
Abstract
Darbepoetin alfa (DA) is a standard treatment for anemia in lower-risk MDS. However, to date there has been no comparative study to investigate the initial dosage. We, thus, conducted a randomized controlled trial to elucidate the optimal initial dosage of DA. International Prognostic Scoring System low or intermediate-1 risk MDS patients with hemoglobin levels ≤9.0 g/dL, serum erythropoietin levels ≤500 mIU/mL, and red blood cell transfusion dependency were enrolled. Patients were randomized to receive DA either at 60, 120, or 240 μg/week for 16 weeks followed by continuous administration with dose adjustment up to 48 weeks. Of 17, 18, and 15 patients in the 60, 120, and 240 μg DA groups included in the efficacy analysis, 64.7, 44.4, and 66.7 %, respectively, achieved the primary endpoint (major or minor erythroid response), while 17.6, 16.7, and 33.3 % achieved major erythroid responses in the initial 16-week period. No clinically significant safety concerns were identified. DA reduced the transfusion requirements effectively and safely in transfusion-dependent, lower-risk MDS patients. Given the highest achievement rate of the major erythroid response in the 240 μg group and the absence of dose-dependent adverse events, 240 μg weekly is the optimal initial dosage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myelodysplastic syndromes (MDS) consist of a diverse group of clonal hematopoietic stem cell disorders characterized by cytopenia associated with ineffective hematopoiesis and progression to acute myeloid leukemia (AML). Common clinical symptoms of MDS include anemia, infections, and bleeding associated with cytopenia. Among those, symptoms of anemia such as dizziness, shortness of breath, and fatigue are especially frequent. Aggravation of anemia is a great stress on the circulatory system due to the reduction of oxygen supply. Regular red blood cell (RBC) transfusions are the major supportive care for anemic patients with MDS. Chronic transfusion therapy inevitably leads to secondary iron overload, which can cause significant damage to many organs such as the liver, heart, and endocrine system. A poor prognosis associated with an increased transfusion volume has been reported in transfusion-dependent patients with MDS [1]. Although excessive iron may be removed by iron chelation, gastrointestinal and/or renal adverse reactions sometimes prevent continuous treatment.
Specific MDS treatment strategies based on the risk stratification by the International Prognostic Scoring System (IPSS) [2] are recommended in the National Comprehensive Cancer Network (NCCN) Guidelines [3]. Treatment to improve cytopenia is recommended for patients with IPSS low or intermediate-1 risk. For patients with intermediate-2 or high risk, the guidelines recommend radical treatment with a hematopoietic stem cell transplant or treatment to decrease the blast count to delay the progression to AML.
Recombinant human erythropoietin (rHuEPO) used for the treatment of MDS-related anemia has shown a significantly higher erythroid response rate than placebo or best supportive care [4–7]. Clinical trials of darbepoetin alfa (DA), a long-acting erythropoiesis-stimulating agent (ESA), have also been conducted [8–15]. A meta-analysis reported a significantly higher erythroid response with rHuEPO compared to a controlled drug and a comparable erythroid response between rHuEPO and DA [16, 17]. Patients with MDS are known to have higher serum erythropoietin (EPO) levels than healthy adults [18]. Higher serum EPO levels are inversely associated with a patient’s response to ESAs [19]. A transfusion volume of less than two units per month has been reported to be associated with a greater improvement in anemia [20, 21]. Based on these study reports, the NCCN Guidelines recommend ESAs as a first-line treatment for MDS patients with IPSS low or intermediate-1 risk, symptomatic anemia, and a serum EPO level of ≤500 mIU/mL [3]. The recommended DA dosage is 150–300 µg once weekly, based on reports of single-arm clinical trials [8, 11].
Meanwhile, no prospective randomized controlled trial to determine the optimal ESA dosage has yet been conducted. Moreover, the efficacy and safety of ESAs in Japanese or Korean patients with MDS have not been evaluated. We thus conducted a randomized controlled trial in Japanese and Korean transfusion-dependent patients to determine the optimal dosage for DA treatment. DA pharmacokinetics, patient survival, and AML progression were also investigated.
Methods
Eligibility criteria
The main inclusion criteria for patients in this study were: age of 20 years or older, IPSS low or intermediate-1 risk MDS, RBC transfusion dependency with an RBC transfusion-free period of less than 56 consecutive days within a 112-day period, serum EPO levels of ≤500 mIU/mL, hemoglobin (Hb) levels of ≤9 g/dL, and creatinine levels of ≤2.0 mg/dL. Patients with cardiac problems, previous thrombotic events, a concurrent active infection or a chronic inflammatory disease, anemia caused by other conditions than MDS, and previous or concurrent active malignancies were excluded. This study was conducted in accordance with the Declaration of Helsinki. The institutional review boards of the participating centers approved the protocol, and informed consent was obtained from all patients before beginning any study-related procedures. This study has been registered at www.clinicaltrials.gov as NCT01497145.
Study design
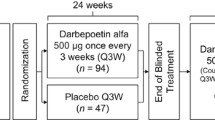
This was a multi-center (42 sites), randomized, open-label, phase 2, comparative study conducted in Japan and Korea. Patients were randomized via a dynamic allocation method and were assigned to each DA cohort (60, 120, or 240 μg DA) in a 1:1:1 ratio after stratification by ethnic groups (Japanese, Korean) and serum EPO levels (<100 mIU/mL or ≥100 mIU/mL) (Fig. 1).

Study design. DA darbepoetin alfa
Procedures
In the initial dose evaluation phase, DA was administered at a dose of 60, 120, or 240 μg subcutaneously weekly for 16 weeks. In the extended treatment evaluation phase (week 17–48), the DA dose was adjusted to maintain the major or minor erythroid response (based on changes in Hb levels) at the discretion of the investigator if patients showed (1) a major erythroid response (defined as not requiring an RBC transfusion for at least 56 consecutive days RBC transfusion independent) with a maximum increase in the Hb level of ≥1.0 g/dL above baseline, or (2) a minor erythroid response (defined as a ≥50 % reduction in RBC transfusion requirement during a 56-consecutive-day period as compared to baseline). If a patient did not exhibit a major or minor erythroid response, the DA dose was increased every 8 weeks up to a highest dose of 240 μg. When a patient showed a continuous major or minor erythroid response during the study period, the dosing frequency may be changed from a weekly to a biweekly schedule after week 17 with a doubled dose. If the Hb level exceeded 11.0 g/dL during the treatment period, DA was temporally discontinued. Patients who achieved neither a major nor a minor erythroid response after a 16-week treatment period with DA at 240 μg weekly were withdrawn from the study. Other ESAs or other drugs to treat MDS were prohibited during the study period. Granulocyte colony-stimulating factor was prohibited except for use for the treatment of infections.
Blood samples were obtained weekly before DA administration. For pharmacokinetic analysis, blood samples were obtained at weeks 1, 9, and 17 before DA administration, and 4, 24, 48, 72, 96, and 168 h after DA administration at week 1. Serum DA concentrations were measured using the Quantikine® IVD® Erythropoietin ELISA kit (R&D Systems; Minneapolis, MN, USA).
Anti-DA antibodies were tested at week 1, 17, and at the end of the study.
Efficacy and safety measurements
The primary efficacy endpoint in this study was the proportion of patients with a major or minor erythroid response after 16 weeks of treatment. The proportion of patients with a major erythroid response after 16 weeks of treatment, the proportion of patients with a major or minor erythroid response after 48 weeks of treatment, and changes in Hb level were assessed as the secondary endpoints. The Cancer Therapy Evaluation Program Common Toxicity Criteria version 4.0 was used to report adverse events. Anti-DA antibody expression was also assessed. The 1-year overall and AML-free survival rates of patients were investigated in an outcome survey.
Data sets and statistical analysis
The target sample size was 45 patients (15 patients for each dose group). In previous clinical studies, the erythroid response rates in patients treated with DA doses of 120 and 240 μg were 40 and 63 %, respectively [10, 11]. Based on these data, we assumed that the erythroid response rate in patients treated with 60 μg DA would be 25 %. Basing the calculations of the expected erythroid response rates on 15 patients per group, the erythroid response rate can be evaluated with an accuracy of ±12 % standard error. Regarding the safety of DA, we calculated that the probability that an adverse reaction with an incidence of 5 % would occur in at least 1 of 45 patients treated with DA is 90 %. We thus concluded that we would be able to detect any adverse reactions with a relatively high incidence in our patient group. In our study, the Safety Analysis Set was defined as patients who received at least 1 dose of DA, whereas the Per Protocol Set (PPS) excluded patients who received DA doses for less than 12 weeks.
For the safety analysis, adverse events were coded according to the Japanese version 16.1 of the Medical Dictionary for Regulatory Activities, and the number of patients for each adverse event was summarized by dose group. All analyses were performed using the SAS software (version 9.2).
For the pharmacokinetic analysis, the patients’ baseline serum DA concentration was first subtracted from the post-dose serum concentrations to adjust for baseline EPO levels. Then, pharmacokinetic parameters were calculated based on the corrected concentrations using the noncompartment model analysis (WinNonlin software version 6.1, Pharsight Corp.; Mountain View, CA, USA). The maximum DA concentration (C max), the time to reach the maximum concentration (t max), and the area under the concentration–time curve from time zero to the last sampling point (168 h) (AUC0–168) were assessed. Since the terminal phase could not be reliably identified, only these parameters were calculated.
Overall survival was defined as the time from the date of treatment with the first DA dose to the date of death. Patients who did not die were censored at the last date of follow-up. AML-free survival was defined as the time from the date of treatment with the first DA dose to the date at which the patient’s progression to AML was observed. Patients who did not progress to AML were censored at the last date of follow-up. The overall survival and AML-free survival rates were estimated by the Kaplan–Meier method.
Results
Patients
A total of 52 patients (17, 18, and 17 patients receiving 60, 120, and 240 μg DA, respectively), were randomized from December 2011 to February 2013 and received DA. Two patients in the 240 μg group who received an insufficient number of DA doses were excluded from PPS. Of 30 patients (10 patients in each dose group) whose serum DA concentration was measured, 2 in 60 μg group and 1 in 240 μg group were excluded from pharmacokinetic analysis set because their pharmacokinetic parameters could not be calculated. This set included 4, 5, and 5 Japanese patients, and 4, 5, and 4 Korean patients receiving 60, 120, and 240 μg DA, respectively. Of the 52 enrolled patients, 1 in 60 μg group did not provide consent to the outcome survey. The patient disposition is shown in Fig. 2. No significant differences in the demographic factors and disease characteristics were observed among the patients receiving different DA doses (Table 1).

Patient disposition. DA darbepoetin alfa, PPS per protocol set
Efficacy
The proportion of patients achieving an erythroid response during the initial dose evaluation phase (the primary endpoint) was 64.7 % (11 of 17 patients) in the 60 μg group, 44.4 % (8 of 18 patients) in the 120 μg group, and 66.7 % (10 of 15 patients) in the 240 μg group. The erythroid response rate was similar across the three groups (Fig. 3).

Major or minor erythroid response during initial dose evaluation phase (PPS). DA darbepoetin alfa, PPS per protocol set
The proportion of patients achieving a major erythroid response during the initial dose evaluation phase was 17.6 % (3 of 17 patients) in the 60 μg group, 16.7 % (3 of 18 patients) in the 120 μg group, and 33.3 % (5 of 15 patients) in the 240 μg group. The response rate was higher in the 240 μg group than in the other groups (Fig. 3). During the initial dose evaluation phase, the mean Hb levels continued to rise over the first 2 weeks of DA treatment and remained between 8.6 and 9.1 g/dL thereafter in the 240 μg group, whereas they were slightly lower in the 60 μg group (7.6–8.1 g/dL) and the 120 μg group (8.1–8.4 g/dL) (Fig. 4).

Plot of mean hemoglobin concentrations over time in the initial dose evaluation phase (PPS). DA darbepoetin alfa, PPS per protocol set
Dose escalation was allowed at week 17 and thereafter, which resulted in major erythroid responses in 1 patient with no response in the 60 μg group, and in 2 patients with minor erythroid responses each in the 60 and 120 μg groups.
The proportion of patients who experienced a temporal treatment discontinuation because of a Hb level of >11 g/dL in the initial dose evaluation phase was 11 % (2 patients) in the 120 μg group, and 23.5 % (4 patients) in the 240 μg group. The Hb level did not exceed 12.0 g/dL in either of these patients.
Of the 41 patients who started the extended treatment evaluation phase, the DA dosing frequency was adjusted from once weekly to biweekly in 5 patients. In 2 of these patients, the dosing frequency was subsequently readjusted to once weekly because their Hb levels decreased during the biweekly regimen. The Hb levels recovered in both patients, and they completed the study. The rest of these patients completed the study with their Hb levels maintains with 120 or 240 μg biweekly administration of DA.
As an explanatory assessment, the prognostic factors for DA efficacy were investigated. Our results indicate that patients with high Hb levels, low EPO levels, low serum ferritin levels, and those requiring low RBC transfusions volume tended to show higher major or minor erythroid response. On the other hand, age or bone marrow (BM) blast proportion was not associated with the patients’ major or minor erythroid response rate (Table 2). In terms of major erythroid response, similar trend was observed (data not shown).
Safety
In this study, adverse events leading to death included pneumonia and acute respiratory distress syndrome in 1 patient (6 %) each in the 60 μg group, and febrile neutropenia, pneumonia, and septic shock in 1 patient (6 %) each in the 120 μg group. These events were considered not related to DA treatment.
Other serious adverse events occurred in 14 (27 %) of 52 patients. A causal relationship with DA could not be ruled out in 1 patient with abulia who received 240 μg DA.
During the initial dose evaluation phase, adverse events occurred in 39 (75 %) patients. Nasopharyngitis had the highest incidence and occurred in 11 (21 %) patients. Other adverse events that occurred in at least 10 % of patients included diarrhea in 8 patients (15 %) and headache in 6 patients (12 %).
From week 1 to week 48, adverse events occurred in 48 (92 %) of 52 patients (Table 3). Hypertension was reported in 6 (11.5 %) patients [3 (17.6 %) in the 60 μg group, 1 (95.6 %) in the 120 μg group, and 2 (11.8 %) in the 240 μg group]. No remarkable differences in the incidence of adverse events or adverse reactions between the different groups were noted during the initial dose evaluation phase or from week 1 to week 48.
The anti-DA antibody test was negative in all patients.
Pharmacokinetic evaluation
Serum DA concentration profiles were similar in patients receiving 60 or 120 μg DA until 72 h after administration of the drug. Thereafter, the concentration increased in patients receiving 60 μg DA. A higher concentration profile was observed in the 240 μg group compared to the 60 and 120 μg groups (Supplementary Fig. 1). The mean t max were 82.84, 73.36, and 60.84 h in the 60, 120, and 240 μg groups, respectively. No dose proportionality was observed for C max and AUC0–t for 60–240 μg DA (Supplementary Table 1). No remarkable changes in serum DA trough concentration in any group were observed throughout the treatment period. There was no relationship between the serum DA trough concentration at week 17 and efficacy (minor erythroid response and major erythroid response) in the initial-dose evaluation phase (data not shown).
Survival and AML progression
The outcome survey was conducted within a median follow-up period of 316 days (range 1–600 days) from the start of treatment with DA. At the time of the survey, death was reported in 7 (14 %) of 51 patients (1, 3, and 3 patients in the 60, 120, and 240 μg DA groups, respectively). The one-year survival rate was 84.5 % (Fig. 5a). The causes of death were pneumonia in 2 patients, and cardiac failure, geromarasmus, subdural haematoma, AML, and septic shock in 1 patient each.

Overall and AML-free survival. The graphs depict overall patient survival (a), and AML-free survival (b). AML acute myeloid leukemia
Progression to AML was reported in 2 patients receiving 240 μg DA. The proportion of patients who did not progress to AML within 1 year was 96.0 % (Fig. 5b). The proportion of the total number of patients who progressed to AML 1 year after treatment initiation was 3.9 % (2 of 51 patients). One of them was a 69-year-old woman with refractory anemia with excess blasts (RAEB) by the French-American-British (FAB) classification and RAEB-1 by the 2008 World Health Organization (WHO) classification. At screening, BM blast proportion was 2.4 %, and peripheral blood blast proportion was 1.0 %. The patient started treatment with 240 μg DA. At week 16, her peripheral blood blast proportion increased to 5.0 %. The patient withdrew from the study at week 17 in accordance with the prespecified criteria for withdrawal (lack of efficacy). Progression to AML type M5 (FAB classification) was confirmed 3 days after the withdrawal with BM blast proportion of 87.8 %. Another was a 70-year-old man with RAEB by the FAB classification and RAEB-1 by the 2008 WHO classification. At the screening examinations, BM blast proportion was 7.1 %. The patient started treatment with 240 μg DA. After treatment initiation, peripheral blood blast proportion remained at 0 %. The patient was hospitalized for pneumonia on day 75, and DA administration was discontinued in week 12 (day 79). The patient was removed from the study on day 119 in accordance with the prespecified criteria for withdrawal (lack of efficacy). On day 148, progression to AML (type unknown) was confirmed by BM blast proportion of 51.4 %. The patient died due to AML 200 days after the initial study treatment.
Discussion
Recommended DA dosage
This study is the first prospective randomized controlled trial aiming to determine an optimal initial DA dosage in MDS patients with IPSS low or intermediate-1 risk, an EPO level of ≤500 mIU/mL, and who were RBC transfusion-dependent.
The proportion of patients with major or minor erythroid response in the initial dose evaluation phase was comparable between the groups, whereas the major erythroid response was observed more frequently in the 240 µg group.
From a clinical perspective, major erythroid response is more important than minor erythroid response since eliminating RBC transfusion dependency is more significant for patients than decreasing the transfusion volume. Hb levels started to increase 2 weeks after starting treatment and remained around 9.0 g/dL in patients receiving 240 µg DA, whereas no apparent increase in Hb levels was noted in the 60 or 120 µg groups. Our safety analysis revealed no dose-dependent adverse events. Hb levels exceeded 11 g/dL in 4 patients in 240 µg, but returned to 11 g/dL or lower shortly after treatment interruption. No persistent increase in Hb levels was reported. Although our analysis revealed no dose dependency for the major or minor erythroid responses, we considered a DA dose of 240 µg once weekly to be the most appropriate initial dose based on the major erythroid response rates and the changes in Hb levels. The response rate of 66.7 % in the 240 µg group is comparable to rates reported in MDS patients in Europe (61.0 and 66.6 %) with IPSS low or intermediate-1 risk and RBC transfusion dependency who received DA at 300 µg once weekly [11, 23].
Predictive factors for DA efficacy
Patients with low serum EPO levels and low transfusion requirements have been shown to have a relatively high response rate [24]. This trend was also noted in this study. In addition, the response rate was high in patients with high Hb and low serum ferritin levels. All the 3 patients with the IPSS-R [22] very low category showed responses. These data indicate that patients with milder MDS may have a greater erythroid response to DA.
Although this was not an endpoint of this study, we examined the relationship between a change in absolute reticulocyte (Ret) count and erythroid response. Bowen et al. evaluated the prognostic factors for drug efficacy in 21 low-risk MDS patients who received single doses of rHuEPO and G-CSF. They reported that an increase in Rets count by >30 × 109/L on day 7 was a promising prognostic factor [25]. In our study, 8 of 50 patients had an increase in the Ret count exceeding 30 × 109/L at week 2 (equivalent to day 7 in Bowen’s study), and all of them achieved a major or minor erythroid response during the initial dose evaluation phase. The absolute Ret increment (mean) at week 2 was 15 × 109/L in patients with a major or minor erythroid response and 2.3 × 109/L in patients with no response during the initial dose evaluation phase, suggesting that the change in Ret count in the initial 1-week period may be a good predictor of erythroid responses.
Serum ferritin levels
This study included patients who were already dependent on RBC transfusion. A retrospective study suggested that early treatment with ESAs in patients with IPSS low or intermediate-1 risk MDS may delay the need for RBC transfusion [26]. Elimination of RBC transfusion dependency and delayed transfusion may decrease the risk of transfusion-related iron overload and delay the use of an iron chelate.
In this study, 13 patients used iron chelate (Deferoxamine Mesylate in 2, Deferasirox in 11) during the study period. Although not preplanned, changes in serum ferritin [(pre − post)/pre × 100] were evaluated in 37 patients excluding the 13 chelate users. The median serum ferritin level changed from 502.4 to 400.1 ng/mL (−26.4 %; median duration of treatment, 329.5 days) in patients who did not require RBC transfusion anymore (n = 14 with major erythroid response), whereas it changed from 936.9 to 1445.1 ng/mL (43.9 %; median duration of treatment, 277 days) in patients who continued to receive transfusions (minor erythroid responders and non-responders) (Supplementary Table 2). The serum ferritin level decreased without iron chelate in patients with a major erythroid response, suggesting that the risk of iron overload may be diminished.
Safety
Hypertension is a known adverse reaction to ESAs [27]. Hypertension was reported in 6 of 52 patients (11.5 %) in this study. A causal relationship to DA was ruled out in 4 and deemed possible in 2 patients. Since this study enrolled patients who were dependent on RBC transfusion, hypertension reported as an adverse event was mostly associated with a temporary increase in Hb level due to transfusion. No excessive Hb increase (>12 g/dL) caused by DA and associated hypertension was reported in our study, as the treatment with DA was interrupted when the Hb level exceeded 11 g/dL.
Pharmacokinetics
This is the first study to investigate the pharmacokinetics of DA in patients with MDS. Nonlinear pharmacokinetics was observed for DA doses in the range of 60–240 μg. The exposure (C max and AUC0–t ) in the 60 μg DA group was higher compared to that in the 120 μg DA group. We could not determine the mechanism of the nonlinear pharmacokinetics, but we hypothesize that it might have been caused by the greater inter-patient variability in the 60 μg DA group compared to the other two groups.
Prognosis
Clinical studies in patients with chemotherapy-induced anemia reported adverse effects of ESAs (shortened survival time, advanced cancer and increased recurrence risk, higher incidence of thromboembolisms, and higher mortality) on the patients’ prognosis [28–32]. Since these studies used a target Hb level of ≥12.0 g/dL, a significant increase in Hb levels may be one of the reasons for this poor prognosis. The NCCN guidelines states that “the long term use of ESA in MDS patients compared to either randomized controls or historical controls have shown no negative impact of such treatment on survival or AML evolution” and “a target Hb level ≤12 g/dL” [3].
In this study, progression to AML was reported in 2 patients with RAEB-1 receiving 240 μg DA. Causal relationships to DA were not clear. Since 4 out of 5 patients classified as RAEB-1 at baseline happened to be randomized into 240 μg DA group [i.e., 0 (0 %), 1 (5.6 %), and 4 (23.5 %) patients in 60, 120, and 240 μg DA group], the progression to AML in this study might have been affected by baseline status of subjects.
Although the different sample sizes and study designs did not allow a direct comparison, the one-year survival rate and cumulative proportion of patients whose disease progressed to AML 1 year after starting treatment in our study were not significantly different from past clinical studies [33, 34].
Past studies suggested that treatment with ESAs may improve the prognosis of MDS patients [35, 36]. The comparison of the Kaplan–Meier curves of survival time between responders and non-responders in this study indicated a longer survival time in responders. Reasons for the different survival times may include (1) possible disease progression of MDS in non-responders, (2) prevention of progressive iron overload, and (3) improved cardiac function and performance status by improving anemia in responders who were not dependent on transfusions anymore or whose transfusion volume was decreased because of ESAs, as discussed in previous studies.
Conclusion
In conclusion, the efficacy and safety of DA in red blood cell transfusion-dependent patients with IPSS low or intermediate-1 risk MDS were demonstrated in the first randomized controlled trial of this kind. A weekly dose of 240 µg DA was shown to be an appropriate initial dose with a high treatment efficacy.
References
Malcovati L, Della Porta MG, Cazzola M. Predicting survival and leukemic evolution in patients with myelodysplastic syndrome. Haematologica. 2006;91:1588–90.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88.
NCCN Clinical Practice Guidelines in Oncology. Myelodysplastic syndromes, Ver.1.2016.
Italian Cooperative Study Group for rHuEpo in Myelodysplastic Syndromes, Ferrini PR, Grossi A, Vannucchi AM, Barosi G, Guarnone R, Piva N, et al. A randomized double-blind placebo-controlled study with subcutaneous recombinant human erythropoietin in patients with low-risk myelodysplastic syndromes. Br J Haematol. 1998; 103:1070–4.
Thompson JA, Gilliland DG, Prchal JT, Bennett JM, Larholt K, Nelson RA, et al. Effect of recombinant human erythropoietin combined with granulocyte/macrophage colony-stimulating factor in the treatment of patients with myelodysplastic syndrome. GM/EPO MDS Study Group. Blood. 2000;95:1175–9.
Greenberg PL, Sun Z, Miller KB, Bennett JM, Tallman MS, Dewald G, et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood. 2009;114:2393–400.
Casadevall N, Durieux P, Dubois S, Hemery F, Lepage E, Quarré MC, et al. Health, economic, and quality-of-life effects of erythropoietin and granulocyte colony-stimulating factor for the treatment of myelodysplastic syndromes: a randomized, controlled trial. Blood. 2004;104:321–7.
Musto P, Lanza F, Balleari E, Grossi A, Falcone A, Sanpaolo G, et al. Darbepoetin alpha for the treatment of anaemia in low-intermediate risk myelodysplastic syndromes. Br J Haematol. 2005;128:204–9.
Patton JF, Sullivan T, Mun Y, Reeves T, Rossi G, Wallace JF. A retrospective cohort study to assess the impact of therapeutic substitution of darbepoetin alfa for epoetin alfa in anemic patients with myelodysplastic syndrome. J Support Oncol. 2005;3:419–26.
Stasi R, Abruzzese E, Lanzetta G, Terzoli E, Amadori S. Darbepoetin alfa for the treatment of anemic patients with low- and intermediate-1-risk myelodysplastic syndromes. Ann Oncol. 2005;16:1921–7.
Mannone L, Gardin C, Quarre MC, Bernard JF, Vassilieff D, Ades L, et al. Groupe Francais des Myelodysplasies. High-dose darbepoetin alpha in the treatment of anaemia of lower risk myelodysplastic syndrome results of a phase II study. Br J Haematol. 2006;133:513–9.
Giraldo P, Nomdedeu B, Loscertales J, Requena C, de Paz R, Tormo M, et al; Aranesp in Myelodysplastic Syndromes (ARM) Study Group. Darbepoetin alpha for the treatment of anemia in patients with myelodysplastic syndromes. Cancer. 2006; 107:2807–16.
Gabrilove J, Paquette R, Lyons RM, Mushtaq C, Sekeres MA, Tomita D, et al. Phase 2, single-arm trial to evaluate the effectiveness of darbepoetin alfa for correcting anaemia in patients with myelodysplastic syndromes. Br J Haematol. 2008;142:379–93.
Gotlib J, Lavori P, Quesada S, Stein RS, Shahnia S, Greenberg PL. A Phase II intra-patient dose-escalation trial of weight-based darbepoetin alfa with or without granulocyte-colony stimulating factor in myelodysplastic syndromes. Am J Hematol. 2009;84:15–20.
Oliva EN, Nobile F, Alimena G, Specchia G, Danova M, Rovati B, et al. Darbepoetin alfa for the treatment of anemia associated with myelodysplastic syndromes: efficacy and quality of life. Leuk Lymphoma. 2010;51:1007–14.
Ross SD, Allen IE, Probst CA, Sercus B, Crean SM, Ranganathan G. Efficacy and safety of erythropoiesis-stimulating proteins in myelodysplastic syndrome: a systematic review and meta-analysis. Oncologist. 2007;12:1264–73.
Moyo V, Lefebvre P, Duh MS, Yektashenas B, Mundle S. Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Ann Hematol. 2008;87:527–36.
Aul C, Arning M, Runde V, Schneider W. Serum erythropoietin concentrations in patients with myelodysplastic syndromes. Leuk Res. 1991;15:571–5.
Hellström-Lindberg E. Efficacy of erythropoietin in the myelodysplastic syndromes: a meta-analysis of 205 patients from 17 studies. Br J Haematol. 1995;89:67–71.
Hellström-Lindberg E, Negrin R, Stein R, Krantz S, Lindberg G, Vardiman J, et al. Erythroid response to treatment with G-CSF plus erythropoietin for the anaemia of patients with myelodysplastic syndromes: proposal for a predictive model. Br J Haematol. 1997;99:344–51.
Hellström-Lindberg E, Gulbrandsen N, Lindberg G, Ahlgren T, Dahl IM, Dybedal I, Scandinavian MDS Group, et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol. 2003;120:1037–46.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Villegas A, Arrizabalaga B, Fernández-Lago C, Castro M, Mayans JR, González-Porras JR, et al. Darbepoetin alfa for anemia in patients with low or intermediate-1 risk myelodysplastic syndromes and positive predictive factors of response. Curr Med Res Opin. 2011;27:951–60.
Santini V, Schemenau J, Levis A, Balleari E, Sapena R, Adès L, et al. Can the revised IPSS predict response to erythropoietic-stimulating agents in patients with classical IPSS low or intermediate-1 MDS? Blood. 2013;122:2286–8.
Bowen D, Hyslop A, Keenan N, Groves M, Culligan D, Johnson P, et al. Predicting erythroid response to recombinant erythropoietin plus granulocyte colony-stimulating factor therapy following a single subcutaneous bolus in patients with myelodysplasia. Haematologica. 2006;91:709–10.
Park S, Kelaidi C, Sapena R, Vassilieff D, Beyne-Rauzy O, Coiteux V, et al. Early introduction of ESA in low risk MDS patients may delay the need for RBC transfusion: a retrospective analysis on 112 patients. Leuk Res. 2010;34:1430–6.
Kelaidi C, Fenaux P. Darbepoetin alfa in anemia of myelodysplastic syndromes: present and beyond. Expert Opin Biol Ther. 2010;10:605–14.
Henke M, Laszig R, Rübe C, Schäfer U, Haase KD, Schilcher B, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet. 2003;362:1255–60.
Overgaard J, Hoff CM, Hansen HS, Specht L, Overgaard M, Grau C, et al. Danish Head and Neck Cancer Group (DAHANCA). Randomized study of darbepoetin alfa as modifier of radiotherapy in patients with primary squamous cell carcinoma of the head and neck (HNSCC): final outcome of the DAHANCA 10 trial [Abstract]. J Clin Oncol. 2009;27:6007.
Leyland-Jones B, Semiglazov V, Pawlicki M, Pienkowski T, Tjulandin S, Manikhas G, et al. Maintaining normal hemoglobin levels with epoetin alfa in mainly nonanemic patients with metastatic breast cancer receiving first-line chemotherapy: a survival study. J Clin Oncol. 2005;23:5960–72.
Luksenburg H, Weir A, Wager R. FDA Briefing document: safety concerns associated with Aranesp (darbepoetin alfa) Amgen, Inc. and Procrit (epoetin alfa) Ortho Biotech, L.P., for the treatment of anemia associated with cancer chemotherapy. ODAC 2004. Available from http://www.fda.gov/ohrms/dockets/ac/04/briefing/4037b2_04_fda-aranesp-procrit.htm. Accessed 3 Oct 2014.
Smith RE Jr, Aapro MS, Ludwig H, Pintér T, Smakal M, Ciuleanu TE, et al. Darbepoetin alfa for the treatment of anemia in patients with active cancer not receiving chemotherapy or radiotherapy: results of a phase III, multicenter, randomized, double-blind, placebo-controlled study. J Clin Oncol. 2008;26:1040–50.
Golshayan AR, Jin T, Maciejewski J, Fu AZ, Bershadsky B, Kattan MW, et al. Efficacy of growth factors compared to other therapies for low-risk myelodysplastic syndromes. Br J Haematol. 2007;137:125–32.
Revlimid Capsules 5 mg [Guide to Proper Use]. Tokyo: Celgene K.K. 2011. Available from http://www.revlimid-japan.jp/professional/product/pdf/guide/guide_rev_mds_201008.pdf. Accessed 9 Oct 2014.
Park S, Grabar S, Kelaidi C, Beyne-Rauzy O, Picard F, Bardet V, et al. GFM group (Groupe Francophone des Myélodysplasies). Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood. 2008;111:574–82.
Jädersten M, Malcovati L, Dybedal I, Della Porta MG, Invernizzi R, Montgomery SM, et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol. 2008;26:3607–13.
Acknowledgments
We would like to thank all of the participating patients and their families as well as investigators, research nurses, study coordinators, and operations staff.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest and sources of funding
Kyowa Hakko Kirin Co., Ltd. sponsored this clinical study and was responsible for medical monitoring and auditing. R. Shimazaki is an employee of and owns stock in Kyowa Hakko Kirin Co., Ltd. J. H. Jang, H. Harada, H. Shibayama, H. J. Kim, and K. Mitani were principle investigators for this trial. HJ Kim and K Mitani were coordinating investigators of this trial and received consulting fees from Kyowa Hakko Kirin Co., Ltd. K. Sawada was the medical adviser of this trial and received consulting fees from Kyowa Hakko Kirin Co., Ltd. H. Harada has received grant from Nippon-Shinyaku Co., Ltd., Novartis Pharma outside the submitted work, and personal fees from Nippon-Shinyaku Co., Ltd., Novartis Pharma, and Celgene K. K. outside the submitted work. H. Shibayama has received personal fee from Kyowa Hakko Kirin Co., Ltd. outside the submitted work. K. Mitani has received grant from Kyowa Hakko Kirin Co., Ltd., Chugai Pharmaceutical Co., Ltd., Novartis Pharma, Bristol-Myers Squibb outside the submitted work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Jang, J.H., Harada, H., Shibayama, H. et al. A randomized controlled trial comparing darbepoetin alfa doses in red blood cell transfusion-dependent patients with low- or intermediate-1 risk myelodysplastic syndromes. Int J Hematol 102, 401–412 (2015). https://doi.org/10.1007/s12185-015-1862-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-015-1862-5