacs JS (2017) Secreted heat shock protein 90 promotes prostate cancer stem cell heterogeneity. Oncotarget 8(12):19323–19341
Paatero AO, Kellosalo J, Dunyak BM, Almaliti J, Gestwicki JE, Gerwick WH, Taunton J, Paavilainen VO (2016) Apratoxin kills cells by direct blockade of the Sec61 protein translocation channel. Cell Chem Biol 23(5):561–566
Park MA, Yacoub A, Rahmani M, Zhang G, Hart L, Hagan MP, Calderwood SK, Sherman MY, Koumenis C, Spiegel S et al (2008) OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells. Mol Pharmacol 73(4):1168–1184
Pouyssegur J, Shiu RP, Pastan I (1977) Induction of two transformation-sensitive membrane polypeptides in normal fibroblasts by a block in glycoprotein synthesis or glucose deprivation. Cell 11(4):941–947
Rodina A, Wang T, Yan P, Gomes ED, Dunphy MP, Pillarsetty N, Koren J, Gerecitano JF, Taldone T, Zong H et al (2016) The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538(7625):397–401
Roy J, Wycislo KL, Pondenis H, Fan TM, Das A (2017) Comparative proteomic investigation of metastatic and non-metastatic osteosarcoma cells of human and canine origin. PLoS One. 12(9):e0183930
Saibil H (2013) Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 14(10):630–642
Schiffman JD, Breen M (2015) Comparative oncology: what dogs and other species can teach us about humans with cancer. Philos Trans R Soc Lond Ser B, Biol Sci 370(1673)
Shemesh N, Jubran J, Dror S, Simonovsky E, Basha O, Argov C, Hekselman I, Abu-Qarn M, Vinogradov E, Mauer O et al (2021) The landscape of molecular chaperones across human tissues reveals a layered architecture of core and variable chaperones. Nat Commun 12(1):2180
Shen DW, Pouliot LM, Hall MD, Gottesman MM (2012) Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev 64(3):706–721
Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, Zhao R, Puravs E, Tra J, Michael CW et al (2003) Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem 278(9):7607–7616
Shiu RP, Pouyssegur J, Pastan I (1977) Glucose depletion accounts for the induction of two transformation-sensitive membrane proteinsin rous sarcoma virus-transformed chick embryo fibroblasts. Proc Natl Acad Sci U S A 74(9):3840–3844
Simpson S, Dunning MD, de Brot S, Grau-Roma L, Mongan NP, Rutland CS (2017) Comparative review of human and canine osteosarcoma: morphology, epidemiology, prognosis, treatment and genetics. Acta Vet Scand 59(1):71
Skorupski KA, Uhl JM, Szivek A, Allstadt Frazier SD, Rebhun RB, Rodriguez CO Jr (2016) Carboplatin versus alternating carboplatin and doxorubicin for the adjuvant treatment of canine appendicular osteosarcoma: a randomized, phase III trial. Vet Comp Oncol 14(1):81–87
Szasz I, Koroknai V, Patel V, Hajdu T, Kiss T, Adany R, Balazs M (2021) Cell proliferation is strongly associated with the treatment conditions of an er stress inducer new anti-melanoma drug in melanoma cell lines. Biomedicines 9(2)
Szewczyk M, Lechowski R, Zabielska K (2015) What do we know about canine osteosarcoma treatment? Rev Vet Res Commun 39(1):61–67
Thornburg CC, Cowley ES, Sikorska J, Shaala LA, Ishmael JE, Youssef DT, McPhail KL (2013) Apratoxin H and apratoxin A sulfoxide from the red sea cyanobacterium Moorea producens. J Nat Prod 76(9):1781–1788
Vail DM, MacEwen EG (2000) Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Invest 18(8):781–792
Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, Multhoff G, Arispe N, De Maio A (2008) Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol 180(6):4299–4307
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059):1081–1086
Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H et al (2006) Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127(4):803–815
Weijing C, Ratnayake R, Wang M, Chen Q-Y, Raisch K, Dang LH, Law B, Luesch H (2021) Inhibition of cotranslational translocation by apratoxin S4: effects on oncogenic receptor tyrosine kinases and the fate of transmembrane proteins produced in the cytoplasm. Curr Res Pharmacol Drug Discov 2
Weinman MA, Ramsey SA, Leeper HJ, Brady JV, Schlueter A, Stanisheuski S, Maier CS, Miller T, Ruby CE, Bracha S (2021) Exosomal proteomic signatures correlate with drug resistance and carboplatin treatment outcome in a spontaneous model of canine osteosarcoma. Cancer Cell Int 21(1):245
Wong WL, Brostrom MA, Kuznetsov G, Gmitter-Yellen D, Brostrom CO (1993) Inhibition of protein synthesis and early protein processing by thapsigargin in cultured cells. Biochem J 289(Pt 1):71–79
Yang S, Xiao H, Cao L (2021) Recent advances in heat shock proteins in cancer diagnosis, prognosis, metabolism and treatment. Biomed Pharmacother 142:112074
Yun CW, Kim HJ, Lim JH, Lee SH (2019) Heat shock proteins: agents of cancer development and therapeutic targets in anti-cancer therapy. Cells 9(1)
Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW, Shaw YJ, Kulp SK, Chen CS (2004) From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer Res 64(12):4309–4318
Zimmermann R, Eyrisch S, Ahmad M, Helms V (2011) Protein translocation across the er membrane. Biochim Biophys Acta 1808(3):912–924
Funding
This work was supported by an Interdisciplinary Research Grant from the Division of Health Sciences at Oregon State University (MM, SB, JEI); an OSU Undergraduate Research, Innovation, Scholarship & Creativity (URISC) award; and NIH/NCCIH T32AT010131 (DRM).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Consent for publication
All authors have consented to publication.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mattos, D.R., Weinman, M.A., Wan, X. et al. Canine osteosarcoma cells exhibit basal accumulation of multiple chaperone proteins and are sensitive to small molecule inhibitors of GRP78 and heat shock protein function. Cell Stress and Chaperones 27, 223–239 (2022). https://doi.org/10.1007/s12192-022-01263-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-022-01263-3