
Abstract
Patients with indolent non-Hodgkin lymphoma (iNHL) typically respond to first-line immunochemotherapy, but relapse is common. Treatment options for relapsed iNHL include chemotherapy ± rituximab and rituximab monotherapy. Lenalidomide plus rituximab (R2) is an immunomodulatory regimen that enhances rituximab-mediated cytotoxicity and improves clinical activity in iNHL. AUGMENT was a double-blind phase III randomized trial of R2 vs. rituximab + placebo (R-placebo) in patients with relapsed/refractory follicular lymphoma or marginal zone lymphoma who were not refractory to rituximab. The primary endpoint was progression-free survival (PFS). Data reported here focus on Japanese patients from AUGMENT and reflect 36 patients (n = 18, each group). PFS was superior in the R2 group, HR = 0.32 (95% CI 0.11–0.96). Median PFS was not reached (95% CI 19.7–NE) in the R2 group vs. 16.5 months (95% CI 11.3–30.6) in the R-placebo group. Grade 3/4 adverse events were more frequent in patients treated with R2 (67%) than with R-placebo (22%), primarily attributable to increased neutropenia (50% vs 17%). R2 resulted in significantly longer median PFS than R-placebo in Japanese patients with R/R iNHL, and the efficacy and the safety profile of R2 were similar to those reported in the global population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Indolent non-Hodgkin lymphoma (iNHL) is mostly of B-cell origin [1] and is generally highly responsive to initial therapy, but most patients experience relapse and require multiple lines of treatment [1,2,3,4]. The most common types of iNHL are follicular lymphoma (FL) and marginal zone lymphoma (MZL), and the proportion of FL (15%/14%) and MZL (8%/8%) in NHL is similar in US/Japanese populations [5]. Incidence rates of NHL, including FL and MZL, in Japan have more than doubled since the early 1990s [5], and although survival for patients with iNHL has improved, relapse continues to be a problem [6].
Currently, there is a broad range of treatment options for patients with relapsed/refractory (R/R) FL, with different efficacy/toxicity profiles [7,8,9]. Patients with R/R MZL are managed using the same options as in patients with R/R FL, with the exception of those with the localized mucosa-associated lymphoid tissue (MALT) subtype [10]. Rituximab is approved as monotherapy by the US Food and Drug Administration and the Japan’s Pharmaceuticals and Medical Devices Agency for CD20 + B-cell NHL [11, 12] and re-treatment with rituximab monotherapy is an option for patients after relapse on a rituximab-containing therapy [8, 12]. Lenalidomide is an IMiD immunomodulatory agent that causes degradation of the transcription factors Aiolos and Ikaros [13, 14] leading to anti-lymphoma and immune-modulation activities. In preclinical studies, lenalidomide restored the response of tumor-infiltrating lymphocytes [15] and increased natural killer cell count and functional activity [16, 17]. Combining lenalidomide + rituximab (R2) has resulted in enhanced antibody-dependent cell-mediated cytotoxicity, immune synapse formation, monocyte-mediated killing, and direct cytotoxicity against FL cells compared with either treatment alone [18].
R2 has shown high response rates in multiple phase II studies in patients with R/R and previously untreated iNHL [16, 19,20,21,22,23]. R2 has also shown similar efficacy and favorable safety profile compared to rituximab-containing chemotherapy in a phase III study of patients with advanced, previously untreated FL [24].
The goal of the AUGMENT phase III study was to prospectively compare the efficacy and safety of R2 vs rituximab + placebo (R-placebo) in patients with R/R iNHL. Recently, results of the AUGMENT study showed statistically superior efficacy, as measured by the primary endpoint—progression-free survival (PFS)—and other secondary endpoints, of R2 vs R-placebo in patients with R/R iNHL [25]. Considering the different treatment landscapes for iNHL globally, subgroup analysis was performed in Japanese patients enrolled in the AUGMENT study, and the results are presented here.
Patients and methods
Eligibility criteria
Patients with MZL or FL (grades 1–3a [transformed FL excluded]) requiring treatment per investigator assessment and aged ≥ 18 years were eligible. Eligibility also included previous treatment with ≥ 1 prior chemotherapy, immunotherapy, or chemoimmunotherapy and ≥ 2 previous doses of rituximab; documented relapsed, refractory, or progressive disease (PD), and not rituximab-refractory. Full details of all inclusion and exclusion criteria are reported in the primary AUGMENT publication [25].
Treatment protocol
A brief summary of the patient population and study method is provided here; for full details, please refer to the primary AUGMENT publication [25]. This was a single-country analysis from the AUGMENT trial, a multicenter, double-blind, randomized, phase III trial designed to evaluate the efficacy and safety of R2 vs R-placebo in Japanese patients with R/R iNHL (NCT01938001). Patients were randomly assigned in a 1:1 ratio to receive R2 or R-placebo and stratified according to previous rituximab treatment (yes or no), time since last anti-lymphoma therapy (≤ 2 vs > 2 years), and histology (FL vs MZL). Treatment was given for a maximum of 12 cycles or until relapse, PD, withdrawal of consent, or unacceptable toxicity. R2 dosing consisted of oral lenalidomide 20 mg/day (10 mg if creatinine clearance was 30–59 mL/min) on days 1–21, plus intravenous rituximab 375 mg/m2 on days 1, 8, 15, and 22 of cycle 1 and day 1 of cycles 2–5, in 28-day cycles. R-placebo was given on the same dosing schedule. Upon treatment completion or discontinuation, patients were followed for efficacy, subsequent anti-lymphoma therapies, and second primary cancers for up to 5 years.
The study was designed and conducted to adhere to Good Clinical Practice per the International Conference on Harmonisation Guideline E6 requirements and in accordance with the ethical principles outlined in the Declaration of Helsinki. The study was conducted in accordance with applicable national, state, and local laws per each study site’s Institutional Review Board, independent ethics committee, and pertinent regulatory authorities. All patients provided written informed consent before any study-related procedures were performed.
Study endpoints
The primary endpoint was PFS, as assessed by the independent review committee (IRC, i.e., central review) per 2007 International Working Group (IWG 2007) criteria [26], but without positron emission tomography imaging. Secondary endpoints included overall response rate (ORR), complete response (CR), duration of response (DOR), overall survival (OS), event-free survival (EFS), time to next anti-lymphoma treatment (TTNLT), and safety. Primary efficacy analyses were conducted in the intent-to-treat (ITT) population, defined as all patients randomized into the trial, regardless whether they received study treatment or not. All efficacy responses were assessed by a blinded, independent central review assessment of radiologic data using the 2007 IWG criteria, and progression or relapse was confirmed based on computed tomography/magnetic resonance imaging scans. Bone marrow biopsy was required to confirm a CR.
The safety analysis population was defined as all patients who received at least one dose of study treatment. Adverse events (AEs) were classified using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03, except for tumor flare reaction, which was graded using the NCI CTCAE version 3.0, and tumor lysis (assessed per Cairo–Bishop Criteria) [27].
Statistical methods
The primary objective of AUGMENT was to show superior PFS in the R2 group compared with the R-placebo group, using a one-sided α = 0.025 level. The study was not designed with sufficient power to detect differences in an exploratory single-country subgroup analysis such as presented here. PFS and other time-to-event data were characterized by the Kaplan–Meier procedure [28]. A hazard ratio (HR) with a two-sided 95% CI was estimated using the stratified Cox proportional hazard regression model. For binary secondary efficacy endpoints, the number and percentage of patients were tabulated by treatment group, and a stratified Cochran–Mantel–Haenszel test to adjust for possible confounding effects of the stratification factors was performed. Prespecified subgroup analysis of PFS was also performed, with HR estimated from a Cox proportional hazard regression model.
Results
Patient population
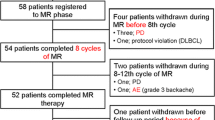
From October 28, 2014, through June 6, 2016, a total of 36 Japanese patients were enrolled into the study, with 18 patients each in the R2 and R-placebo groups. Baseline characteristics were similar in the two groups (Table 1). Median age was 61 years (range 44–83). Thirty-five (97%) patients had FL and only 1 (3%) patient had MZL (R-placebo group, MALT subtype). Of the patients with FL, 25% had a high-risk Follicular Lymphoma International Prognostic Index score. Nine (25%; R2, n = 4; R-placebo, n = 5) patients had ≥ 3 prior systemic anti-lymphoma treatments. Relapse or progression within 2 years of their initial diagnosis had occurred in 13 (36%) patients, and 1 (3%) R2 patient was refractory to last regimen. The Japanese patient population was similar to the global AUGMENT population [25]; however, some differences (Japanese vs global) were noted, including Eastern Cooperative Oncology Group Performance Status 0 (92% vs 68%), high tumor burden (39% vs 51%), MZL histology (3% vs 18%), B symptoms (0% vs 8%), prior rituximab treatment (100% vs 84%), and refractory to last regimen (3% vs 16%).
Treatment completion and dose intensity
In the ITT population, the full treatment course was completed in 61% of patients in the R2 group and 83% of patients in the R-placebo group. In the safety population, the median number of lenalidomide/placebo treatment cycles was 12 (range, 4–12) for R2 and 12 (range 6–12) for R-placebo. The median relative dose intensity of rituximab was 96% in the R2 group and 98% in the R-placebo group. The median relative dose intensity of lenalidomide was 91% and the average daily dose was 17.30 mg (interquartile range, 4.90 mg from Q3–Q1 [20–15.10 mg]) in the R2 group.
In the R2 vs R-placebo groups, treatment discontinuations occurred in 39% vs 17% of patients, respectively. The most common reasons for discontinuation of lenalidomide or placebo were AE (17%) in the R2 group and PD (17%) in the R-placebo group. One patient in the R2 group withdrew from the study and discontinued rituximab. AEs leading to dose reduction/interruption of lenalidomide or placebo occurred in 44%/67% vs 11%/33% of patients in the R2 and R-placebo groups, respectively. AEs leading to dose interruption of rituximab occurred in 56% of R2 vs 50% of R-placebo patients. AEs leading to early discontinuation of lenalidomide or placebo occurred in 17% of R2 vs 0% of R-placebo patients.
In the R2 vs R-placebo group, the median time to first dose reduction of lenalidomide or placebo was 106 days (range, 12–288) vs 186 days (range, 78–294) and median time to first dose interruption was 58 days (range 6–261) vs 113 days (range, 29–304). Among patients in the R2 group, neutropenia was the most common reason for lenalidomide dose reduction (28%), interruption (44%), and discontinuation (11%).
Efficacy
The primary endpoint of PFS by IRC was superior in the R2 group vs the R-placebo group (HR 0.32; 95% CI 0.11–0.96). Median PFS as assessed by IRC in the R2 vs R-placebo group was not reached (NR) (95% CI 19.7–NE) vs 16.5 months (95% CI 11.3–30.6; Fig. 1). PFS as assessed by investigator also showed HR in favor of R2 (HR 0.62; 95% CI 0.23–0.1.67); median PFS by investigator was NR (95% CI 16.7–NE) vs 19.3 months (95% CI 13.9–NE) for the R2 and R-placebo groups, respectively.

Progression-free survival, as assessed by IRC in the ITT population
Best ORR as assessed by IRC was higher in the R2 group, 94% (95% CI 73–100) compared with the R-placebo group, 56% (95% CI 31–79), with CR rates of 17% (95% CI 4–41) and 11% (95% CI 1–35), respectively (Table 2). Best response rates as assessed by investigator were also higher in the R2 vs R-placebo group. OS results were immature, with 1 death reported (R2 group; arrhythmia; Fig. 2).

Overall survival in the ITT population
DOR (HR 0.4; 95% CI 0.13–1.25), EFS (HR 0.36; 95% CI 11.3–30.6), and TTNLT (HR 0.49; 95% CI 0.12–2.00) all had HRs in favor of R2. Median DOR, median EFS, and median TTNLT were not reached in the R2 group compared with 19.0 months, 16.5 months, and 37.1 months, respectively, in the R-placebo group (Supplementary Fig. 1–3). The median follow-up was 33.0 months.
Safety and tolerability
All 36 patients received treatment and were included in the safety population. Eighteen patients in the R2 group (100%) and 17 in the R-placebo group (94%) had AEs that occurred within 28 days after last dose (Table 3). Any-grade AEs that occurred more frequently (≥ 20% difference) in the R2 vs R-placebo group included infusion-related reaction (72% vs 44%), neutropenia (61% vs 33%), constipation (56% vs 11%), diarrhea (39% vs 0%), nasopharyngitis (39% vs 28%), rash (39% vs 17%), rash maculopapular (28% vs 0%), and increased alanine aminotransferase (28% vs 0%). More patients in the R2 group (67%) had at least one grade 3/4 AE compared with the R-placebo group (22%). Grade 3/4 AEs reported in > 1 patient in either group (R2 vs R-placebo) included neutropenia (50% vs 17%), decreased lymphocyte count (17% vs 11%), and decreased white blood cell count (6% vs 11%). One patient died on study (R2 group, arrhythmia, cycle 7, related to lenalidomide) and 1 patient had a second primary malignancy (SPM) (R2 group, invasive carcinoid tumor of the gastrointestinal tract). Time to onset of SPM was 19.6 months.
Discussion
R2 in Japanese patients from AUGMENT demonstrated superior efficacy compared with R-placebo and reduced the risk of progression by 68% (HR 0.32; 95% CI 0.11–0.96) compared with R-placebo. Median PFS was not reached in the R2 group compared with 16.5 months in the R-placebo group. Superior efficacy of R2 was also evident in the secondary endpoints of response rate, DOR, EFS, and TTNLT.
AEs were more common in the R2 group, largely due to higher rates of grade 3/4 neutropenia. Two patients in the R2 group discontinued lenalidomide due to neutropenia; however, neutropenia was predictable and manageable primarily through dose modifications and growth factor support. Growth factor support was used in 4 (22%) and 3 (17%) patients in the R2 and R-placebo groups, respectively. There were no instances of febrile neutropenia in either treatment arm.
The data for the Japanese subgroup were generally consistent with the global AUGMENT study [25]. There were no clinically relevant imbalances between the Japanese subgroup and the global population. R2 efficacy results were similar between the Japanese subgroup and global population, including median PFS HR (0.32 vs 0.46), PFS probability at 2 years (69% vs 58%), and ORR/CR (94%/17% vs 78%/34%). Additionally, tolerability of R2 was similar in the Japanese subgroup and global population. Any grade R2 associated AEs that occurred with a ≥ 25% difference between global vs Japanese patients included constipation (26% vs 56%), infusion-related reaction (15% vs 72%), and rash (11% vs 39%); however, R2 associated grade 3/4 AEs were similar between the Japanese and global populations, including rates of grade 3/4 neutropenia which occurred in 50% in both populations. In the R-placebo control arms, safety and efficacy data in Japanese patients were also similar to data in the global population. There was only one reported histological transformation in the Japanese population (R2 group).
Current Japanese guidelines for treating patients with relapsed iNHL, not refractory to rituximab, include rituximab monotherapy, bendamustine ± rituximab, fludarabine ± rituximab, multi-agent chemotherapy ± rituximab, radiotherapy, radioimmunotherapy, and other options; however, relative superiority of these options is unclear [9]. Similar to the global AUGMENT data, the data presented here demonstrated that R2 offered a clinically meaningful improvement compared with R-placebo in Japanese patients and had a tolerable safety profile. Although this was not a comparative study with other chemotherapies, these results suggest that R2 should be a new treatment option for Japanese patients with R/R iNHL.
References
Swerdlow SH CE, Harris NL, Jafie ES, Pileri SA, Stein H, et al. World Health Organization classification of tumors of haematopoietic and lymphoid tissues, ed. Lyon. 2008: IARC Press.
Arcaini L, Merli M, Volpetti S, Rattotti S, Gotti M, Zaja F. Indolent B-cell lymphomas associated with HCV infection: clinical and virological features and role of antiviral therapy. Clin Dev Immunol. 2012;2012:638185.
Sousou T, Friedberg J. Rituximab in indolent lymphomas. Semin Hematol. 2010;47:133–42.
Arcaini L, Rattotti S, Gotti M, Luminari S. Prognostic assessment in patients with indolent B-cell lymphomas. Sci World J. 2012;2012:107892.
Chihara D, Ito H, Matsuda T, Shibata A, Katsumi A, Nakamura S, et al. Differences in incidence and trends of haematological malignancies in Japan and the United States. Br J Haematol. 2014;164:536–45.
Chihara D, Ito H, Izutsu K, Hattori M, Nishino Y, Ioka A, et al. Advance and stagnation in the treatment of patients with lymphoma and myeloma: Analysis using population-based cancer registry data in Japan from 1993 to 2006. Int J Cancer. 2015;137:1217–23.
Dreyling M, Ghielmini M, Rule S, Salles G, Vitolo U, Ladetto M. Newly diagnosed and relapsed follicular lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27:v83–v90.
Izutsu K. Treatment of follicular lymphoma. J Clin Exp Hematop. 2014;54:31–7.
Izutsu K. JSH practical guidelines for hematological malignancies, 2018: II. Lymphoma-1. Follicular lymphoma (FL). Int J Hematol. 2019; 110:11–19.
Dreyling M, Thieblemont C, Gallamini A, Arcaini L, Campo E, Hermine O, et al. ESMO Consensus conferences: guidelines on malignant lymphoma. part 2: marginal zone lymphoma, mantle cell lymphoma, peripheral T-cell lymphoma. Ann Oncol. 2013;24:857–77.
Zucca E, Conconi A, Laszlo D, Lopez-Guillermo A, Bouabdallah R, Coiffier B, et al. Addition of rituximab to chlorambucil produces superior event-free survival in the treatment of patients with extranodal marginal-zone B-cell lymphoma: 5-year analysis of the IELSG-19 randomized study. J Clin Oncol. 2013;31:565–72.
Igarashi T, Ohtsu T, Fujii H, Sasaki Y, Morishima Y, Ogura M, et al. Re-treatment of relapsed indolent B-cell lymphoma with rituximab. Int J Hematol. 2001;73:213–21.
Gandhi AK, Kang J, Havens CG, Conklin T, Ning Y, Wu L, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4 (CRBN.). Br J Haematol. 2014;164:811–21.
Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–35.
Ramsay AG, Clear AJ, Kelly G, Fatah R, Matthews J, Macdougall F, et al. Follicular lymphoma cells induce T-cell immunologic synapse dysfunction that can be repaired with lenalidomide: implications for the tumor microenvironment and immunotherapy. Blood. 2009;114:4713–20.
Fowler NH, Davis RE, Rawal S, Nastoupil L, Hagemeister FB, McLaughlin P, et al. Safety and activity of lenalidomide and rituximab in untreated indolent lymphoma: an open-label, phase 2 trial. Lancet Oncol. 2014;15:1311–8.
Wu L, Adams M, Carter T, Chen R, Muller G, Stirling D, et al. Lenalidomide enhances natural killer cell and monocyte-mediated antibody-dependent cellular cytotoxicity of rituximab-treated CD20+ tumor cells. Clin Cancer Res. 2008;14:4650–7.
Chiu H, Trisal P, Bjorklund C, Carrancio S, Torano EG, Guarinos C, et al. Combination lenalidomide-rituximab immunotherapy activates anti-tumour immunity and induces tumour cell death by complementary mechanisms of action in follicular lymphoma. Br J Haematol. 2019.
Chaudhry M, Nastoupil L, Samaniego F, Neelapu SS, Hagemeister FB, Fanale MA, et al. Treatment with combination of lenalidomide and rituximab achieves durable responses in a long term follow up of patients with indolent non-hodgkin's lymphoma. Hematol Oncol. 2017;35:215–6.
Kiesewetter B, Willenbacher E, Willenbacher W, Egle A, Neumeister P, Voskova D, et al. A phase 2 study of rituximab plus lenalidomide for mucosa-associated lymphoid tissue lymphoma. Blood. 2017;129:383–5.
Leonard JP, Jung SH, Johnson J, Pitcher BN, Bartlett NL, Blum KA, et al. Randomized trial of lenalidomide alone versus lenalidomide plus rituximab in patients with recurrent follicular lymphoma: CALGB 50401 (Alliance). J Clin Oncol. 2015;33:3635–40.
Martin P, Jung SH, Pitcher B, Bartlett NL, Blum KA, Shea T, et al. A phase II trial of lenalidomide plus rituximab in previously untreated follicular non-Hodgkin's lymphoma (NHL): CALGB 50803 (Alliance). Ann Oncol. 2017;28:2806–12.
Tuscano JM, Dutia M, Chee K, Brunson A, Reed-Pease C, Abedi M, et al. Lenalidomide plus rituximab can produce durable clinical responses in patients with relapsed or refractory, indolent non-Hodgkin lymphoma. Br J Haematol. 2014;165:375–81.
Morschhauser F, Fowler NH, Feugier P, Bouabdallah R, Tilly H, Palomba ML, et al. Rituximab plus lenalidomide in advanced untreated follicular lymphoma. N Engl J Med. 2018;379:934–47.
Leonard JP, Trneny M, Izutsu K, Fowler NH, Hong X, Zhu J, et al. AUGMENT: a phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J Clin Oncol. 2019;37:1188–99.
Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86.
Cairo MS, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3–11.
Kaplan EL, Meier P. Non-parametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–81.
Acknowledgements
All authors performed and/or designed the research study, analyzed the data, contributed to the development and revision of the manuscript, and approved the final version for submission. The authors thank the patients, their family members, and collaborators from participating institutions and Celgene Corporation. Thank you to following investigators for their participation in data collection at each site: Drs. Kenichi Ishizawa, Yoko Okitsu, Satoshi Ichikawa, Kunihiro Tsukasaki, Takayuki Yoshino, Kiyohiko Hatake, Dai Maruyama, and Masafumi Taniwaki. Medical writing support was provided by Benjamin Levine, PhD of Bio Connections LLC, and funded by Celgene Corporation; the authors were fully responsible for content and editorial decisions for this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
KI reports grants from Celgene during the conduct of the study; grants and personal fees from Celgene, Eisai, MSD, Takeda, Janssen, Mundipharma, Chugai, Astra Zeneca, Abbvie, Bayer, and Zenyaku outside the submitted work; personal fees from Bristol Myers Squib, Dainihon Sumitomo, Nihon Mediphysics, and Kyowa Hakko Kirin outside the submitted work; and grants from Ono, Gilead, Solasia, Symbio, Astellas Amgen, and Daiichi Sankyo outside the submitted work. YM reports research funding from Ono Pharmaceutical and lecture fees from BMS, Novartis, and Pfizer outside the submitted work. NF reports grants from Celgene K.K. during the conduct of the study; grants and personal fees from Eisai and Takeda outside the submitted work; grants from Abbvie, Bayer, Gilead and Solasia Pharma outside the submitted work; and personal fees from Celgene, Chugai, Janssen, Kyowa Hakko Kirin, Mochida, Mundi, Nippon Shinyaku, Ono and Zenyaku outside the submitted work. GY reports personal fees from Celgene, Kyowa Hakko Kirin, Takeda, Bristol-Myers Squibb, Mundipharma, and Janssen outside the submitted work; and grants and personal fees from Chugai Pharma and Eisai outside the submitted work. HN reports grants from Celgene Corporation during the conduct of the study; grants and personal fees from Janssen Pharmaceutical K. K, Celgene Corporation, Mundipharma K.K., Bayer Yakuhin, Takeda Pharmaceutical, Kyowa Hakko Kirin, Esai, Bristol-Myers Squibb, Ono Pharmaceutical, Gilead Sciences, Zenyaku Kogyo, AstraZeneca, SymBio Pharmaceuticals, Otsuka Pharmaceutical, and Roche; grants from Abbvie G.K., Solasia Pharma K.K., HUYA Bioscience International, IQVIA Japan K.K.; and personal fees from Sanofi K.K. outside the submitted work. TO reports employment and stockholder with Celgene K.K. during the conduct of the study. SK reports employment and stockholder with Celgene during the conduct of the study. PF reports employment, stockholder, and received personal fees from Celgene during the conduct of the study. SM reports employment with Celgene K.K. and stockholder of Celgene. KT reports grants and personal fees from Celgene during the conduct of the study; personal fees from Zenyaku, HUYA Bioscience International, Yakult, Daiichi Sanky, Bristol-Myers Squibb, Meiji Seika Kaisha, Solasia Pharma, and Verastem outside the submitted work; grants from Janssen and Abbvie outside the submitted work; and grants and personal fees from Eisai, Takeda, Mundipharma, Kyowa Hakko Kirin, Chugai Pharma, and Ono Pharmaceutical outside the submitted work. YT, TJ, TI, TKo, and TKi report no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1
Duration of response, as assessed by IRC (TIF 131 kb)
Supplementary Fig. 2
Event-free survival, as assessed by IRC (TIF 132 kb)
Supplementary Fig. 3
Time to next anti-lymphoma treatment (TIF 130 kb)
About this article

Cite this article
Izutsu, K., Minami, Y., Fukuhara, N. et al. Analysis of Japanese patients from the AUGMENT phase III study of lenalidomide + rituximab (R2) vs. rituximab + placebo in relapsed/refractory indolent non-Hodgkin lymphoma. Int J Hematol 111, 409–416 (2020). https://doi.org/10.1007/s12185-019-02802-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-019-02802-y