
Abstract
Congenital thrombophilia which is characterized by deficiencies in proteins such as antithrombin (AT), protein C (PC) and protein S (PS), is a major cause of venous thromboembolism (VTE). A total of 130 patients with VTE were evaluated for congenital thrombophilia based on the activity of AT, PC, or PS. Fifteen VTE patients with congenital AT deficiency (11.5 %), 16 with congenital PC deficiency (12.3 %) and eight with congenital PS deficiency (6.2 %) were diagnosed using DNA analysis. The frequency of congenital AT deficiency was significantly higher in subjects with pregnancy-related and idiopathic VTE than in those with VTE due to other causes, and congenital PC and PS deficiency were frequently associated with idiopathic VTE. Among the groups examined, the plasma levels of AT were the lowest in subjects with pregnancy-related VTE. Although our findings may have been influenced by some unintentional bias, congenital thrombophilia is nevertheless a major cause of VTE in pregnant patients as well as in young or middle-aged patients without any underlying diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Venous thromboembolism (VTE) such as deep vein thrombosis (DVT) and pulmonary embolism (PE) is associated with a high mortality and morbidity and increased hospitalization duration and costs [1, 2]. Therefore, the early diagnosis or prophylaxis of VTE is important. The mechanism of VTE onset differs based on the underlying disease or condition. VTE is commonly caused by any of a number of pathological conditions, such as pregnancy [3, 4], surgery (e.g., orthopedic surgery) [5, 6], trauma [7], malignancy [8], bed rest [7], long travel [9], autoimmune diseases [10], drugs (e.g., contraceptives) [11], dehydration, and thrombophilia [12].
Although factor V Leiden and prothrombin G20210A mutations [13] are common risk factors for VTE in Europe and North America, few reports have examined these risk factors in a Japanese population. Congenital deficiencies in natural anticoagulants such as antithrombin (AT), protein C (PC), or protein S (PS), have often been reported in Japan [14, 15]. AT mainly inhibits activated factor X and thrombin, activated PC (APC) mainly inhibits activated factor VIII and V, and PS works as a cofactor for APC. The K196E mutation is the PS mutation most frequently reported as the cause of VTE in Japanese people [16]. AT and PS levels are frequently decreased in pregnant women and patients with multiple organ failure or liver dysfunction, while PC and PS levels are decreased in patients treated with warfarin. Therefore, a DNA analysis to determine the AT, PC, or PS mutation status is required to determine the cause or mechanism of the onset of VTE. In acquired thrombophilia, antiphospholipid antibody syndrome (APS) [17] is major risk factor for VTE, and several assays for detecting anti-phospholipid antibody are required to diagnose this syndrome.
In the present study, the levels of natural anticoagulants and APS were examined in 130 patients with DVT or PE to determine the relationship between thrombophilia and VTE.
Materials and methods
A total of 130 patients with DVT or PE (median age, 45.5 years; 25th–75th percentile, 31.0–66.0 years; 76 females and 54 males) were examined to determine the causes of DVT or PE between January 1, 1985, and December 31, 2014, at Mie University Hospital. Most of the patients were residents of Mie prefecture. The causes of DVT or PE were pregnancy in 17 patients, surgery in 14, bed rest in 11, malignant diseases in 10, autoimmune diseases in 7, drugs in 4, infection in 3, long travel in 3, others in 7 and idiopathic onset in 54. DVT was diagnosed using echography or venography. PE or cerebral vascular disease was diagnosed using computed tomography (CT) or magnetic resonance imaging (MRI), and cerebral venous sinus thrombosis (CVST) was diagnosed based on MRI, magnetic resonance venography (MRV), or cerebral angiography (CAG). The diagnosis of APS was made based on the established criteria and depending on the thrombotic symptoms or pregnancy complications and presence of antiphospholipid antibodies [18].
The study protocol was approved by the Human Ethics Review Committee of the Mie University School of Medicine and a signed consent form was obtained from each subject. This study was faithfully carried out in compliance with the Declaration of Helsinki.
Measurement of the AT, PC, PS and antiphospholipid antibody concentrations
Plasma was prepared from citrate-stabilized blood (9 volumes of blood and 1 volume of 3.2 % sodium citrate). The blood was centrifuged at 2000g for 15 min, and the supernatant was stored at −80 °C before use. The free PS antigen (reference value; 60–150 %)concentration was measured using a monoclonal antibody-based enzyme-linked immunosorbent assay (ELISA) with the Asserachrom free PS kit (Diagnostica Stago, Asnières, France). The plasma PS (reference value; 60–150 %) and PC (reference value; 70–140 %) activity levels were measured via the clotting time method using a STA®-Staclot® Protein S and STA®-Staclot® Protein C kit (Diagnostica Stago). The plasma PC (reference value; 69–144 %) antigen concentration was measured via a latex agglutination test using a LPIA-ACE PC kit (Mitsubishi Chemical Medience Corporation, Tokyo, Japan). The plasma AT activity (reference value; 83–118 %) was measured via a synthetic substrate assay using a Chromorate ATIII (C) kit (Mitsubishi Chemical Medience Corporation). Lupus anticoagulant (LA), dilute Russell’s viper venom time (DRVVT) was measured via the clotting time method using a Gradipore LA test (Gradipore, Sydney, Australia). Activated partial thromboplastin time (APTT)-LA was measured using PTT-LA (Roche Diagnostica, Basel, Schweiz). The titers of anti-cardiolipin-β2 glycoprotein I (ACL-β2GPI) and anti-cardiolipin IgG antibodies were measured with an ELISA kit (Yamasa Co, Tokyo, Japan) and MESACUP caldiolin test (MBL, Nagoya, Japan), respectively. The PC, PS, and AT activities were evaluated at the onset of VTE and during stable phase.
DNA analysis of AT, PC, and PS
Genomic DNA was prepared from peripheral blood leukocytes using a QIAamp DNA Blood Mini Kit (QIAGEN) in accordance with the manufacturer’s instructions. Each exon and exon/intron boundary of the gene was amplified from genomic DNA using a polymerase chain reaction (PCR), as previously described. The PCR products were directly sequenced using a Big-Dye Terminator Cycle Sequencing Kit and Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) [19].
Multiplex ligation-dependent probe amplification (MLPA) analysis of AT
An MLPA analysis was performed in accordance with the manufacturer’s instructions using the SALSA MLPA P227 Serpin C1 kit (MRC-Holland, Amsterdam, Netherlands). The MLPA analysis detects deletions/duplications of all 7 exons of the SERPINC1 gene using specifically designed probes. Briefly, the sample DNA (50–250 ng) is denatured for 5 min at 98 °C, and then cooled to 25 °C. The sample is then incubated at 95 °C for 1 min and hybridized at 60 °C for 16–20 h. The ligation mix is successively added, and the sample is incubated at 54 °C for 15 min (for ligation) and at 98 °C for 5 min to inactivate the ligation enzyme. The ligation products are subjected to PCR amplification using the common primer set. The amplification products were run on the 3130 Genetic Analyzer using the GeneScan 500 LIZ size standard. The data were analyzed using the GeneMapper 4.0 software program. Dosage quotient analyses were performed based on comparisons between the patient and reference control DNA samples using the Microsoft Excel software program. A 30–60 % decrease in the relative peak area of the amplification product indicated the presence of a heterozygous deletion of the corresponding exon. The MLPA analysis was repeated three times for all samples.
The DNA analyses were performed in subjects with AT <70 % and PC <60 %, or PS activity <60 % or low of activity/antigen ratio (<0.6).
Statistical analysis
The data are expressed as the median (25th–75th percentile). The differences between the groups were examined for statistical significance using the Mann–Whitney U test. A p value <0.05 denoted the presence of a statistically significant difference.
Results
The DNA analyses were performed in patients with an AT activity <70 % (n = 20) or PC activity <60 % (n = 22), or PS activity <60 % or low of activity/antigen ratio (n = 15) at stable state. Fifteen (11.5 %) subjects were found to have congenital AT deficiency, 16 (12.3 %) congenital PC deficiency, 8 (6.2 %) congenital PS deficiency, and 1 (#15 in Table 2 and #8 in Table 3) was found to be combined heterozygous for PC and PS (Tables 1, 2, 3). Among the 15 subjects with congenital AT deficiency including eight mutations, the median age (25th–75th percentile) was 26.0 years (23.0–30.0 years), and there were 13 families and 7 patients with pregnancy-related VTE (Table 1). Subject #8 demonstrated a deletion of exon 1, and this abnormality was detected using the multiplex ligation-dependent probe amplification (MLPA) assay (Fig. 1).

MLPA electropherogram of subject #8 (top) and a control subject (bottom). The peaks of 7 exons are shown by open columns with the exon number. The non-highlighted peaks correspond to control genes (healthy volunteer). The X-axis represents the fragment size in base pairs. From the MLPA electropherogram, we can see that the peak height of Exon 1 in subject #8 was almost half that observed in the control subject
Among the 16 subjects with congenital PC deficiency including 9 mutations, the median age (25th–75th percentile) was 40.0 years (32.5–60.0 years). Three patients had pregnancy-related VTE (Table 2).
Among the 8 subjects with congenital PS deficiency including 7 mutations, the median age (25th–75th percentile) was 39.0 years (30.5–50.0 years). One patient had pregnancy-related VTE (Table 3).
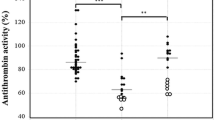
Among the 7 subjects with APS, the median age (25th–75th percentile) was 54.0 years (33.5–72.8 years), and the underlying diseases were ovarian cancer in 1 and SLE in 1, with 4 idiopathic (Table 4). The relationship between thrombophilia and several situations causing VTE is shown in Table 5. Among the examined groups, the plasma levels of AT were the lowest in subjects with pregnancy-related VTE [median (25th–75th percentile): 59.1 % (55.8–76.4 %); Fig. 2]. The frequency of congenital thrombophilia was markedly high in those with pregnancy-related VTE (n = 10, 58.8 %) and those with idiopathic VTE (n = 24, 44.4 %). There were no significant differences in the PC or PS activities among the situations causing VTE (Figs. 3, 4).

Plasma AT activities according to the underlying disease or state. AT antithrombin. **p < 0.01; *p < 0.05

Plasma PC activities according to the underlying disease or state

Plasma PS activities according to the underlying disease or state
Discussion
In the present study, the frequencies of congenital AT, PC, and PS deficiency in patients with VTE were 11.5, 12.3 and 6.2 %, respectively. However, as these patients were chosen to examine their thrombophilia, our results might have been influenced by bias. A previous Japanese study also reported a high frequency of congenital AT deficiency (8.1 %), congenital PC deficiency (9.8 %), and congenital PS deficiency (16.2 %) in a population of 173 DVT patients [25]. The frequency of congenital AT, PC, and PS deficiencies must be determined in order adequately describe the relationship between thrombophilia and VTE. The age at the onset of VTE in the subjects with congenital AT deficiency was relatively young, and about half of the patients with congenital AT deficiency had pregnancy-related VTE, suggesting that the thrombogenicity may be higher with congenital AT deficiency than with congenital PC or PS deficiency, and that thrombophilia is involved in the onset of thrombosis during pregnancy. In the subjects with congenital AT deficiency, eight mutations had been previously reported (Table 1) [19–26], and four families showed the same mutation (AT Glasgow) [23], suggesting that these families shared a distant ancestor. Subject #8 demonstrated a deletion of exon 1 that could not be detected by the usual sequencing methods. An MLPA assay [39] was therefore conducted in this patient.
Among the subjects with congenital PC deficiency, six of the nine mutations had been previously reported (Table 2) [27–32]. The mutations p.Gly334Ser (PC Nagoya-II) [28], p.Arg211Trp (PC-Tochigi, PC Osaka-1) [30], and p.Gly423valfsX82 (PC Nagoya) [31] were observed in three, two, and five families, respectively, suggesting that these families also shared a distant ancestor. Given that subjects, homozygous for p.Gly334Ser [28] rarely experienced VTE which was rate onset, this mutation might correlate a low risk for VTE.
Among the subjects with congenital PS deficiency, six of seven PS mutations had been previously reported (Table 3) [33–38]. The mutation p.Lys196Glu [34] was observed in four families, and mildly decreased PS activity was observed in two patients with only this mutation, suggesting that the p.Lys196Glu mutation might correlate with a relative low risk for VTE. However, markedly decreased PS activity was observed in two patients with a combined heterozygous status, indicating that multiple PS mutations or complications with AT or PC mutations may be associated with a high risk for VTE. In contrast, a mildly decreased PS activity was observed in two patients with only the p.Lys196Glu mutation [34].
Although the frequency of congenital PS deficiency in this VTE study was lower than in previous reports [25], the frequency of PS Tokushima was high in Mie Prefecture [40].
Regarding the relationship between congenital thrombophilia and several situations causing VTE, the frequency of congenital thrombophilia was markedly high in subjects with pregnancy-related and idiopathic VTE, suggesting that congenital thrombophilia is a major cause of VTE in these groups (Table 5). Especially, the frequency of congenital AT deficiency was significantly higher in the subjects with pregnancy-related and idiopathic VTE than in those with VTE due to other causes, and congenital PC and PS deficiency were frequently associated with idiopathic VTE. In addition, the pregnancy-related VTE subjects and those without any underlying disease tended to be younger than those with VTE due to surgery, bed rest, malignant diseases and autoimmune diseases. Therefore, the activity of AT, PC, and PS should be examined in young or middle-aged patients with pregnancy-related or idiopathic VTE. Although APS was observed in three patients with autoimmune diseases, it was also observed in patients with other diseases, as well as in those without any underlying diseases at all (Table 4). Antiphospholipid antibody levels should be examined in VTE patients with autoimmune diseases and idiopathic VTE. Plasma AT levels were significantly lower in subjects with pregnant-related VTE than in those with VTE due to other causes, and acquired AT deficiency has been reported to be frequently associated with VTE in pregnant women [4], suggesting that decreased levels of AT are involved in the onset of VTE in this particular population (Fig. 2). There were no significant differences in the PC or PS activities among the situations causing VTE, although these activities tended to be low all-around (Figs. 3, 4).
In conclusion, congenital thrombophilia is a major cause of VTE in pregnant patients and young or middle-aged patients without underlying diseases.
References
Sevitt S. Fatal road accidents. Injuries, complications, and causes of death in 250 subjects. Br J Surg. 1968;55:481–505.
Spyropoulos AC, Lin J. Direct medical costs of venous thromboembolism and subsequent hospital readmission rates: an administrative claims analysis from 30 managed care organizations. J Manag Care Pharm. 2007;13:475–86.
van Vlijmen EF, Veeger NJ, Middeldorp S, Hamulyák K, Prins MH, Kluin-Nelemans H, et al. The impact of a male of female thrombotic family history on contraceptive counselling: a cohort study. J Thromb Haemost. 2016 (in press).
Kamimoto Y, Wada H, Ikejiri M, Nakatani K, Sugiyama T, Osato K, et al. High frequency of decreased antithrombin level in pregnant women with thrombosis. Int J Hematol. 2015;102:253–8.
Albayati MA, Grover SP, Saha P, Lwaleed BA, Modarai B, Smith A. Postsurgical inflammation as a causative mechanism of venous thromboembolism. Semin Thromb Hemost. 2015;41:615–20.
Aota T, Naitoh K, Wada H, Yamashita Y, Miyamoto N, Hasegawa M, et al. Elevated soluble platelet glycoprotein VI is a useful marker for DVT in postoperative patients treated with edoxaban. Int J Hematol. 2014;100:450–6.
García-Olivares P, Guerrero JE, Keough E, Galdos P, Carriedo D, Murillo F, et al. PROF-ETEV study investigators.: clinical factors associated with inappropriate prophylaxis of venous thromboembolic disease in critically ill patients. A single day cross-sectional study. Thromb Res. 2016;143:111–7.
Posch F, Königsbrügge O, Zielinski C, Pabinger I, Ay C. Treatment of venous thromboembolism in patients with cancer: a network meta-analysis comparing efficacy and safety of anticoagulants. Thromb Res. 2015;136:582–9.
Bartholomew JR, Schaffer JL, McCormick GF. Air travel and venous thromboembolism: minimizing the risk. Cleve Clin J Med. 2011;78:111–20.
Yusuf HR, Hooper WC, Grosse SD, Parker CS, Boulet SL, Ortel TL. Risk of venous thromboembolism occurrence among adults with selected autoimmune diseases: a study among a US cohort of commercial insurance enrollees. Thromb Res. 2015;135:50–7.
Zöller B, Ohlsson H, Sundquist J, Sundquist K. Family history of venous thromboembolism is a risk factor for venous thromboembolism in combined oral contraceptive users: a nationwide case-control study. Thromb J. 2015;13:34.
Meyer MR, Witt DM, Delate T, Johnson SG, Fang M, Go A, et al. Thrombophilia testing patterns amongst patients with acute venous thromboembolism. Thromb Res. 2015;136:1160–4.
Mannucci PM, Franchini M. Classic thrombophilic gene variants. Thromb Haemost. 2015;114:885–9.
Yokoyama K, Kojima T, Sakata Y, Kawasaki T, Tsuji H, Miyata T, et al. A survey of the clinical course and management of Japanese patients deficient in natural anticoagulants. Clin Appl Thromb Hemost. 2012;18:506–13.
Neki R, Miyata T, Fujita T, Kokame K, Fujita D, Isaka S, et al. Nonsynonymous mutations in three anticoagulant genes in Japanese patients with adverse pregnancy outcomes. Thromb Res. 2014;133:914–8.
Adachi T. Protein S and congenital protein S deficiency: the most frequent congenital thrombophilia in Japanese. Curr Drug Targets. 2005;6:585–92.
Kelchtermans H, Pelkmans L, de Laat B, Devreese KM. IgG/IgM antiphospholipid antibodies present in the classification criteria of the antiphospholipid syndrome: a critical review of their association with thrombosis. J Thromb Haemost. 2016 (in press).
Miyakis S, Lockshin MD, Atsumi T, Branch DW, Brey RL, Cervera R, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295–306.
Olds RJ, Lane DA, Caso R, Panico M, Morris HR, Sas G, et al. Antithrombin III Budapest: a single amino acid substitution (429Pro to Leu) in a region highly conserved in the serpin family. Blood. 1992;79:1206–12.
Erdjument H, Lane DA, Ireland H, Panico M, DiMarzo V, Blench I, et al. Formation of a covalent disulfide-linked antithrombin complex by an antithrombin variant, antithrombin Northwick Park. J Biol Chem. 1987;262:13381–4.
Koide T, Odani S, Takahashi K, Ono T, Sakuragawa N. Antithrombin III Toyama; replacement of arginine 47 by cysteine in hereditary abnormal antithrombin III that lacks heparin-binding ability. Proc Natl Acad Sci USA. 1984;81:289–93.
Katayama K, Hashimoto N, Tanaka Y, Ozawa T, Emi Y, Ikeda T, et al. Identification of a novel amino acid deletion mutation and a very rare single nucleotide variant in a Japanese family with type I antithrombin deficiency. Thromb Res. 2005;116:215–21.
Erdjument H, Lane DA, Panico M, diMarzo V, Morris HR. Single amino acid substitutions in the reactive site of antithrombin leading to thrombosis. Congenital substitution of arginine 393 to cysteine in antithrombin Northwick Park and to histidine in antithrombin Glasgow. J Biol Chem. 1988;263:5589–93.
Muta T, Okamura T, Kawamoto M, Ichimiya H, Yamanaka M, Wada Y, et al. Successful therapy with argatroban for superior mesenteric vein thrombosis in a patient with congenital antithrombin deficiency. Eur J Haematol. 2005;75:167–70.
Miyata T, Sato Y, Ishikawa J, Okada H, Takeshita S, Sakata T, et al. Prevalence of genetic mutations in protein S, protein C and antithrombin genes in Japanese patients with deep vein thrombosis. Thromb Res. 2009;124:14–8.
Ozawa T, Takikawa Y, Niiya K, Fujiwara T, Suzuki K, Sato S, et al. Antithrombin Morioka (Cys 95-Arg): a novel missense mutation causing type I antithrombin deficiency. Thromb. Haemost. 1997;77:403.
Lind B, Koefoed P, Thorsen S. Symptomatic type 1 protein C deficiency caused by a de novo Ser270Leu mutation in the catalytic domain. Br J Haematol. 2001;113:642–8.
Yamamoto K, Matsushita T, Sugiura I, Takamatsu J, Iwasaki E, Wada H, et al. Homozygous protein C deficiency: identification of a novel missense mutation that causes impaired secretion of the mutant protein C. J Lab Clin Med. 1992;119:682–9.
Miyata T, Sakata T, Zheng YZ, Tsukamoto H, Umeyama H, Uchiyama S, et al. Genetic characterization of protein C deficiency in Japanese subjects using a rapid and nonradioactive method for single-stand conformational polymorphism analysis and a model building. Thromb Haemost. 1996;76:302–11.
Matsuda M, Sugo T, Sakata Y, Murayama H, Mimuro J, Tanabe S, et al. A thrombotic state due to an abnormal protein C. N Engl J Med. 1988;319:1265–8.
Yamamoto K, Tanimoto M, Emi N, Matsushita T, Takamatsu J, Saito H. Impaired secretion of the elongated mutant of protein C (protein C-Nagoya). Molecular and cellular basis for hereditary protein C deficiency. J Clin Invest. 1992;90:2439–46.
Miyata T, Zheng YZ, Sakata T, Tsushima N, Kato H. Three missense mutations in the protein C heavy chain causing type I and type II protein C deficiency. Thromb Haemost. 1994;71:32–7.
Ikejiri M, Shindo A, Ii Y, Tomimoto H, Yamada N, Matsumoto T, et al. Frequent association of thrombophilia in cerebral venous sinus thrombosis. Int J Hematol. 2012;95:257–62.
Hayashi T, Nishioka J, Shigekiyo T, Saito S, Suzuki K. Protein S Tokushima: abnormal molecule with a substitution of Glu for Lys-155 in the second epidermal growth factor-like domain of protein S. Blood. 1994;83:683–90.
Ikejiri M, Tsuji A, Wada H, Sakamoto Y, Nishioka J, Ota S, et al. Analysis three abnormal Protein S genes in a patient with pulmonary embolism. Thromb Res. 2010;125:529–32.
Tsuda H, Urata M, Tsuda T, Wakiyama M, Iida H, Nakahara M, et al. Four missense mutations identified in the protein S gene of thrombosis patients with protein S deficiency effect on secretion and anticoagulant activity of protein S. Thromb Res. 2000;105:233–9.
Espinosa-Parrilla Y, Yamazaki T, Sala N, Dahlbäck B, de Frutos PG. Protein S secretion differences of missense account for phenotypic heterogeneity. Blood. 2000;95:173–9.
Kinoshita S, Iida H, Inoue S, Watanabe K, Kurihara M, Wada Y, et al. Protein S and protein C gene mutations in Japanese deep vein thrombosis patients. Clin Biochem. 2005;38:908–15.
Lee ST, Kim HJ, Kim DK, Schuit RJ, Kim SH. Detection of large deletion mutations in the SERPINC1 gene causing hereditary antithrombin deficiency by multiplex ligation-dependent probe amplification (MLPA). J Thromb Haemost. 2008;6:701–3.
Ikejiri M, Wada H, Sakamoto Y, Ito N, Nishioka J, Nakatani K, et al. The association of protein S Tokushima-K196E with a risk of deep vein thrombosis. Int J Hematol. 2010;92:302–5.
Acknowledgments
This work was supported in part by a Grant-in-Aid from the Ministry of Health, Labour and Welfare of Japan for Blood Coagulation Abnormalities and the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest in association with this study.
About this article

Cite this article
Ikejiri, M., Wada, H., Yamada, N. et al. High prevalence of congenital thrombophilia in patients with pregnancy-related or idiopathic venous thromboembolism/pulmonary embolism. Int J Hematol 105, 272–279 (2017). https://doi.org/10.1007/s12185-016-2111-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-2111-2