
Abstract
Seventy-kilodalton heat shock proteins (Hsp70s) are molecular chaperones essential for maintaining cellular homeostasis. Apart from their indispensable roles in protein homeostasis, specific Hsp70s localize at the plasma membrane and bind to specific lipids. The interaction of Hsp70s with lipids has direct physiological outcomes including lysosomal rescue, microautophagy, and promotion of cell apoptosis. Despite these essential functions, the Hsp70-lipid interactions remain largely uncharacterized. In this study, we characterized the interaction of HspA1A, an inducible Hsp70, with five phospholipids. We first used high concentrations of potassium and established that HspA1A embeds in membranes when bound to all anionic lipids tested. Furthermore, we found that protein insertion is enhanced by increasing the saturation level of the lipids. Next, we determined that the nucleotide-binding domain (NBD) of the protein binds to lipids quantitatively more than the substrate-binding domain (SBD). However, for all lipids tested, the full-length protein is necessary for embedding. We also used calcium and reaction buffers equilibrated at different pH values and determined that electrostatic interactions alone may not fully explain the association of HspA1A with lipids. We then determined that lipid binding is inhibited by nucleotide-binding, but it is unaffected by protein-substrate binding. These results suggest that the HspA1A lipid-association is specific, depends on the physicochemical properties of the lipid, and is mediated by multiple molecular forces. These mechanistic details of the Hsp70-lipid interactions establish a framework of possible physiological functions as they relate to chaperone regulation and localization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Seventy-kilodalton heat shock proteins (Hsp70s) are molecular chaperones essential for cellular homeostasis and survival under both physiological and pathological conditions (Daugaard et al. 2007; Lindquist and Craig 1988). The molecular chaperone activities of Hsp70s depend on their ability to interact with hydrophobic stretches of proteins in an ATP-dependent manner (Bukau and Horwich 1998).
In addition to their indispensable functions in protein-chaperoning and cell signaling, Hsp70s also function at the plasma membrane (PM) and several organelle membranes, as well as in the extracellular environment (Broquet et al. 2003; Gehrmann et al. 2008; Kirkegaard et al. 2010; Lancaster and Febbraio 2005a, b). Several members of the Hsp70 family, including HspA1A, the major heat-inducible Hsp70 in humans and mice, are translocated to the PM, are actively secreted from a variety of viable cells (Lancaster and Febbraio 2005a; Mambula et al. 2007; Multhoff 2007), and bind to specific lipids as well as other surface molecules like acidic glycans (De Maio 2011, 2014; Harada et al. 2014).
It has been postulated that the interaction between Hsp70s and lipids is a critical step of their membrane-associated functions. This interaction was first showed by Guidon and Hightower (Guidon and Hightower 1986a, b). This initial report was followed by many investigators showing that several members of the Hsp70 protein family, including HspA1A, as well as several bacterial Hsp70 (DnaK) proteins interact specifically with multiple glycolipids and phospholipids. These include phosphatidylserine (PS), bis-(monoacylglycero)-phosphate (BMP), globoyltriaosyl-ceramide (Gb3), sulfo-glycolipids, and anandamide (Arispe and De Maio 2000; Arispe et al. 2002, 2004; Armijo et al. 2014; Broquet et al. 2003; Browne et al. 2007; Chen et al. 2005; Gehrmann et al. 2008; Harada et al. 2007, 2014, 2015; Kirkegaard et al. 2010; Mahalka et al. 2014; McCallister et al. 2015; Oddi et al. 2009; Petersen et al. 2010; Whetstone and Lingwood 2003). These reports demonstrate that, although Hsp70s do not contain known lipid-binding domains, they interact with specific lipids and this interaction is evolutionarily conserved.
The interaction of Hsp70s with cellular membranes and lipids has direct physiological outcomes, and extracellular or membrane-bound Hsp70s regulate many vital processes. These processes include among others activation of the immune system (De Maio 2011, 2014; Gehrmann et al. 2004; Henderson 2010; Multhoff et al. 1998; Multhoff and Hightower 1996; Vega et al. 2008), cytoprotection of injured neural and other cells, suppression of tumor growth (Brown 2007; Chen et al. 2005; Gehrmann et al. 2008; Henderson 2010; Lancaster et al. 2004; Schmitt et al. 2007; Tytell 2005), channel formation (Arispe and De Maio 2000; Arispe et al. 2004), and promotion of apoptosis (Broquet et al. 2003; Gastpar et al. 2005; Gehrmann et al. 2008; Multhoff and Botzler 1998; Multhoff et al. 1998; Multhoff and Hightower 1996; Schilling et al. 2009). Intracellularly, Hsp70s’ interaction with lipids prevents cell death by stabilizing the lysosomal membrane (Kirkegaard et al. 2010), regulates cell survival by mediating microautophagy (Sahu et al. 2011), and acts as cytosolic carrier of the endogenous cannabinoid lipid anandamide (Oddi et al. 2009).
Several reports have described the biochemical properties of Hsp70 lipid binding. These reports revealed that the binding of HspA1A to PS and other lipids depends on the lipid environment and that the protein is most probably using different mechanisms and different regions to bind to particular lipids (Arispe et al. 2004; Armijo et al. 2014; Harada et al. 2007, 2015; Kirkegaard et al. 2010; Mahalka et al. 2014; Mamelak and Lingwood 2001; Mamelak et al. 2001; McCallister et al. 2015; Schilling et al. 2009; Whetstone and Lingwood 2003). Yet, the molecular mechanism of the interaction between HspA1A and different anionic lipids remains elusive. Based on these reports, we hypothesized that HspA1A lipid binding is dictated by the physiochemical properties of the lipid head group and is mediated by multiple types of molecular forces and binding sites. To test these hypotheses, we generated recombinant HspA1A and determined how its binding to five phospholipids is affected by the presence of ions and molecules important for the HspA1A chaperone function.
Materials and methods
Lipids, chemicals, and reagents
Phosphatidylcholine [1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC; PC)], phosphatidylethanolamine [1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (PE)], phosphatidylserine [1,2-dioleoyl-sn-glycero-3-phospho-l-serine (sodium salt) (DOPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (sodium salt) (POPS), 1,2-dipalmitoyl-sn-glycero-3-phosphoserine (sodium salt) (DPPS; PS)], phosphatidic acid [1,2-dipalmitoyl-sn-glycero-3-phosphate (sodium salt) (PA)], and phosphatidylglycerol [1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (sodium salt) (PG)] were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Other common chemicals and reagents, e.g., antibiotics, buffers, and growth media, were obtained from Fisher Scientific or Sigma-Aldrich (St. Louis, MO).
Generation of recombinant DNA clones
The mouse cDNA clone containing the hspA1A gene sequence, accession number BC054782, was purchased from OpenBiosystems (GE Dharmacon). HspA1A, as a typical Hsp70, contains an N-terminal 44-kDa nucleotide binding domain (NBD), an 18-kDa substrate-binding domain (SBD), and a C-terminal region of variable length. The NBD and SBD are connected via a hydrophobic linker. Clones corresponding to the full-length (FL) HspA1A gene and its NBD (amino acids 1–395; includes the linker) and SBD (amino acids 384–641; includes the linker and the C-terminal lid domain) fragments were generated by the polymerase chain reaction (PCR) method using specific oligonucleotide primers. The primer sequences used as well as the restriction enzymes (NdeI and XhoI; underlined nucleotides) that were incorporated for directional cloning of the genes are given below. FL-forward primer was CCGCATATGATGGCCAAGAACACGGCG, and FL-reverse primer was CAGCTCGAGATCCACCTCCTCGATGGT, CAACTCGAG GTCCAGCAGCAGCAGGTC for the NBD fragment (paired with the FL forward primer), and CCGCATATGAAGTCGGAGAACGTGCAG for the SBD fragment (paired with the FL reverse primer).
The amplified DNA fragments were then cloned into the protein expression vector pET-22b + (Novagen) using the Rapid DNA Ligation Kit (Roche) following the manufacturer’s protocol. The ligation mixtures were later transformed in Escherichia coli strain DH5α cells (Life Technologies), the positive colonies were verified by PCR, and the intact open reading frames were verified by DNA sequencing.
Generation and purification of recombinant proteins
Purified plasmid DNA of sequence-verified recombinant clones was subsequently transformed into BL21(DE3) E. coli cells (Life Technologies). A single colony was then added to 15 mL of Luria-Bertani (LB) broth with ampicillin (100 μg/mL) and grown until an OD of between 0.8 and 1.0 was reached. Recombinant protein production was induced using 1 mM (final concentration) of Isopropyl β-d-1-thiogalactopyranoside (IPTG) at 25 °C for 14–16 h. The cultures were pelleted by centrifugation, and the cells were lysed in a lysis buffer containing 50 mM sodium phosphate, pH 7.4, and 300 mM sodium chloride. During lysis, phenylmethylsulfonyl fluoride (PMSF) (1 mM), lysozyme (0.5 mg/mL), and Triton-X (1 %) were added, and the lysates were sonicated until optically clear. After sonication, the lysates were rotated at 4 °C for 30 min and were centrifuged at 10,000×g for 5 min. The supernatant, containing the soluble Hsp70 proteins, was mixed with Cobalt agarose beads (Pierce), equilibrated in the same buffer and rotated, at 4 °C for 1 h. The samples were then centrifuged at 700×g for 2 min, and the beads were washed 3× with the same buffer to remove proteins that did not interact with the cobalt beads. Finally, the recombinant proteins were eluted from the beads by incubation with equal volume of lysis buffer containing 150 mM imidazole. The elutions were then dialyzed extensively against a 25 mM Tris–HCl or 25 mM HEPES, pH 7.4, buffer using Amicon Ultra centrifugal filters.
The protein concentration was determined using the Coomassie Blue Plus Protein Assay Reagent (Pierce) following the protocol supplied by the manufacturer. Protein purity was assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Separated proteins were detected by staining with Simply Blue™ Safe Stain (Life Sciences). For Western blotting, separated proteins were transferred to nitrocellulose (Protran; Whatman) and blocked with 5 % milk powder, in 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, and 0.05 % Tween 20 (TBST) for 1 h at room temperature. Western blots were probed with a polyclonal anti-His antibody (Thermo Scientific; 1:2000 in TBST) overnight at 4 °C. The secondary antibody, peroxidase-conjugated goat anti-rabbit immunoglobulin (Thermo Scientific; 1:10000 in TBST), was incubated with the nitrocellulose for 1 h at room temperature. Bound antibody was visualized with the Pierce ECL Western Blotting Substrate (Pierce). All gels and signals were collected using the Omega Lum C system from Aplegen.
To determine the effect of the histidine-tag on the lipid-binding properties of HspA1A, a tag-free mouse recombinant protein (ADI-SPP-502-F) was purchased from Enzo Life Sciences.
Lipid binding assays
The proteins were then tested for their ability to bind to lipids using the well-established lipid vesicle sedimentation (LVS) assay (Narayan and Lemmon 2006). This method involves the quantitative pelleting of liposomal vesicles from a lipid/protein mixture. The extent of lipid-protein binding was then evaluated by comparing the amount of protein that remained in the supernatant to the amount of protein that was pelleted. The assay requires that the protein does not precipitate, and its precipitation is not promoted by the presence of the lipid due to nonspecific binding. Therefore, we used proteins that do not precipitate and several control-lipids (Narayan and Lemmon 2006). Control lipids are lipids that have similar or identical hydrophobic tails to lipids that Hsp70s bind but have different head groups and do not bind or show minimal binding to HspA1A (for example PC or PE are control lipids for PS).
In all experiments, PC, the major constituent of eukaryotic plasma membranes, showed very low basal binding to both proteins, and thus, it was used as a backbone lipid. Next, liposomal vesicles that resemble physiological membranes were generated by mixing PC with the lipid of interest in ratios that parallel those in cellular membranes (van Meer et al. 2008). Specifically, the specified molar ratios used were PC/PE (70:30), PC/PS (80:20), PC/PA (90:10), and PC/PG (90:10). The lipid mixtures were dried under vacuum for approximately 40 min and then hydrated at room temperature for 1 h in a buffer containing 25 mM HEPES, pH 7.4, and 100 mM NaCl (HBS) and vortexed frequently. Within 30 min of hydration, the samples were probe-sonicated for 10 × 1-s bursts. After 1-h hydration, multilayered vesicles were formed. To generate the final uniform population of small, unilamellar vesicles, the samples were subjected to 8 cycles of freezing in liquid nitrogen and thawing in a bath sonicator (45 °C, 10 min sonics) until optically clear. The binding reactions contained a fixed concentration (1 μM) of Hsp70 in a total reaction volume of 100 μl and were incubated at 30 °C for 30 min.
After the incubation period, the samples were transferred to ultracentrifuge tubes and ultracentrifuged at 166,000×g at 25 °C for 40 min. After centrifugation, 85 μl of the supernatant was removed and saved in microcentrifuge tubes. The remaining 15 μl of supernatant at the interface of the pellet was discarded. The pellets were then resuspended in equal volume of HBS. Equal volumes of supernatant and pellet fractions, which contained unbound and bound to liposomes proteins, respectively, were separated on an SDS-PAGE as described above. The gels were stained as described above, and relative protein amounts were quantified by densitometry using the UltraQuant Omega Lum C (Aplegen) software. All experiments were repeated at least three times using different batches of protein, except the ones that used the Enzo protein.
The LVS method performed in HBS buffer alone reveals whether a protein associates with a lipid in a quantitative manner. However, it does not discriminate between peripheral binding and embedding. Therefore, in some reactions, 500 mM KCl was included. In these reactions containing the salt, the hydration and reaction buffer, HBS was supplemented with equal concentrations of KCl to ensure liposomal integrity. In the pH experiments apart from HBS, MES [2-(N-morpholino)ethanesulfonic acid] at pH 5.5 and 4.5 was used. In these experiments instead of the standard lipid concentration (1 mM), a much lower lipid concentration was used (0.2 mM), because at lower pH values and high lipid concentrations, the binding was 100 % and it was impossible to determine the effects of pH using this assay. The reactions for ATP, ADP, and protein substrate contained 0.5 mM of both MgCl2 and KCl. These reactions were incubated at 30 °C for 1 h to allow testing of whether the order of addition mattered. Specifically, reactions were included where the lipid vesicles and the molecule (nucleotide or substrate) being tested were added simultaneously from the beginning of the reaction incubation time. Another set of reactions was also included where the lipid vesicles were included in the reactions from the beginning and the molecule being tested was added 30 min into the reaction time and incubated together for an additional 30 min.
To determine the extent of liposomal aggregation because of the addition of CaCl2 reactions containing liposomes with different lipids at the specified ratios (see above) that were used, the aggregation (turbidity of the solution) was measured at 340 nm at 15, 30, and 60 min of incubation at 30 °C in the presence or absence of CaCl2.
Statistical tests
Data were graphed and analyzed using SigmaPlot (version 10.0, Systat Software Inc.). The same program was also used to calculate mean binding and standard deviations. Statistical significance was determined by an unpaired t test. A P value <0.05 was considered statistically significant.
Results
Generation, expression, and purification of full length HspA1A gene products and domain fragments
The polymerase chain reaction was employed to amplify the full-length (FL) HspA1A gene sequence and two fragments corresponding to the nucleotide-binding domain (NBD) and substrate-binding domain (SBD) using specifically designed oligonucleotide primers. Then, the amplified products were cloned, expressed, and purified (Fig. 1). The apparent molecular weights of the proteins were determined by SDS-PAGE (Fig. 1a). Transfer of the purified protein products to nitrocellulose and probing with anti-His antibody demonstrated equal reactivity with all products, showing a single major immunoreactive species in each case (Fig. 1b). For the FL recombinant proteins, we also determined that they prevent protein aggregation, promote protein refolding, and hydrolyze ATP (as described in McCallister et al. 2015).

Purified recombinant HspA1A protein and its major domains. SDS-polyacrylamide gel electrophoresis of the His-tagged HspA1A and its domain fragments visualized with a Coomassie Blue staining and b Western blot using a rabbit polyclonal anti-His antibody. m molecular weight marker, FL full-length HspA1A, NBD protein fragment corresponding to the nucleotide-binding domain, SBD protein fragment corresponding to the substrate-binding domain
Effects of potassium chloride (KCl) on HspA1A lipid binding
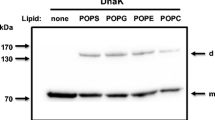
The LVS method using HBS buffer alone does not discriminate between peripheral binding and protein embedding within the lipid bilayer. To determine whether HspA1A embeds in the lipid bilayer or binds to lipid vesicles peripherally, lipid binding was assessed in the absence (0 mM KCl) and presence of high KCl concentrations (500 mM) (Fig. 2). The expectation was that if HspA1A is peripherally associated with liposomes containing a particular lipid, then high levels of salt would decrease the amount of protein bound to the vesicles because high salt concentrations disrupt electrostatic and minimally hydrophobic interactions, but they will not affect proteins embedded in the bilayer (Ben-Tal et al. 1997; Hanshaw et al. 2008).

HspA1A binds to several anionic lipids and embeds in the lipid bilayer. A single concentration of liposomes (1 mM total lipid used but approximately 0.5 mM is available in the outer layer of the lipid bilayer) composed of different lipid mixtures in specified molar ratio (see figure and “Materials and Methods”) was incubated with 1 μM of protein in the absence or presence of 500 mM KCl. After centrifugation, the extent of lipid-protein binding was determined by comparing the amount of protein that remained in the supernatant to the amount of protein that was pelleted. a Representative SDS-PAGE gel electrophoresis of the supernatant (S) and pellet (P) fractions of HspA1A with liposomes composed of different PC and PS lipid species. b Quantification of the binding of HspA1A to liposomal vesicles composed of different lipid mixtures in a specified molar ratio (see figure and Materials and Methods). The graphs are expressed as percentage of protein bound to lipid vesicles (Y-axis). NL no lipid controls. For clarity, only the lipid of interest is shown in the figure. Error bars represent standard deviations for three independent experiments. Star denotes statistical significance (α = 0.05). The p value obtained by comparing the binding at the presence of potassium was PG-KCl, p value = 0.0052
We first determined that HspA1A binds minimally and peripherally to vesicles composed of DOPC, POPC, DPPC, and DPPC/DPPE (Fig. 2). This result is consistent with previous reports showing that HspA1A does not specifically interact with PC and PE, two neutral lipids (Armijo et al. 2014; Harada et al. 2007; Mamelak et al. 2001; McCallister et al. 2015). We also observed that HspA1A binds to vesicles containing 20 % DOPS, POPS, or DPPS in the absence of 500 mM of KCl. However, the binding to vesicles, containing 20 % DOPS and POPS, decreases drastically in the presence of 500 mM KCl, while it is retained for vesicles composed of 20 % DPPS (Fig. 2a). These observations suggest that the protein embeds maximally in vesicles composed of 20 % DPPS, while it associates peripherally with vesicles containing 20 % of either DOPS or POPS (Fig. 2a). These data are consistent with the study of Armijo et al. (2014), which showed that embedding of HspA1A in the lipid bilayer increases with the saturation level of PS.
We next quantified the binding of HspA1A to vesicles composed of DPPC/DPPS (80:20; abbreviated as PC and PS in the remaining of the text, respectively), DPPC/DPPA (90:10; PA), and DPPC/DPPG (90:10; PG) in the absence and presence of 500 mM KCl. This assay determined that the binding to PS was reduced by 3 %, the binding to PA was increased by approximately 3 %, and the binding to PG vesicles was reduced by 10 % after the addition of 500 mM KCl (Fig. 2b). The maintenance of HspA1A binding to PS was further established with the addition of 500 mM Na2CO3 (pH 11.0) in the reaction mixture (Supplementary Fig. 1). These relatively small changes suggest that almost all vesicle-associated HspA1A is embedded in the lipid bilayer composed of PS and PA, while ∼80 % of the vesicle-associated HspA1A is embedded in bilayers composed of PG.
Lipid-binding properties of the FL HspA1A and its major domains
To determine which region of the HspA1A is responsible for binding to lipids, the LVS assay with a single concentration of lipids was used. This quantitative assay used the full length HspA1A (FL) protein and two polypeptides corresponding to the two major domains, the NBD and SBD (Fig. 1).
The results showed that the NBD binds to liposomal vesicles much higher than the SBD (Fig. 3b). Based on the almost equal amounts of protein bound, it is evident that the HspA1A PS association is mediated mainly by the NBD region of the protein. Differently, the binding of the NBD to both PA and PG is significantly lower as compared to the FL protein (Fig. 3b). To further characterize the binding of the two domains to PS, PA, and PG, the LVS was performed using an increasing concentration of lipids (Fig. 3c, d). These experiments revealed that the NBD-PS association highly increased as the lipid concentration increased, while this increase was much less profound for PA and PG. In contrast, the SBD increased slightly or showed no change with the increase in lipid concentration. These results reveal that the NBD-lipid association is higher and more specific than the SBD-lipid association.

HspA1A binds to anionic lipids mainly by its nucleotide-binding domain. HspA1A, 1 μM, was incubated with a single or multiple concentration of liposomes composed of different lipid mixtures (see figure and Materials and Methods for ratios; FL full length, NBD nucleotide-binding domain, SBD substrate-binding domain). For clarity, only the lipid of interest is shown in the figure. The protein-liposome mixture was centrifuged, and after centrifugation, the extent of lipid-protein binding was determined by comparing the amount of protein that remained in the supernatant to the amount of protein that was pelleted. a Representative SDS-PAGE gel electrophoresis of the supernatant (S) and pellet (P) fractions of FL, NBD, and SBD protein fragments with liposomes composed of different lipids. b–d Quantification of the binding between the FL, NBD, and SBD of HspA1A with different lipids using a single (b) and several (c, d) lipid concentrations. The graphs are expressed as percentage of protein bound to lipid vesicles (Y-axis). NL no lipid controls. Error bars represent standard deviations for three independent experiments. Stars denote statistical significance (α = 0.05). The p values obtained by comparing the FL to the NBD or SBD binding values were PC-SBD, p value = 0.0067; PE-SBD, p value = 0.0467; PS-SBD, p value = 0.0013; PA-NBD, p value = 0.0094; PA-SBD, p value = 0.0034; PG-NBD, p value = 0.0104; PG-SBD, p value = 0.0040
These data suggest that in the case of PS, the NBD mediates most of the lipid association, while in the case of PA and PG, the FL protein (containing both domains) is required for membrane association. However, the LVS method as used does not discriminate between peripheral binding and embedding. Therefore, we accessed the binding of both domains in the presence of 500 mM KCl to determine whether they embed in or interact peripherally with the membrane. The results showed that the binding of both domains significantly decreased in the presence of 500 mM KCl (Fig. 4a–c). These data revealed that although the FL HspA1A embeds in the lipid bilayer (Fig. 2), its individual domains (NBD and SBD) interact with the liposomal membrane peripherally. Furthermore, in competition experiments, the FL HspA1A competed the NBD-lipid association (Fig. 4d, e). Together, these observations suggest that in all lipids used, the FL HspA1A is necessary for membrane binding and insertion.

The nucleotide and substrate-binding domains of HspA1A bind peripherally to liposomal vesicles composed of different lipids. The vesicle sedimentation assay was performed by incubating a single concentration of liposomes composed of different lipid mixtures in a specified molar ratio (see figure and Materials and Methods) with 1 μM of protein in the absence or presence of 500 mM KCl. a Representative SDS-PAGE gel electrophoresis of the supernatant (S) and pellet (P) fractions of NBD and SBD with liposomes. b–c Quantification of the binding of NBD and SBD to liposomal vesicles composed of different lipid mixtures in a specified molar ratio (see figure and Materials and Methods). The graphs are expressed as percentage of protein bound to lipid vesicles (Y-axis). Error bars represent standard deviations for three independent experiments. Stars denote statistical significance (α = 0.05). The p values obtained by comparing the binding at the presence of potassium were for NBD: PS-KCl, p value = 0.0120; PA-KCl, p value = 6.4960e−3; PG-KCl, p value = 7.4404e−3; and for SBD: PS-KCl, p value = 0.0388; PA-KCl, p value = 0.0139; PG-KCl, p value = 0.0185. d-e The FL HspA1A competes with the NBD for binding to lipids. d Representative SDS-PAGE gel electrophoresis of the supernatant (S) and pellet (P) fractions. e Quantification of the binding between the FL HspA1A with liposomes in the presence or absence of the NBD. The p values obtained by comparing the binding of NBD at the presence of FL were PS-NBD, p value = 0.0030; PA-NBD, p value = 0.01212
Effects of reaction buffer pH on HspA1A lipid binding
To determine whether pH alters the lipid-binding properties of HspA1A, the LVS assay was performed using reaction buffers equilibrated in three different pH values: 7.4, 5.5, and 4.5 (Fig. 5). In these experiments, both histidine-tagged and tag-free recombinant HspA1A were used. These control experiments revealed that the tag did not interfere with the lipid binding (Fig. 5a). We then quantified the effect of the pH on HspA1A lipid binding (Fig. 5b) and found that HspA1A binding to PC slightly increased at pH 5.5 but was not significantly different between pH 7.4 and 4.5. However, the binding between HspA1A and PS or PA increased by approximately 5–10 % as the reaction pH became more acidic. Differently, the association between HspA1A and PG increased by approximately 50 % at pH 4.5. These results suggest that the total protein charge, as well as the amino acid, and lipid head group protonation states are important determinants for the HspA1A-association to all lipids tested and this is more pronounced for PG.

HspA1A lipid binding increases as the reaction pH becomes acidic. The vesicle sedimentation assay was performed by incubating a single concentration of liposomes composed of different lipid mixtures in a specified molar ratio (see figure and Materials and Methods) with 1 μM of protein using reaction buffers with different pH. a Representative SDS-PAGE gel electrophoresis of the supernatant (S) and pellet (P) fractions of his-tagged and tag-free HspA1A with two concentrations of liposomes composed of DPPC/DPPS (80:20) at different pH values. b Quantification of the binding of HspA1A to liposomal vesicles. The graphs are expressed as percentage of protein bound to lipid vesicles (Y-axis). For clarity, only the lipid of interest is shown in the figure. NL no lipid controls. Error bars represent standard deviations for three independent experiments. Stars denote statistical significance (α = 0.05). The p values obtained by comparing the binding at pH 7.4 to the binding at pH 5.5 and 4.5 were PS-5.5, p value = 0.0004; PS-4.5, p value = 0.0294; PA-5.5, p value = 0.0440; PA-4.5, p value = 0.0225; PG-5.5, p value = 0.0329; PG-4.5, p value = 0.0152
Effects of low concentrations of calcium, magnesium, and potassium on HspA1A lipid binding
To determine whether calcium and other ions known to affect protein-lipid interactions are important for the molecular chaperone function of HspA1A, the LVS assay was performed by including in the reaction various concentrations of CaCl2, MgCl2, and KCl (Fig. 6a). These results show that binding of HspA1A to PS is unaffected by low concentrations of these cations. Additionally, these results reveal that calcium is not required for binding; thus, HspA1A does not use a calcium bridge to bind to anionic lipids.

The interaction of HspA1A with anionic lipids is not altered by low concentrations of Ca2+, Mg2+, and K+ and does not depend solely on electrostatic interactions. a The vesicle sedimentation assay was performed by incubating a single concentration of liposomes composed of DPPC/DPPS (80:20) with 1 μM of protein in the absence or presence of 0.5 and 1 mM of CaCl2, MgCl2, or KCl. b The same assay was performed in the presence of 10 mM CaCl2 for liposomes composed of different lipid mixtures in a specified molar ratio (see figure and Materials and Methods). For clarity, only the lipid of interest is shown in the figure. Error bars represent standard deviations for three independent experiments. Stars denote statistical significance (α = 0.05). The p values obtained by comparing the binding in the presence of calcium were PS-CaCl2, p value = 0.0500; PG-CaCl2, p value = 0.0020
Furthermore, to assess the importance of electrostatic interactions involved in the binding of HspA1A to PS, PA, and PG, 10 mM of CaCl2 was used (Fig. 6b). The expectation was that because mid-range concentrations of calcium disrupt electrostatic interactions and calcium may be used to bridge the protein and the lipid, a decrease in the average percent of protein bound would indicate that HspA1A binds to lipids either using largely electrostatic interactions or calcium bridges (Hanshaw et al. 2008). Our results showed that in the presence of 10 mM calcium, binding of HspA1A to PS decreased by approximately 5 % and binding to PG decreased by approximately 10 %, whereas binding to PA was largely unaffected (Fig. 6b).
To control for spontaneous liposomal aggregation caused by the CaCl2 alone, which may interfere with protein-lipid association, the extent of aggregation was measured (Supplementary Fig. 2). No increase in liposomal aggregation was observed under these conditions. Therefore, we propose that Ca2+ ions, under the conditions and lipid ratios used, are masking the charge of the lipid head as was showed by a recent report (Matsunaga et al. 2015). In turn, this assumption suggests that the observed small but consistent changes observed for PS and PG (Fig. 6b) are due to the disruption of electrostatic interactions between positively charged regions of the protein and the negatively charged lipid head.
Effects of ATP, ADP, and protein substrate on the lipid-binding properties of HspA1A
To determine whether molecules important for the chaperone function and oligomerization state of HspA1A alter its lipid binding properties nucleotides (ATP, ADP, and ATPγS) as well as protein substrate [peptide NIVAKKK (Takenaka et al. 1995)] were included in the LVS assay in a buffer containing both Mg2+ and K+ (Fig. 7a).

The interaction of HspA1A and lipids is inhibited by the presence of ATP, ADP, ATPγS but not by the presence of a particular protein substrate. The vesicle sedimentation assay was performed by incubating a single concentration of liposomes composed of different lipid mixtures in a specified molar ratio (see figure and Materials and Methods) with 1 μM of protein in the absence or presence of 1 mM nucleotide or 1 μM substrate. Two sets of reactions were used. In the first one, labeled as simultaneously, the lipid vesicles and the nucleotide or substrate were added simultaneously in the reaction and incubated with the protein for 1 h. In the second set of reactions, labeled as lipid first, the lipid vesicles were incubated with the protein for 30 min, and then the nucleotide or substrate was added and incubated together for an additional 30 min. a Representative SDS-PAGE gel electrophoresis of the supernatant (S) and pellet (P) fractions. b–e Quantification of the effect of the presence of nucleotide or substrate in the binding of HspA1A to lipids. For clarity, only the lipid of interest is shown in the figures. Error bars represent standard deviations for three independent experiments. Stars denote statistical significance (α = 0.05). The p values obtained by comparing the binding at the presence of ATP (sim simultaneously and lf lipid first) were PS-sim, p value = 0.0025463; PS-lf, p value = 0.0184; PA-sim, p value = 0.008957; PA-lf, p value = 0.0304; PG-sim, p value = 0.0324; PG-lf, p value = 0.0224. The p values obtained by comparing the binding at the presence of ADP (sim simultaneously and lf lipid first) were PS-sim, p value = 0.0034; PS-lf, p value = 0.0063; PA-sim, p value = 0.0117; PA-lf, p value = 0.0182; PG-sim, p value = 0.0342; PG-lf, p value = 0.0223. The p value obtained by comparing the binding at the presence of ATPγS was p = 1.0543e−05. The p value obtained by comparing the binding at the presence of protein substrate (lf lipid first) was PA-lf, p value = 0.0394
These experiments showed that in the presence of both ATP and ADP, the interaction of HspA1A with PC was not affected. However, binding of HspA1A to PS significantly decreased by almost 40 % when ATP or ADP and the lipid were added simultaneously. Differently, when HspA1A was pre-incubated with PS liposomes, the reduction in binding was less drastic (15 %) (Fig. 7b, c). Binding of HspA1A to PA and PG significantly decreased when ATP or ADP was added simultaneously and when the protein was pre-incubated with the lipid vesicles (Fig. 7b, c). To determine whether the observed effect was due to the binding or hydrolysis of ATP, a nonhydrolyzable ATP analog, ATPγS was used (Fig. 7d). These experiments revealed that both ATP and ATPγS inhibit PS binding indicating that ATP binding rather than hydrolysis affects the interaction of HspA1A with lipids as has been also showed by Arispe et al. (2002). These data suggest that binding of ATP or ADP to HspA1A inhibits its association with all anionic lipids tested. However, these results do not reveal whether the nucleotides affect the protein binding to the liposomes, its membrane insertion, or both.
In contrast to the major changes induced by the presence of nucleotides, the presence of a particular peptide had minimal effects on the binding of HspA1A to the lipids tested. Specifically, the observed binding to PS or PG was decreased only by 2–3 % when the substrate was present (Fig. 7e). Differently, binding of HspA1A to PC was maximally increased by 4 %, while binding to PA was increased by 2 % when the substrate and the lipid were incubated simultaneously with HspA1A and decreased by 10 % when the lipid was pre-incubated with HspA1A and the substrate was added later.
Discussion
Our quantitative results show that HspA1A associates with the negatively charged lipids PS, PA, and PG, while its interaction with PC and PE is weak and probably nonspecific (Fig. 2). Consistent with previous reports (Arispe et al. 2002; Armijo et al. 2014; Harada et al. 2007, 2015; Kirkegaard et al. 2010; Mamelak and Lingwood 2001; McCallister et al. 2015), our results suggest that the association of Hsp70s with anionic lipids is specific. Nevertheless, the binding of Hsp70s to negatively charged lipid head groups could be the result of random physical attraction between positive amino acids and the negative lipid head group. However, several observations argue against this suggestion. First, both aromatic and positive amino acids have been implicated in the association between Hsp70s with BMP, PS, and SGC (Kirkegaard et al. 2010; Mahalka et al. 2014; Mamelak and Lingwood 2001; Sahu et al. 2011). Second, HspA1A embeds in the lipid bilayer composed of PS, PA, PG [Fig. 2b and (Armijo et al. 2014)], and SGC (Harada et al. 2015). Third, the binding of Hsp70s to lipids is not directly related to the charge of the lipid head group (Schilling et al. 2009). Fourth, addition of 10 mM CaCl2 (Fig. 6b) has small effects on binding. Fifth, acidic pH increases the HspA1A binding to most lipids tested (Fig. 5b). These observations strongly suggest that, although important, electrostatic interactions alone are insufficient to fully explain the observed binding. Therefore, the most plausible interpretation is that HspA1A lipid binding is specific and depends on the chemical nature of the lipid head group. Lipid-specific binding is also supported by the different theoretical binding models (McCallister et al. 2015) and the lipid-specific differences observed by us (Figs. 2b, 5b, and 6b) and others (Armijo et al. 2014; Harada et al. 2014, 2015; Mahalka et al. 2014; Mamelak and Lingwood 2001; Mamelak et al. 2001).
Arispe and De Maio (2000) and later Armijo et al. (2014) showed that HspA1A embeds in the lipid bilayer of PS liposomes. Our data expand this original observation to include PA and PG (Fig. 2). However, the three lipids do not show identical patterns because approximately 20 % of the PG associated HspA1A is peripherally bound (Fig. 2b). Furthermore, the binding as well as the embedding of HspA1A with liposomes containing PS increased as the fatty acid chains changed from unsaturated to saturated (Fig. 2a). This result is consistent with the study of Armijo et al. (2014) and further suggests that association of HspA1A with membranes increases as the membrane fluidity decreases. Together, these observations imply that HspA1A-membrane association would be increased in membrane regions enriched in saturated lipids, such as lipid rafts, and in cellular membranes containing a high number of saturated lipids. These suggestions are consistent with the presence of HspA1A and other Hsp70s at the plasma membrane of stressed and cancer cells, which have increased membrane rigidity (Baenke et al. 2013; De Maio 2014; Kirkegaard et al. 2010; Lancaster and Febbraio 2005a; Mambula et al. 2007; Multhoff 2007; Schilling et al. 2009).
Another observation from our study is the differential involvement of each protein domain to the association with lipids, which once more depends on the lipid tested. For example, the NBD of HspA1A shows higher binding than the SBD region for PS, PA, and PG (Fig. 3). However, the binding to NBD is much higher in the case of PS than in the cases of PA or PG (Fig. 3). These results suggest that in the case of PS, the NBD mediates most of the lipid association, while in the case of PA and PG, the FL protein is necessary. However, competition experiments showed that the FL protein competes the NBD (Fig. 4d, e). Furthermore, although the FL protein embeds in the lipid bilayer of PS liposomes, the NBD binds peripherally to the vesicles (Fig. 4). Taken together, these data suggest that for all lipids tested, including PS, the FL HspA1A is essential for membrane binding and insertion.
Although these results are congruent with previous reports on different Hsp70s showing that both protein domains are important for the association of the chaperone with lipids and this association is lipid-specific (Mamelak and Lingwood 2001; Mamelak et al. 2001) (Kirkegaard et al. 2010; Mamelak and Lingwood 2001; Sahu et al. 2011) (Armijo et al. 2014; Harada et al. 2007, 2014, 2015; Mahalka et al. 2014), they do not directly show which part of the protein embeds in the lipid bilayer. However, Armijo et al. (2014) showed that in the case of PS, the SBD region of HspA1A embeds within the lipid bilayer. Additionally, the association of HspA1A with PS, PA, and PG liposomes (Fig. 7), the channel activity and liposomal aggregation of HspA1A (Arispe and De Maio 2000; Arispe et al. 2002, 2004), the NBD-dependent oligomerization within SGC liposomes, and the binding to endosomes are all altered by ATP and ADP (Harada et al. 2015; Sahu et al. 2011). The above data strongly support the idea that the NBD region is accessible to the nucleotides and thus must be oriented outside of the liposomal membrane. Furthermore, the NBD binds to lipids quantitatively more than the SBD, but neither the NBD nor the SBD embeds in the lipid bilayer (Figs. 2, 3, and 4). Therefore, we theorize that the NBD region of HspA1A interacts with the lipid bilayer of all lipids studied causing a conformational change that inserts the SBD region within the lipid bilayer.
Another observation from our data is that HSPA1A-lipid association is reduced in the presence of nucleotides (ATP, ADP, and ATPγS), while a protein peptide has no major effect on it (Fig. 7). These results are consistent with several previous reports showing that ATP and ADP alter the association of HspA1A with lipids by affecting liposomal aggregation, altering HspA1A channel activity, and reducing membrane insertion and lipid-induced oligomerization (Arispe and De Maio 2000; Harada et al. 2015; Mamelak and Lingwood 2001). However, whether this reduction is due to loss of binding, loss of embedding, or both, as well as whether it is the result of direct competition all remain unclear.
The observed reduction of HspA1A-lipid association could be the result of either direct competition of the nucleotide and the lipid for the same binding sites, or change of the protein conformation and oligomerization states caused by nucleotide binding. Conceptually, the first explanation of direct competition for the same binding sites could explain the observed lipid-binding reduction (Fig. 7b, c) because of the much higher affinities of HspA1A for either nucleotide (nM range) (Arakawa et al. 2011; Buxbaum and Woodman 1996) as compared to its affinities for the lipids used (μM range) (McCallister et al. 2015). Also theoretically, the HspA1A-lipid association (binding and insertion) could be affected by changes in oligomerization caused by ATP, which favors Hsp70 monomers, and ADP, which favors oligomerization (Malinverni et al. 2015; Thompson et al. 2012). Additionally, ATP or ADP binding results into two distinct HspA1A conformations, which could also alter lipid binding.
However, direct competition is not favored in the case of HspA1A-PS association, because when the protein is pre-incubated with PS liposomes, the reduction caused by the nucleotide is much smaller, showing that they do not directly compete (Fig. 7b, c). Moreover, two independent reports have showed that the binding of Hsp70s to BMP and SGC (Kirkegaard et al. 2010; Mamelak and Lingwood 2001) is mediated by amino acids different than the ones known to bind to ATP or ADP. These two arguments favor the second explanation suggesting that changes of the conformation and oligomerization protein states caused by nucleotide-binding (Young 2010; Zhuravleva and Gierasch 2011) are responsible for the observed reduction in lipid binding. The second explanation is also favored by the fact that HspA1A embeds in PS liposomes via its SBD region (Armijo et al. 2014), and the finding that ATP and ADP strongly inhibit sulfatide-induced formation of the HspA1A oligomers only in the presence of KCl (Harada et al. 2015). Although these observations favor the conformation/oligomerization explanation, it remains unclear how both ATP and ADP (or ATPγS) result in lipid-binding reduction. One explanation could be that binding of ATP alters lipid binding by disassociating HspA1A oligomers, while ADP inhibits membrane insertion by causing a conformational change of the monomers.
In the context of the literature and the results discussed in this study, we conclude that (a) HspA1A lipid binding has specificity and is, in part, dictated by the physiochemical properties of the lipid and (b) multiple types of molecular forces are differentially involved in HspA1A-lipid association. Based on these, we infer that multiple lipid-binding sites exist on HspA1A and the protein binds to different lipids using different molecular mechanisms.
It has been postulated that lipid binding provides Hsp70s with the necessary specificity to localize and function at different membranes and pathways during cellular stress (De Maio 2011, 2014; McCallister et al. 2015). It is known that binding of HspA1A to BMP and HspA8 to PS prevents permeabilization of the lysosomal membrane and mediates microautophagy by late endosomes, respectively (Kirkegaard et al. 2010; Sahu et al. 2011). Also, HspA1A binds to several lipids involved at various stages of endocytosis (PA, PS, BMP, PI(5)P, and PI(3,4,5)P3) (McCallister et al. 2015; van Meer et al. 2008; Yang et al. 2008). Therefore, the ability of HspA1A to associate with lipids may allow the protein to participate in multiple stages of the endocytic pathway, e.g., vesicle formation, targeting, and fusion, which require precise regulation and numerous changes in lipid composition (Cho and Stahelin 2005; Yang et al. 2008). HspA1A may also interact with proteins that will be released, e.g., PrPC, a GPI anchor protein, and enhance their membrane localization and release (Wang et al. 2011). Furthermore, it has been postulated that HspA1A is directed to the membrane via its interaction with Gb3, where it recognizes and binds PS (Armijo et al. 2014; Gehrmann et al. 2008). At the membrane under particular conditions (increased lipid saturation and concentration), HspA1A may be inserted to the membranes, oligomerize, and either form channels, be secreted, or both. Extracellular HspA1A may differentially interact with SGC and other lipids, as well as acidic glycans and protein receptors at the surface of neighbor cells. Because binding of proteins to glycans, receptors, and lipids has been implicated in cell recognition, communication, and signaling of several systems including the immune, these interactions may allow HspA1A to participate in the immune response by activating specific cell types such as natural killer cells. Although attractive, these speculations require experimental validation, which will determine the conditions that favor HspA1A-lipid association and the amino acid residues that mediate this association.
References
Arakawa A, Handa N, Shirouzu M, Yokoyama S (2011) Biochemical and structural studies on the high affinity of Hsp70 for ADP. Protein Sci 20:1367–1379. doi:10.1002/pro.663
Arispe N, De Maio A (2000) ATP and ADP modulate a cation channel formed by Hsc70 in acidic phospholipid membranes. J Biol Chem 275:30839–30843. doi:10.1074/jbc.M005226200
Arispe N, Doh M, De Maio A (2002) Lipid interaction differentiates the constitutive and stress-induced heat shock proteins Hsc70 and Hsp70. Cell Stress Chaperones 7:330–338
Arispe N, Doh M, Simakova O, Kurganov B, De Maio A (2004) Hsc70 and Hsp70 interact with phosphatidylserine on the surface of PC12 cells resulting in a decrease of viability. FASEB J 18:1636–1645
Armijo G et al (2014) Interaction of heat shock protein 70 with membranes depends on the lipid environment. Cell Stress Chaperones. doi:10.1007/s12192-014-0511-x
Baenke F, Peck B, Miess H, Schulze A (2013) Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 6:1353–1363. doi:10.1242/dmm.011338
Ben-Tal N, Honig B, Miller C, McLaughlin S (1997) Electrostatic binding of proteins to membranes. Theoretical predictions and experimental results with charybdotoxin and phospholipid vesicles. Biophys J 73:1717–1727
Broquet AH, Thomas G, Masliah J, Trugnan G, Bachelet M (2003) Expression of the molecular chaperone Hsp70 in detergent-resistant microdomains correlates with its membrane delivery and release. J Biol Chem 278:21601–21606
Brown IR (2007) Heat shock proteins and protection of the nervous system. Ann N Y Acad Sci 1113:147–158
Browne CL, Swan JB, Rankin EE, Calvert H, Griffiths S, Tytell M (2007) Extracellular heat shock protein 70 has novel functional effects on sea urchin eggs and coelomocytes. J Exp Biol 210:1275–1287
Bukau B, Horwich AL (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92:351–366
Buxbaum E, Woodman PG (1996) Binding of ATP and ATP analogues to the uncoating ATPase Hsc70 (70 kDa heat-shock cognate protein). Biochem J 318(Pt 3):923–929
Chen S, Bawa D, Besshoh S, Gurd JW, Brown IR (2005) Association of heat shock proteins and neuronal membrane components with lipid rafts from the rat brain. J Neurosci Res 81:522–529
Cho W, Stahelin RV (2005) Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct 34:119–151. doi:10.1146/annurev.biophys.33.110502.133337
Daugaard M, Rohde M, Jaattela M (2007) The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 581:3702–3710
De Maio A (2011) Extracellular heat shock proteins, cellular export vesicles, and the Stress Observation System: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones 16:235–249
De Maio A (2014) Extracellular Hsp70: export and function. Curr Protein Pept Sci 15:225–231
Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, Multhoff G (2005) Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res 65:5238–5247
Gehrmann M, Brunner M, Pfister K, Reichle A, Kremmer E, Multhoff G (2004) Differential up-regulation of cytosolic and membrane-bound heat shock protein 70 in tumor cells by anti-inflammatory drugs. Clin Cancer Res 10:3354–3364
Gehrmann M et al (2008) Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS ONE 3, e1925
Guidon PT Jr, Hightower LE (1986a) The 73 kilodalton heat shock cognate protein purified from rat brain contains nonesterified palmitic and stearic acids. J Cell Physiol 128:239–245
Guidon PT Jr, Hightower LE (1986b) Purification and initial characterization of the 71-kilodalton rat heat-shock protein and its cognate as fatty acid binding proteins. Biochemistry 25:3231–3239
Hanshaw RG, Stahelin RV, Smith BD (2008) Noncovalent keystone interactions controlling biomembrane structure. Chemistry (Weinheim Bergstr Ger) 14:1690–1697
Harada Y, Sato C, Kitajima K (2007) Complex formation of 70-kDa heat shock protein with acidic glycolipids and phospholipids. Biochem Biophys Res Commun 353:655–660
Harada Y, Garenaux E, Nagatsuka T, Uzawa H, Nishida Y, Sato C, Kitajima K (2014) Interaction of 70-kDa heat shock protein with glycosaminoglycans and acidic glycopolymers. Biochem Biophys Res Commun 453:229–234. doi:10.1016/j.bbrc.2014.05.137
Harada Y, Sato C, Kitajima K (2015) Sulfatide-Hsp70 interaction promotes Hsp70 clustering and stabilizes binding to unfolded protein. Biomolecules 5:958–973. doi:10.3390/biom5020958
Henderson B (2010) Integrating the cell stress response: a new view of molecular chaperones as immunological and physiological homeostatic regulators. Cell Biochem Funct 28:1–14
Kirkegaard T et al (2010) Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 463:549–553
Lancaster GI, Febbraio MA (2005a) Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem 280:23349–23355
Lancaster GI, Febbraio MA (2005b) Mechanisms of stress-induced cellular HSP72 release: implications for exercise-induced increases in extracellular HSP72. Exerc Immunol Rev 11:46–52
Lancaster GI, Moller K, Nielsen B, Secher NH, Febbraio MA, Nybo L (2004) Exercise induces the release of heat shock protein 72 from the human brain in vivo. Cell Stress Chaperones 9:276–280
Lindquist S, Craig EA (1988) The heat-shock proteins. Annu Rev Genet 22:631–677
Mahalka AK, Kirkegaard T, Jukola LT, Jaattela M, Kinnunen PK (2014) Human heat shock protein 70 (Hsp70) as a peripheral membrane protein. Biochim Biophys Acta 1838:1344–1361. doi:10.1016/j.bbamem.2014.01.022
Malinverni D, Marsili S, Barducci A, De Los Rios P (2015) Large-Scale Conformational Transitions and Dimerization Are Encoded in the Amino-Acid Sequences of Hsp70 Chaperones. PLoS Comput Biol 11, e1004262. doi:10.1371/journal.pcbi.1004262
Mambula SS, Stevenson MA, Ogawa K, Calderwood SK (2007) Mechanisms for Hsp70 secretion: crossing membranes without a leader. Methods 43:168–175
Mamelak D, Lingwood C (2001) The ATPase domain of hsp70 possesses a unique binding specificity for 3'-sulfogalactolipids. J Biol Chem 276:449–456. doi:10.1074/jbc.M006732200
Mamelak D et al (2001) Hsp70s contain a specific sulfogalactolipid binding site. Differential aglycone influence on sulfogalactosyl ceramide binding by recombinant prokaryotic and eukaryotic hsp70 family members. Biochemistry 40:3572–3582
Matsunaga S, Yamada T, Kobayashi T, Kawai M (2015) Scanning tunneling microscope observation of the phosphatidylserine domains in the phosphatidylcholine monolayer. Langmuir 31:5449–5455. doi:10.1021/acs.langmuir.5b00859
McCallister C, Siracusa MC, Shirazi F, Chalkia D, Nikolaidis N (2015) Functional diversification and specialization of cytosolic 70-kDa heat shock proteins. Sci Rep 5:9363. doi:10.1038/srep09363
Multhoff G (2007) Heat shock protein 70 (Hsp70): membrane location, export and immunological relevance. Methods 43:229–237
Multhoff G, Botzler C (1998) Heat-shock proteins and the immune response. Ann N Y Acad Sci 851:86–93
Multhoff G, Hightower LE (1996) Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones 1:167–176
Multhoff G, Botzler C, Issels R (1998) The role of heat shock proteins in the stimulation of an immune response. Biol Chem 379:295–300
Narayan K, Lemmon MA (2006) Determining selectivity of phosphoinositide-binding domains. Methods 39:122–133
Oddi S et al (2009) Molecular identification of albumin and Hsp70 as cytosolic anandamide-binding proteins. Chem Biol 16:624–632. doi:10.1016/j.chembiol.2009.05.004
Petersen NH, Kirkegaard T, Olsen OD, Jaattela M (2010) Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle (Georgetown, Tex) 9(12):2305–2309
Sahu R et al (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20:131–139
Schilling D et al (2009) Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J 23:2467–2477. doi:10.1096/fj.08-125229
Schmitt E, Gehrmann M, Brunet M, Multhoff G, Garrido C (2007) Intracellular and extracellular functions of heat shock proteins: repercussions in cancer therapy. J Leukoc Biol 81:15–27
Takenaka IM, Leung SM, McAndrew SJ, Brown JP, Hightower LE (1995) Hsc70-binding peptides selected from a phage display peptide library that resemble organellar targeting sequences. J Biol Chem 270:19839–19844
Thompson AD, Bernard SM, Skiniotis G, Gestwicki JE (2012) Visualization and functional analysis of the oligomeric states of Escherichia coli heat shock protein 70 (Hsp70/DnaK). Cell Stress Chaperones 17:313–327. doi:10.1007/s12192-011-0307-1
Tytell M (2005) Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int J Hyperth 21:445–455
van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124
Vega VL et al (2008) Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol 180:4299–4307
Wang GH, Zhou XM, Bai Y, Yin XM, Yang LF, Zhao D (2011) Hsp70 binds to PrPC in the process of PrPC release via exosomes from THP-1 monocytes. Cell Biol Int 35:553–558. doi:10.1042/CBI20090391
Whetstone H, Lingwood C (2003) 3'Sulfogalactolipid binding specifically inhibits Hsp70 ATPase activity in vitro. Biochemistry 42:1611–1617
Yang JS et al (2008) A role for phosphatidic acid in COPI vesicle fission yields insights into Golgi maintenance. Nat Cell Biol 10:1146–1153. doi:10.1038/ncb1774
Young JC (2010) Mechanisms of the Hsp70 chaperone system. Biochem Cell Biol - Biochim Biol Cell 88:291–300
Zhuravleva A, Gierasch LM (2011) Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc Natl Acad Sci U S A 108:6987–6992
Acknowledgments
This work was supported by start-up funds from California State University, Fullerton, a California State Mini Grant, and a grant from CSU Program for Education and Research in Biotechnology to NN. CM was supported by a Howard Hughes Medical Institute Scholarship. BK was supported by the Research Careers Preparatory Program at CSUF. The authors thank Dr. Dimitra Chalkia and Kyle Hess for their valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 323 kb)
Rights and permissions
About this article

Cite this article
McCallister, C., Kdeiss, B. & Nikolaidis, N. Biochemical characterization of the interaction between HspA1A and phospholipids. Cell Stress and Chaperones 21, 41–53 (2016). https://doi.org/10.1007/s12192-015-0636-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-015-0636-6