
Abstract
Purpose of Review
Primary mitochondrial disorders (PMD) are a heterogeneous group of individual genetic multi-systemic diseases that are challenging to diagnose and manage; currently, there is no cure or FDA-approved therapies for these progressive genetic syndromes. Among the many organs that may be affected by mitochondrial disorders, the heart is one of the most common, given its high energy requirements, leading to mitochondrial cardiomyopathies.
Recent Findings
Mitochondrial cardiomyopathies are due to underlying genetic defects in genes involved in mitochondrial functioning. These genes, which can be of nuclear or mitochondrial DNA, are either directly involved in the electron transport chain and oxidative phosphorylation or play a role in other mitochondrial pathways such as mitochondrial DNA (mtDNA) replication or maintenance of the inner mitochondrial membrane. Due to the high degree of variability and complexity, current therapeutic strategies are inadequately effective in treating mitochondrial cardiomyopathies. Further research, including longitudinal prospective natural history studies and large-scale randomized clinical trials, is warranted to determine the most effective therapeutic and pharmacologic strategies to address mitochondrial cardiomyopathies.
Summary
In this review, we present our current understanding of mitochondrial cardiomyopathies, diagnostic tools, and management.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myocytes heavily rely on mitochondria for bioenergetic demands and energy consumption. Defects in mitochondrial function, therefore, impact the physiological functioning of myocytes. Primary mitochondrial disorders are due to pathogenic variants in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) and many of these have been known to involve cardiac function and/or structure. [1•, 2] While primary mitochondrial disorders may cause electrical disturbances, including Wolff-Parkinson-White, supraventricular tachycardia, bundle branch block, and other arrhythmias, this review will focus on mitochondrial cardiomyopathies. Disruption in bioenergetic mechanisms has recently been shown to contribute to several forms of heart failure. [3] While traditional cardiology focuses on general causes of pathologies causing heart conditions, the role of primary mitochondrial disorders and their impact on structure and function is recently becoming more apparent. We present a review of our current clinical understanding of cardiomyopathies known to be caused by primary mitochondrial disorders including clinical features, diagnosis, and management.
Primary Mitochondrial Disorders
Primary mitochondrial disorders (PMD) are a group of heterogeneous disorders often affecting multiple organ systems, especially those with the highest energy requirements including but not limited to the brain, skeletal muscle, endocrine, renal, ophthalmologic, sensorineural hearing, and cardiac conduction system and cardiac muscle. [4•] Disease manifestations may occur at any age and rarely can affect single rather than multiple organs, including isolated familial hypertrophic cardiomyopathy. [5] PMD, therefore, generally need coordinated multi-specialty consultations for thorough evaluation and management. Pathogenic variants in the genes that contribute to mitochondrial machinery cause insufficient energy production needed for the normal functioning of all cells, in addition to the creation of excess reactive oxygen species (ROS). The clinical course can be difficult to predict; some PMDs are progressive with high morbidity and mortality early in life, while others have a slowly progressive course with long periods of stability.
The prevalence of primary mitochondrial disorders is currently estimated to be about 1:6000, with an estimated carrier frequency of a pathogenic variant in mtDNA at 1:200. [6] Mitochondrial proteins are under the control of two genomes, nDNA and mtDNA. The maternally inherited mtDNA is present in hundreds to thousands of copies per mitochondrion, with numerous mitochondrion per cell; the absolute number varies from organ to organ based on unique energy requirements and demands The mtDNA encodes 37 genes in total; 13 essential polypeptides for the mitochondrial oxidative phosphorylation (OXPHOS) complexes: seven subunits of complex I (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6) one subunit of complex III (cytochrome b, cytb), three subunits of complex IV (COI, COII, and COIII), and two subunits of complex V (ATP6 and ATP8). The mtDNA-encoded hydrophobic polypeptides are translated in situ on mitochondrial ribosomes, which employ 22 tRNAs and 2 rRNAs also coded by the mtDNA. The remainder of genes necessary for mitochondrial function, estimated at > 1000, are nuclear-encoded, with pathogenic variants having an inheritance pattern that can be autosomal dominant, autosomal recessive, or X-linked.
Mitochondrial Involvement in Cardiomyopathies
Cardiomyopathies are a diverse group of pathologies characterized by structural and functional changes in the heart. Based on the American College of Cardiology/American Heart Association (ACC/AHA) stage and New York Heart Association (NYHA) functional class, MOGE(S) nosology encompasses these characteristics, which include morphofunctional phenotype (M), organ (s) involved (O), genetic inheritance pattern (G), etiological annotation (E), and functional status (S). [7] One of the essential first steps is to differentiate whether the origin of cardiomyopathy is mitochondrial. An instrumental identifying feature is the presence of isolated cardiomyopathy versus presentation as part of a multisystem disorder. Cardiomyopathy involving multiple systems with an unknown cause should raise concerns for mitochondrial cardiomyopathy (MCM). [8•, 9] Once the clinical examination and systematic screening of organs have established that cardiomyopathy is a part of a multi-system disorder, the patient should be referred to appropriate specialists, including a cardiac genetics clinic. [1•, 8•, 9,10,11] If the patient presents to a cardiologist, additional findings of central nervous system involvement (global developmental delay, regression, epilepsy), renal involvement, sensorineural hearing loss, diabetes, and/or skeletal myopathy (weakness, fatigue, exercise intolerance) would be a good indication to suspect an underlying primary mitochondrial disorder that requires a multidisciplinary approach.
Physiologic stressors such as infection, fever, fasting/starvation, dehydration, surgery, and anesthesia can trigger symptom onset or decline, sometimes referred to as a “mito crash.” Certain cells, such as neurons, muscle cells (skeletal and cardiac), and the liver, have higher bioenergetic demands and are prominently impacted due to abnormal mitochondrial performance. Due to the high energy requirements of myocytes, cardiac manifestations are a key feature of mitochondrial diseases, with cardiomyopathies being one of the most common. [1•, 12] Twenty to forty percent of children and 30% of adults with mitochondrial disease have been reported to suffer from some form of cardiomyopathy. [13,14,15,16,17].
In a retrospective review of 113 pediatric patients with mitochondrial disease, the prevalence of cardiomyopathy was 40%. The mean presentation age was 33 months. In this cohort, 58% had hypertrophic cardiomyopathy, 29% had dilated cardiomyopathy, and 13% had left ventricular non-compaction. This report further highlighted that the patients with cardiomyopathy had an 18% survival rate to age 16 years as compared to 92% survival in children without cardiomyopathy. [18•].
Several studies on adult patients with mtDNA mutations have reported progressive cardiac disease. Wahbi et al. in 2015 in a retrospective study of 260 adults reported that 30% of the patients with primary mitochondrial disease had cardiac involvement at the time of diagnosis. [19] In another case series of 32 adult patients diagnosed with mitochondrial disease, 69% had mtDNA mutation, and 81% had evidence of cardiac involvement with EKG abnormalities and/or cardiomyopathy (19% hypertrophic; 3% restrictive and 3% left ventricular non-compaction). In the same study on future follow-up, two patients developed hypertrophic cardiomyopathy, and one with NARP developed peripartum dilated cardiomyopathy. [13] When a cardiac disease is present, morbidity and mortality may be increased. [18•].
Cardiac remodeling and subendocardial dysfunction can occur in MD patients without clinical cardiac manifestation. MCM and increased risk of cardiac involvement must be considered in high-risk patients (with higher mutation load and disease burden) who might not have known mitochondrial disease, as cardiac presentation might be the first or even the only clinical manifestation. [5, 20,21,22,23,24] Advanced imaging techniques such as cardiac magnetic resonance (CMR) can be helpful in the early diagnosis of high-risk patients. Bates et al. (2013) in an MRI study of 22 mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) patients (m.3243A > G) without known cardiac involvement reported significant pathological cardiac findings, which included increased left ventricular mass index (LVMI), left ventricular mass to end-diastolic volume ratio (LVM/EDV), and wall thicknesses as compared to controls. [25].
Forms of Cardiomyopathy Associated with Mitochondrial Diseases (Table 1)
The presentation of cardiomyopathy in patients affected by mitochondrial diseases may vary significantly, ranging from being asymptomatic to manifestations such as heart failure, arrhythmias, and sudden cardiac death. [26] MCM most commonly includes hypertrophic cardiomyopathy, dilated cardiomyopathy, left ventricular non-compaction, or restrictive cardiomyopathy (RCM), generally in the absence of valvular disease, coronary artery disease, or hypertension. [1•, 4•, 27•, 28,29,30].
Hypertrophic Cardiomyopathy (HCM)
HCM is the most common form of cardiomyopathy caused due to genetic mutations with a prevalence of 1:500. [31] Due to the high energy demands, the cardiac cells undergo compensatory mitochondrial proliferation due to the mitochondrial defect. HCM has been reported in 40–50% of MCM cases. In addition to HCM being the most common primary cardiomyopathy, it is the most common presentation of MCM involving hypertrophic remodeling with the left ventricular wall thickness of ≥ 15 mm (in adults) in the absence of loading conditions (such as hypertension, valvular disease, etc.) contributing to wall thickening. [32,33,34] In the pediatric population, wall thickening of more than two standard deviations above the mean is a diagnosis of HCM. [32] Some mitochondrial disorders that feature HCM include mitochondrial DNA variants listed in Table 1.
Dilated Cardiomyopathy (DCM)
DCM is a major cause of heart failure, with a reported prevalence of 1:2500. [35, 36] DCM is also one of the major indications for cardiac transplants. It is characterized by dilatation and impaired functioning of one or both ventricles leading to heart failure. The clinical presentation includes signs of congestive heart failure, such as dyspnea, orthopnea, and congestive edema. Patients may also present with either atrial or ventricular arrhythmias and sometimes sudden cardiac death. [35,36,37] DCM accounts for up to 60% of cardiomyopathies among the pediatric population. [38, 39] Weintraub et al. have reported that 35% of DCM are caused due to genetic causes. [40] The exact percent of mitochondrial-related DCM is, however, not exactly known. [40,41,42] Mitochondrial diseases presenting with DCM include but not limited to are MELAS, Maternally inherited diabetes deafness (MIDD), LHON, and Barth syndrome (Table 1).
Restrictive Cardiomyopathy (RCM)
RCM is the least common of major cardiomyopathies and is characterized by stiff ventricular walls leading to diastolic dysfunction, dilated atria, and elevated end-diastolic pressure. [31, 34] Appearance of “granular” echoes on transthoracic echocardiography (TTE) is generally linked to cardiac amyloidosis when diagnosing RCM. However, RCM is also associated with mitochondrial disorders, including but not limited to MELAS, MIDD (Table 1). Specific imaging features on TTE can be a useful tool to screen amyloid deposits from secondary infiltrative cardiomyopathy. [43].
Left Ventricular Non-compaction (LVNC)
LVNC is also known as left ventricular hypertrabeculation or noncompaction cardiomyopathy (NCCM) and is characterized by left ventricular trabeculations, deep intertrabecular recesses along with a thin epicardial layer. LVNC is considered genetic cardiomyopathy by the American Heart Association and has been associated with several mitochondrial diseases (Table 1).
Histiocytoid Cardiomyopathy (HICMP)
HICMP is a rare genetic disorder of the pediatric population characterized by cardiac arrhythmias, Wolff-Parkinson-White (WPW) syndrome, or dilated cardiomyopathy. [44, 45] HICMP has been reported to also be associated with LVNC and MERF. [46] Key histological findings of HICMP include yellow-tan nodules on the epicardium, subendocardium, or cardiac valves consisting of histiocytoid-like cells with foamy granules. These cells have an increased number of normal or dysfunctional mitochondria, and many have called for classifying HICMP as a primary mitochondrial disease, including AHA [31, 44].
Other Mitochondrial Diseases with Cardiomyopathies
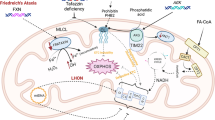
Some other mitochondrial diseases with cardiac pathology include Barth syndrome (OMIM 302,060), Friedreich ataxia (OMIM 229,300), TMEM70-related mitochondrial complex V deficiency (OMIM 614,052), and Sengers syndrome (OMIM 212,350). [26, 47].
Mutations in the gene taffazzin (TAZ) cause Barth syndrome which is an X-linked recessive disorder, seen mainly in males. TAZ is an inner mitochondrial membrane protein that plays a role in the remodeling of cardiolipin. [48] DCM and LVNC are more commonly observed in Barth syndrome as compared to HCM. Boys with Barth syndrome commonly have neutropenia, facial dysmorphic features, and skeletal myopathy, and can have intellectual disability. 3-Methylglutaconic aciduria is a key abnormality on metabolic screening labs (urine organic acids). HCM is a cardiac manifestation of Friedreich ataxia (FA) which is an autosomal recessive disorder due to trinucleotide repeats in the frataxin (FXN) gene. TMEM70-related mitochondrial complex V deficiency, also known as neonatal mitochondrial encephalocardiomyopathy, has severe early onset HCM as one of the key features, and is common in the Romani population with a common founder splice site pathogenic variant. [49].
Sengers syndrome, also known as mitochondrial depletion syndrome 10 (MTDPS10), is an autosomal recessive disorder caused by pathogenic variants in acylglycerol kinase gene (AGK). AGK gene assists in the assembly of the mitochondrial adenosine nucleotide transporter ANT1. [50] Sengers syndrome is characterized by HCM, congenital cataracts, myopathy, and lactic acidosis.
Diagnosis of Mitochondrial Cardiomyopathies
An extensive integrated diagnostic strategy includes a thorough physical exam, patient history, family history, biochemical metabolic screening labs, histopathological studies, functional assays, molecular genetic analysis, and cardiac workup, including EKG and cardiac imaging. Although MCM can be manifested as various forms of cardiomyopathies, HCM is the most common cardiac phenotype in MD. Early stages of MCM include features of heart failure with preserved ejection fraction and worsening diastolic dysfunction. [51] In some cases, cardiac imaging, such as echocardiography and cardiac MRI, is essential for the diagnosis and monitoring of progressive MCM. [25] Some of the tests employed for diagnosing cardiomyopathies are discussed below.
Laboratory Tests and Histology
While development of cardiac manifestations could be due to the underlying primary mitochondrial disorder, the treating clinician should also be on alert for secondary causes of cardiac involvement. Depending on the systems involved, such as renal, endocrine, or gastrointestinal, various lab values could deviate from the normal, sometimes indicating the etiology of cardiomyopathy. Liver enzymes and creatine kinase levels could be elevated on lab investigation. Increased TSH level would indicate endocrinological involvement. Specific assays to interrogate infectious etiologies such as viral or parasitic could also be helpful. Drug abuse, such as alcohol, is one of the agents leading to cardiomyopathy. [40, 52] The thiamine level is an indicator of alcohol abuse. Prognostic stratification could also be determined by BNP and renal function levels. [53, 54].
Metabolic screening labs for mitochondrial dysfunction include lactate (caution: this may be elevated due to poor tissue perfusion), pyruvate, ammonia (to interrogate the urea cycle), acylcarnitine profile (to interrogate the fatty acid oxidation cycle), plasma amino acids, urine organic acids (which can show elevated 3-methlglutaconic acid or Krebs cycle intermediates), and urine amino acids (to check for renal tubular acidosis). Expanded testing can include glutathione (a marker of oxidative stress), coenzymeQ10 level, and newer growth factor-related biomarkers such as FGF-21 and GDF-15.
Functional assays interrogating the respiratory chain function of skeletal muscle (vastus lateralis) have historically been an integral part of diagnosing PMD. These assays include activities of mitochondrial respiratory chain complexes I–IV and citrate synthase. Muscle tissue can also undergo mitochondrial DNA sequencing and deletions, which may detect pathogenic variants not seen in noninvasive testing, such as blood or buccal swab due to tissue heteroplasmy or mutation load, which can differ from tissue to tissue. Histology may show ragged-red fibers, which may stain negative for cytochrome c oxidase (COX) and positive for succinate dehydrogenase (SDH) on skeletal muscle histology, which are classic findings of mitochondrial pathology in PMDs. [1•, 55,56,57].
Electron microscopy has been one of the most informative and direct observational tools in diagnosing mitochondrial proliferation, structure, size, mitochondrial cristae integrity, and foreign inclusions in muscle biopsies. [58, 59] Recent advances in imaging, such as real-time confocal imaging, although they have provided an immense understanding of mitochondrial dynamics, are yet to fully prove clinical utilities in the diagnostic process.
Electrocardiogram (EKG)
EKG is one of the first and most common tests for any suspicion of cardiac etiology. While the findings on EKG are generally non-specific, Limongelli et al. reported progressive changes in the EKG in cardiomyopathy. [13] Findings on the EKG could indicate ventricular hypertrophy, conduction changes such as PR elongation, AV blocks, left bundle branch blocks (LBBB), and pathological Q waves, to highlight a few. [37, 53, 60].
Echocardiography
Echocardiography is an essential tool to investigate the kinetic and structural changes of the ventricles. Most commonly, 2D echocardiography is used but the images can be challenging to obtain due to patient-to-patient variability. Specific findings on echocardiography can be useful for certain cardiomyopathies, such as left ventricular wall thickness > 15 mm is typical of HCM. However, to determine a more accurate size and function of cardiac chambers, an alternative 3D echocardiography might be more reproducible. [61]
Cardiovascular Magnetic Resonance (CMR)
Cardiac involvement is a frequent finding in MD patients. [62] Abnormal CMR findings could include an impaired left ventricular ejection-fraction (LV-EF < 60%), unexplained LV hypertrophy, late-gadolinium-enhancement (LGE)-positive features, higher maximal wall thickness, and concentricity (LV mass to end-diastolic volume). A study by Florian et al. in 2015 was aimed at characterizing the prevalence and pattern of cardiac abnormalities and testing the additional diagnostic value of CMR in mitochondrial disease patients. The cohort (n = 64) included CPEO/KSS (n = 33), MELAS/–like (n = 11), MERRF (n = 3), and other non-specific mitochondrial disease forms (n = 17). The results indicated that 53% of 64 prospectively studied mitochondrial myopathy adult subjects had cardiac MRI abnormalities. Notably, pathological CMR findings indicating cardiac involvement were detected significantly more often than pathological ECG results or elevated cardiac serum biomarkers. [63] CMR has also been reported to detect wall thickness with higher sensitivity than echocardiography. [64] Furthermore, CMR is indicated in the initial workup of DCM as it could provide information on etiology. Inflammation can be suspected based on the enhancement of gadolinium by necrotic or scar tissues, especially if associated with edema and hyperemia. [65, 66] Cardiac magnetic resonance spectroscopy (MRS) is a novel tool which allows assessment of cardiac bioenergetics in vivo and may shed light on abnormal mitochondrial dysfunction and cardiac remodeling.
Endomyocardial Biopsy
A cardiac biopsy is indicated when the treatment is dictated by the diagnosis, for example, in cases of hemochromatosis, sarcoidosis, and myocarditis. [40, 53, 67] Although not routine, biopsies of organs could sometimes reveal a diagnosis of mitochondrial disease. Electron microscopy may reveal abnormal cristae formation, gigantic mitochondria, abnormal inclusions, or onion peeling appearance of the mitochondria. Recently, a case report by Marua et al. reported a diagnosis of Leigh syndrome using an endomyocardial biopsy after skeletal muscle biopsy did not reveal any obvious findings of mitochondrial disorder. [68].
Molecular and Genetic Testing
Genetic testing and counseling are essential components of the diagnostic work-up for MCM, often including the patient’s family members. Pedigree analysis and mode of inheritance pattern are some of the essential first steps in identifying the diagnosis. The maternal inheritance pattern strongly indicates that the presentation could be an MCM due to a pathogenic, maternally inherited mtDNA variant. [51] Advanced molecular workup involving not only nuclear DNA but also mitochondrial DNA may be useful to uncover an underlying mutation. Availability of testing may vary based on location, financial factors including insurance reimbursement, and type of testing. Some centers may offer panel-based testing, including nuclear and mitochondrial DNA genes, while others may offer whole exome or whole genome sequencing. The patient should receive proper counseling and review of the pros and cons of genetic testing. [40, 69, 70].
Clinical Case Presentations of Patients with Mitochondrial Disease and Cardiac Manifestations
Clinical Case #1: Biallelic Pathogenic C1QBP Variants [71]
A 29-year-old male presented four years after ICD placement for septal thickening and normal LVEF, with new onset dyspnea. His echocardiogram showed an LVEF of 15%. He was stabilized on heart failure therapy. One month later, he presented with 20 lb weight loss, anorexia, and diarrhea. On exam, he had CPEO and skeletal muscle weakness. A repeat echocardiogram showed LVEF of 5–10% with concentric hypertrophy and LV thrombi. Despite CICU care, he required ECMO and eventually needed a cardiac transplant. Genetic analysis using whole-exome sequencing (WES) revealed biallelic pathogenic variants in C1QBP (c.612C > G, p.F204L) and a de novo deletion of 17p13.2. Mitochondrial mtDNA analysis on heart explant showed multiple large-scale mtDNA deletions with 33% heteroplasmy. Only 12 patients exhibiting biallelic C1QBP variants have been reported with a high degree of clinical variability. Of the reported cases, skeletal and cardiac myopathies were common in addition to chronic progressive external ophthalmoplegia and lactic acidosis. Forty-one percent of the reported cases were diagnosed with cardiomyopathy in the first decade of life. [72,73,74].
Clinical Case #2: RMND1-Related Mitochondrial Disease [75, 76]
A 12-year-old male presents with a history of global developmental delay, hypotonia, sensorineural hearing loss status post cochlear implants, chronic kidney disease status post-renal transplant, and chronic systolic congestive heart failure. In the neonatal period, he developed respiratory distress; had multiple cardiac arrests, pulmonary hypertension, and pneumothorax; and required extracorporeal membrane oxygenation (ECMO) for 9 days and a ventilator for seventeen days. By 4 months, he had hypotonia and motor delays, and at 9 months, sensorineural hearing loss. Cochlear implants were placed at thirteen and fifteen months. At 18 months, he was diagnosed with failure to thrive, gastroesophageal reflux disease, and feeling aversion, which required the placement of a gastrostomy tube. By age four, he had developed hypertrophic cardiomyopathy from chronic hypertension versus underlying disease. At age six, his cardiomyopathy progressed while he was affected by influenza (ejection fraction decreased from 50 to 20%). At that time, cardiac catheterization was performed as part of the workup for renal transplant. His heart biopsy showed marked cardiomyocyte hypertrophy without fibrosis. However, the electron microscopy showed normal architecture of mitochondria. He received a renal transplant 6 months later. He demonstrated significant improvement after the renal transplant, with improved motor skills, resolution of hypertension, and improvement in cardiomyopathy. WES revealed a previously reported missense mutation c.713A > G, p.(Asn238Ser), and c.1317 + 1G > T splice mutation in gene RMND1. The patient presented several years later with acute on chronic systolic heart failure with worsening renal function and AKI in the setting of chronic kidney disease. He was admitted for treatment of fluid overload, needing diuresis, and milrinone. Shortly after admission, the echocardiography showed stable poor cardiac function with an ejection fraction of 20–25%. He became worse over the next few weeks and his kidneys made modest recovery. His parents elected for compassionate withdraw of care.
Clinical Case #3 (Unpublished): ACAD9 Mutation
A 29-year-old female with concentric LVH of unclear etiology and reported a history of suspected mitochondrial disease in childhood presented with profound shock and lactic acidosis (peak 17), with an axillary Impella. Her hospital course was complicated by acute loss of pulses in the right hand requiring a right axillary cut-down and thrombectomy with the removal of Impella, VA-ECMO, ventilator-dependent respiratory failure requiring tracheostomy, hospital-acquired pneumonia, pulmonary embolism/DVT, and AKI needing intermittent hemodialysis. She showed recovery during her hospital course, her LVEF was 45–50%. She did not tolerate any neurohormonal blockade due to hypotension. Muscle biopsy was obtained and showed myopathy and atrophy with electron microscopy showing mitochondria with widened cristae and dense deposits with scattered mitochondria. Her electron transport chain testing, however, did not show a complex I deficiency. Molecular and genetic analysis using WES showed compound pathogenic/likely-pathogenic variants in ACAD9 (c.1594 C > T (p.R532W) and c.1646 G > A (p.R549Q)), consistent with a diagnosis of ACAD9-related disease. No pathogenic variants were found on mtDNA sequencing. She was started on high-dose riboflavin in addition to dietary changes to reduce long-chain fat intake and consideration of medium-chain fat supplementation.
Clinical Case #4 (Unpublished): MT-TL1 (m.3243A > G): MELAS
A 50-year-old male presented with fatigue and exercise intolerance, thin body habitus, and diabetes mellitus. His mother died 30 years ago of an unknown cause. She was very thin and had adult-onset diabetes and sensorineural hearing loss, fatigue, and exercise intolerance. His younger sister and brother, ages 48 and 46, also had diabetes and sensorineural hearing loss. Both siblings had a history of strokes and epilepsy. Genetic testing on his sister revealed that she harbored the common MT-TL1 pathogenic variant, m.3243A > G, the cause of MELAS. Subsequently, both men were tested and tested positive for MELAS. Within several months, his fatigue progressed from exercise intolerance and needing daily naps to be unable to use stairs and developing dyspnea on any exertion. An echocardiogram revealed a left ventricular ejection fraction (LVEF) of 20%. One and a half years later, he underwent an orthotopic heart transplant. Post-operative complications included nausea from immunosuppressives and weight loss, necessitating the placement of a gastrostomy tube for enteral nutrition. Two and a half years post-transplant, he is now doing well.
Clinical Case #5 (Unpublished): NDUFB11 Mutation
A 3-month-old female presents with a brief resolved unexplained event (BRUE). She was riding in her infant car seat, and her mother heard her cry. The patient could not be woken up and was unresponsive, cyanotic, and with agonal breathing. The mother initiated cardiopulmonary resuscitation (CPR). On arrival of emergency medical services (EMS), the patient was defibrillated three times and ultimately admitted to the intensive care unit. A CMR showed LVNC with heavy trabeculations at the left ventricular apex. The anterior apical and mid-lateral walls were also trabeculated, with the ratio of compacted to non-compacted myocardium at end-diastole at 3.7:1 and 1.6:1, respectively. Moderate to severe left ventricular dilation with mild left ventricular systolic dysfunction (LVEF 45%) was noted. The ventricular septum showed delayed contraction. She underwent a cardiac catheterization complicated by ventricular fibrillation during the procedure requiring chest compressions and defibrillation. An electrophysiology study indicated Wolff-Parkinson-White (WPW) syndrome, and she underwent ablation. She then had an epicardial implantable cardioverter defibrillator (ICD) placed. She was hospitalized for a total of 3 weeks. On further evaluation at the Cardiomyopathy Genetics Clinic, a de novo likely pathogenic variant in NDUFB11 (c.163_170dup; p. Glu57Asp fs*71) was detected using WES. The patient is now seven years old, attends school, and is active, although she has occasional leg pain and fatigue. NDUFB11 is a complex I subunit of the electron transport chain. It is located on the X-chromosome. Pathogenic variants in NDUFB11 have previously been reported with infantile-onset linear skin defects, potentially life-threatening cardiomyopathy, and/or arrhythmia. It has been seen mainly in females as an X-linked dominant disease and is thought to be embryonic lethal in males. Extra-cardiac manifestations include hypotonia, seizures, intellectual disability, brain malformations including agenesis of the corpus callosum and ventriculomegaly, seizures, optic atrophy, anemia, and lactic acidosis. [77,78,79,80,81].
Management of Mitochondrial Cardiomyopathies
Conventional treatment should be initiated if there is evidence of hypertrophic remodeling, even in absence of current symptoms. Beta-blockers or calcium channel blockers in hypertrophied hearts may be used to aid in diastolic filling and are not contraindicated in PMD. ACE inhibitors may prevent early hypertrophic remodeling. Medication management of left ventricular dysfunction may help prevent atrial fibrillation. Cardiac involvement may lead to fatigue, dyspnea on exertion, and exercise intolerance. Pacemaker placement is recommended based on AHA guidelines, and given the unknown natural history of some PMDs, should be done as soon as possible to prevent sudden progression and cardiac death.
The two pathologies of heart failure and arrhythmias are treated with diuretics and vasodilators in warm and wet type, whereas with inotropes in the cold and wet type. [82] Heart failures that are chronic are generally treated with pharmacological agents such as ACE inhibitors, ARBs, beta-blockers, furosemide, ivabradine, mineralocorticoid antagonists, digoxin, and angiotensin receptor neprilysin inhibitor (ARNI). [34, 40] The SGLT2 inhibitors have been established as a strongly recommended treatment for reduced ejection fraction heart failure. Their role, however, in preserved ejection fraction heart failures is yet to be established, and more information is needed to determine if this newer class of medications is efficacious in mitochondrial cardiomyopathies.
Additional interventions include implantable cardioverter defibrillators (ICD), biventricular pacing, mechanical support (ECMO), surgical correction for valvular insufficiency, and in some cases, cardiac transplantation. [34, 40] Cardiac transplantation has been reported in 14% of patients with Barth syndrome. [83] Patients with mitochondrial disease have been reported to generally tolerate solid-organ transplantation except for liver transplantation in POLG-related disease, which needs cautious evaluation. Knowledge of mitochondrial disease as a cause of organ failure during the transplantation procedure is helpful for appropriate consultations but is not an absolute contraindication: the risks and benefits need to be considered along with patient and family wishes and long-term prognostic factors. [76••].
One of the recent approaches to managing patients with DCM is using a combination of genetic and diagnostic testing to determine the positive genotype-negative phenotype and pre-treat patients with medications to avoid developing dilated cardiomyopathy symptoms. Two such drugs have been reported from clinical trials using carvedilol and eplerenone. [84, 85, 86•].
In a randomized, placebo-controlled trial of another drug, elamipretide, in patients with reduced ejection fraction heart failure (ejection fraction ≤ 35%), Daubert et al. (2017) reported a significant decrease in left ventricular end-diastolic volume (− 18 mL; P = 0.009) and end-systolic volume (− 14 mL; P = 0.005) in the highest dose cohort. This was the first study to evaluate the efficacy of elamipretide in heart failure with reduced ejection fraction and demonstrated favorable changes in left ventricular volumes supporting a temporal association and dose–effect relationship. [87].
Furthermore, in 2018, Sabbah et al. performed a randomized control study on left ventricular tissue from dogs and humans with heart failure, comparing them to healthy tissues. The study revealed decreased levels of endothelial nitric oxide synthase, cyclic guanosine monophosphate (cGMP), and peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α, which is a transcription factor that drives mitochondrial biogenesis) in heart failure (both in dog and human tissues). In addition, changes were observed in the regulators of mitochondrial fission and fusion, including fission-1, dynamin-related protein-1, mitofusion-2, dominant optic atrophy-1, and mitofilin. In all instances, the maladaptation was normalized following long-term therapy with elamipretide. [88] Although these findings support the continued development of elamipretide as an innovative therapeutic target, further study of elamipretide is needed to determine long-term safety and efficacy in heart failure management.
Surveillance and Recommendations
As demonstrated in Table 1, there are various genetic causes of mitochondrial cardiomyopathy, and for each disease, there is marked variability. Therefore, surveillance is necessary, with annual cardiology evaluations with an electrocardiogram (for arrhythmias) and echocardiogram. Additional studies may be needed, with guidelines individualized to cardiac status and known genotype. To aid the clinician in management guidelines, there are several available guidelines for mitochondrial disorders, including those from the Mitochondrial Medicine Society. [62, 89••] These recommendations include patient care at a tertiary center with cardiology expertise in mitochondrial disease. For a list of clinics in the United States, please refer to https://www.mitonetwork.org/centers. Baseline assessments should include a standard 12-lead electrocardiogram (EKG) and echocardiogram. Additional monitoring may be needed based on patient symptoms, such as Holter monitoring, for palpitations or high-risk patients based on genetic etiology. Follow-up screening may be determined by the cardiologist while keeping the genetic etiology and risk of developing cardiac manifestations in mind. Follow-up of symptomatic patients (LVEF < 35%, paroxysmal events, LV systolic, or diastolic dysfunction) may require prolonged and more frequent monitoring.
For patients with arrhythmias (SVT, WPW), ablation should be considered. Pacemaker implantation may be indicated to prevent sudden cardiac death and may be combined with an implantable cardioverter defibrillator (ICD). Cardiac MRI may be utilized to obtain more precise imaging. CPET (cardiopulmonary exercise testing) can establish functional capacity, exercise fitness, and conditioning, and to measure response to therapies, caution needs to be taken and the risks vs benefits discussed with the individual patient. Physical therapy may benefit those impacted by PMD, and cardiology may need to dictate any restrictions based on cardiac limitations. Cardiac transplantation is an option based on the multisystemic picture, long-term expected prognosis, and known natural history of the individual mitochondrial disease.
In addition, we advocate that patients and families seek out support from the patient advocacy groups including the United Mitochondrial Disease Foundation (www.umdf.org) and MitoAction (www.mitoaction.org). Clinicians seeking further information may find additional resources through these organizations in addition to the Mitochondrial Medicine Society (www.mitosoc.org).
Conclusion
PMD presents with a highly variable clinical, biochemical, and genetic phenotype, and is extremely challenging to diagnose and manage. There have been consensus recommendations from the Mitochondrial Medicine Society for the diagnosis and management of mitochondrial diseases. [90, 91] According to Binder et al. (2021), given an increased risk of cardiac conduction disease and structural heart disease in PMD patients, a diagnosis of PMD should raise concerns, and patients screened for cardiac abnormalities. [92] As we learn and uncover the pathologies and presentations of MCM, a more updated and integrated recommendation focusing on MCM is warranted.
Various pathologies underlying mitochondrial cardiomyopathies have been reported, the most common of which include mitochondrial proliferation as an adaptive response to energy deficiency. Increased oxidative stress, uncoupled respiratory chain, and uneven mechanical contraction due to misaligned sarcomere are other causes affecting the functioning of myocytes, which could provide additional therapies aimed at these abnormalities specifically.
PMD commonly involves the heart and includes both conduction and/or structural abnormalities such as cardiomyopathy. In some of the PMDs, there is a known genotype–phenotype correlation, such as bundle branch block progressing to complete heart block in those with single large-scale mtDNA deletion syndromes (SLSMDS) including Kearns-Sayre syndrome. Patients with common mtDNA pathogenic variants causing disorders such as MELAS and MERRF (m.3243A > G and m.8344A > G) are at risk for hypertrophic cardiomyopathy and ventricular preexcitation, including asymptomatic family members who also harbor the familial pathogenic variant.
With ever-increasing detailed understanding of the underlying pathologies and diversifying features, MCM might need a categorization befitting its presentation. It remains to be seen whether MCM warrants its nosology akin to MOGE(S) nosology for cardiomyopathies.7
Recent advances in high throughput sequencing, such as next-generation sequencing (NGS) and WES with an additional focus on mtDNA sequencing and deletions, have transformed the diagnostic landscape of PMD. In addition to high-throughput sequencing, further advances in transcriptomics, such as RNA sequencing and proteomics, are expected to revolutionize diagnostic capabilities.
Mitochondrial cardiomyopathies are common in PMD. Initial testing of genetic etiology should include mitochondrial causes. Once diagnosed, patients should have a cardiologist familiar with primary mitochondrial disorders to evaluate and treat these cardiac manifestations.
Cardiac involvement in PMD is treatable, and more likely to be effective when started early, hence the need for cardiac screening in all patients with PMD. Symptoms may not appear until late in the course of cardiac manifestations, and the heart may be involved at any age and may progress slowly. Conversely, cardiac disease may suddenly arise, especially after a stressor such as infection or fasting. Since many patients with PMD are at risk for cardiac manifestations over time, and cardiac involvement may not have symptoms until late in the presentation, surveillance at periodic intervals is recommended. This includes a 12-lead EKG and trans-thoracic echocardiogram after initial diagnosis for baseline assessment and repeated annually unless otherwise specified by a cardiologist familiar with mitochondrial disease. Any further investigations (Holter monitoring, cardiac MRI, etc.) is determined by the cardiologist based on initial findings and the genetic etiology and cardiac risk associated with the patient’s specific mitochondrial disease. PMD patients may derive benefit from addressing cardiac involvement which can improve long-term outcome and quality of life. Natural history studies are needed to determine if other management strategies will be helpful and to determine the overall morbidity and mortality of cardiac involvement in the PMD population.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Tex Heart Inst J. 2013;40(4):385–94. Excellent review of mitochondrial cardiomyopathies.
Lee SR, Kim N, Noh YH, et al. Mitochondrial DNA, mitochondrial dysfunction, and cardiac manifestations. Front Biosci (Landmark Ed). 2017;22(7):1177–94. https://doi.org/10.2741/4541.
Elorza AA, Soffia JP. mtDNA heteroplasmy at the core of aging-associated heart failure. An integrative view of OXPHOS and mitochondrial life cycle in cardiac mitochondrial physiology. Front Cell Dev Biol. 2021;9:625020. https://doi.org/10.3389/fcell.2021.625020.
• Bates MG, Bourke JP, Giordano C, d’Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33(24):3023–33. https://doi.org/10.1093/eurheartj/ehs275. Excellent review of mitochondrial cardiomyopathies.
Taylor RW, Giordano C, Davidson MM, et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol. 2003;41(10):1786–96. https://doi.org/10.1016/s0735-1097(03)00300-0.
Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83(2):254–60. https://doi.org/10.1016/j.ajhg.2008.07.004.
Arbustini E, Narula N, Tavazzi L, et al. The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol. 2014;64(3):304–18. https://doi.org/10.1016/j.jacc.2014.05.027.
• El-Hattab AW, Scaglia F. Mitochondrial cardiomyopathies. Front Cardiovasc Med. 2016;3:25. https://doi.org/10.3389/fcvm.2016.00025. Excellent review of mitochondrial cardiomyopathies.
Finsterer J, Kothari S. Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol. 2014;177(3):754–63. https://doi.org/10.1016/j.ijcard.2014.11.014.
Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67(10):1823–6. https://doi.org/10.1212/01.wnl.0000244435.27645.54.
Bernier FP, Boneh A, Dennett X, Chow CW, Cleary MA, Thorburn DR. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59(9):1406–11. https://doi.org/10.1212/01.wnl.0000033795.17156.00.
Calvo SE, Mootha VK. The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet. 2010;11:25–44. https://doi.org/10.1146/annurev-genom-082509-141720.
Limongelli G, Tome-Esteban M, Dejthevaporn C, Rahman S, Hanna MG, Elliott PM. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail. 2010;12(2):114–21. https://doi.org/10.1093/eurjhf/hfp186.
Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. 2001;49(3):377–83.
Holmgren D, Wahlander H, Eriksson BO, Oldfors A, Holme E, Tulinius M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J. 2003;24(3):280–8. https://doi.org/10.1016/s0195-668x(02)00387-1.
Debray FG, Lambert M, Chevalier I, et al. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics. 2007;119(4):722–33. https://doi.org/10.1542/peds.2006-1866.
Brunel-Guitton C, Levtova A, Sasarman F. Mitochondrial diseases and cardiomyopathies. Can J Cardiol. 2015;31(11):1360–76. https://doi.org/10.1016/j.cjca.2015.08.017.
• Scaglia F, Towbin JA, Craigen WJ, et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114(4):925-31. https://doi.org/10.1542/peds.2004-0718. Excellent review of pediatric mitochondrial cardiomyopathies.
Wahbi K, Bougouin W, Behin A, et al. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Eur Heart J. 2015;36(42):2886–93. https://doi.org/10.1093/eurheartj/ehv307.
Liu Z, Song Y, Li D, et al. The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J Med Genet. 2014;51(3):176–84. https://doi.org/10.1136/jmedgenet-2013-101818.
Merante F, Tein I, Benson L, Robinson BH. Maternally inherited hypertrophic cardiomyopathy due to a novel T-to-C transition at nucleotide 9997 in the mitochondrial tRNA(glycine) gene. Am J Hum Genet. 1994;55(3):437–46.
Shin WS, Tanaka M, Suzuki J, Hemmi C, Toyo-oka T. A novel homoplasmic mutation in mtDNA with a single evolutionary origin as a risk factor for cardiomyopathy. Am J Hum Genet. 2000;67(6):1617–20. https://doi.org/10.1086/316896.
Marin-Garcia J, Goldenthal MJ, Ananthakrishnan R, Pierpont ME. The complete sequence of mtDNA genes in idiopathic dilated cardiomyopathy shows novel missense and tRNA mutations. J Card Fail. 2000;6(4):321–9. https://doi.org/10.1054/jcaf.2000.19232.
Santorelli FM, Tanji K, Manta P, et al. Maternally inherited cardiomyopathy: an atypical presentation of the mtDNA 12S rRNA gene A1555G mutation. Am J Hum Genet. 1999;64(1):295–300. https://doi.org/10.1086/302188.
Bates MG, Hollingsworth KG, Newman JH, et al. Concentric hypertrophic remodelling and subendocardial dysfunction in mitochondrial DNA point mutation carriers. Eur Heart J Cardiovasc Imaging. 2013;14(7):650–8. https://doi.org/10.1093/ehjci/jes226.
Mazzaccara C, Mirra B, Barretta F, Caiazza M, Lombardo B, Scudiero O, Tinto N, Limongelli G, Frisso G. Molecular epidemiology of mitochondrial cardiomyopathy: a search among mitochondrial and nuclear genes. Int J Mol Sci. 2021;22(11):5742. https://doi.org/10.3390/ijms22115742.
• Limongelli G, Masarone D, D’Alessandro R, Elliott PM. Mitochondrial diseases and the heart: an overview of molecular basis, diagnosis, treatment and clinical course. Future Cardiol. 2012;8(1):71-88. https://doi.org/10.2217/fca.11.79. Excellent review of mitochondrial cardiomyopathies.
Mazzaccara C, Limongelli G, Petretta M, et al. A common polymorphism in the SCN5A gene is associated with dilated cardiomyopathy. J Cardiovasc Med (Hagerstown). 2018;19(7):344–50. https://doi.org/10.2459/JCM.0000000000000670.
Limongelli G, Monda E, Tramonte S, et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int J Cardiol. 2020;299:186–91. https://doi.org/10.1016/j.ijcard.2019.06.073.
Blank AC, Breur JMPJ, Fuchs SA, Koop K, Baas AF. Mitochondrial cardiomyopathies In: Baars HF, Doevendans PAFM, Houweling AC, van Tintelen JP (eds). Clinical Cardiogenetics Springer, Cham. 2020.https://doi.org/10.1007/978-3-030-45457-9_11
Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–16. https://doi.org/10.1161/CIRCULATIONAHA.106.174287.
Authors/Task Force m, Elliott PM, Anastasakis A, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–79. https://doi.org/10.1093/eurheartj/ehu284.
Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124(24):e783-831. https://doi.org/10.1161/CIR.0b013e318223e2bd.
Ciarambino T, Menna G, Sansone G, Giordano M. Cardiomyopathies: an overview. Int J Mol Sci. 2021;22(14):7722. https://doi.org/10.3390/ijms22147722
Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies. Br Heart J. Dec 1980;44(6):672–3. https://doi.org/10.1136/hrt.44.6.672
Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93(5):841–2. https://doi.org/10.1161/01.cir.93.5.841.
Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331(23):1564–75. https://doi.org/10.1056/NEJM199412083312307.
Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–76. https://doi.org/10.1001/jama.296.15.1867.
Nugent AW, Daubeney PE, Chondros P, et al. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med. 2003;348(17):1639–46. https://doi.org/10.1056/NEJMoa021737.
Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390(10092):400–14. https://doi.org/10.1016/S0140-6736(16)31713-5.
Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: a translational review of current literature. J Intern Med. 2019;286(4):362–72. https://doi.org/10.1111/joim.12944.
McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123(1):19–26. https://doi.org/10.1172/JCI62862.
Thebault C, Ollivier R, Leurent G, Marcorelles P, Langella B, Donal E. Mitochondriopathy: a rare aetiology of restrictive cardiomyopathy. Eur J Echocardiogr. 2008;9(6):840–5. https://doi.org/10.1093/ejechocard/jen189.
Ruszkiewicz AR, Vernon-Roberts E. Sudden death in an infant due to histiocytoid cardiomyopathy. A light-microscopic, ultrastructural, and immunohistochemical study. Am J Forensic Med Pathol. 1995;16(1):74–80. https://doi.org/10.1097/00000433-199503000-00017.
Cabana MD, Becher O, Smith A. Histiocytoid cardiomyopathy presenting with Wolff-Parkinson-White syndrome. Heart. 2000;83(1):98–9. https://doi.org/10.1136/heart.83.1.98.
Burke A, Mont E, Kutys R, Virmani R. Left ventricular noncompaction: a pathological study of 14 cases. Hum Pathol. 2005;36(4):403–11. https://doi.org/10.1016/j.humpath.2005.02.004.
Jefferies JL. Barth syndrome. Am J Med Genet C Semin Med Genet. 2013;163C(3):198–205. https://doi.org/10.1002/ajmg.c.31372.
Mazurova S, Tesarova M, Magner M, et al. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Med Rep. 2013;114(3):139–53. https://doi.org/10.14712/23362936.2014.16.
Cizkova A, Stranecky V, Mayr JA, et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat Genet. 2008;40(11):1288–90. https://doi.org/10.1038/ng.246.
Guleray N, Kosukcu C, Taskiran ZE, et al. Atypical presentation of Sengers syndrome: a novel mutation revealed with postmortem genetic testing. Fetal Pediatr Pathol. 2020;39(2):163–71. https://doi.org/10.1080/15513815.2019.1639089.
St-Pierre G, Steinberg C, Dubois M, Senechal M. What the cardiologist should know about mitochondrial cardiomyopathy? Can J Cardiol. 2019;35(2):221–4. https://doi.org/10.1016/j.cjca.2018.11.018.
Laonigro I, Correale M, Di Biase M, Altomare E. Alcohol abuse and heart failure. Eur J Heart Fail. 2009;11(5):453–62. https://doi.org/10.1093/eurjhf/hfp037.
Merlo M, Cannata A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20(2):228–39. https://doi.org/10.1002/ejhf.1103.
Rapezzi C, Arbustini E, Caforio AL, et al. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34(19):1448–58. https://doi.org/10.1093/eurheartj/ehs397.
Jha P, Wang X, Auwerx J. Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr Protoc Mouse Biol. 2016;6(1):1–14. https://doi.org/10.1002/9780470942390.mo150182.
Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241(2):236–50. https://doi.org/10.1002/path.4809.
Bourgeois JM, Tarnopolsky MA. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion. 2004;4(5–6):441–52. https://doi.org/10.1016/j.mito.2004.07.036.
Tashiro R, Onoue N, Rikimaru H, et al. Mitochondrial cardiomyopathy with a unique (99m)Tc-MIBI/(123)I-BMIPP mismatch pattern. Intern Med. 2017;56(3):321–5. https://doi.org/10.2169/internalmedicine.56.7525.
Murphy E, Ardehali H, Balaban RS, et al. Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ Res. 2016;118(12):1960–91. https://doi.org/10.1161/RES.0000000000000104.
Lakdawala NK, Winterfield JR, Funke BH. Dilated cardiomyopathy. Circ Arrhythm Electrophysiol. 2013;6(1):228–37. https://doi.org/10.1161/CIRCEP.111.962050.
Badano LP, Miglioranza MH, Edvardsen T, et al. European Association of Cardiovascular Imaging/Cardiovascular Imaging Department of the Brazilian Society of Cardiology recommendations for the use of cardiac imaging to assess and follow patients after heart transplantation. Eur Heart J Cardiovasc Imaging. 2015;16(9):919–48. https://doi.org/10.1093/ehjci/jev139.
Quadir A, Pontifex CS, Lee Robertson H, Labos C, Pfeffer G. Systematic review and meta-analysis of cardiac involvement in mitochondrial myopathy. Neurol Genet. 2019;5(4):e339. https://doi.org/10.1212/NXG.0000000000000339.
Florian A, Ludwig A, Stubbe-Drager B, et al. Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. J Cardiovasc Magn Reson. 2015;17:40. https://doi.org/10.1186/s12968-015-0145-x.
Group JCSJW. Guidelines for diagnosis and treatment of patients with hypertrophic cardiomyopathy (JCS 2012)- Digest Version. Circ J. 2016;80(3):753–74. https://doi.org/10.1253/circj.CJ-66-0122.
Friedrich MG, Sechtem U, Schulz-Menger J, et al. Cardiovascular magnetic resonance in myocarditis: a JACC White Paper. J Am Coll Cardiol. 2009;53(17):1475–87. https://doi.org/10.1016/j.jacc.2009.02.007.
Friedrich MG, Marcotte F. Cardiac magnetic resonance assessment of myocarditis. Circ Cardiovasc Imaging. 2013;6(5):833–9. https://doi.org/10.1161/CIRCIMAGING.113.000416.
Cooper LT, Baughman KL, Feldman AM, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. J Am Coll Cardiol. 2007;50(19):1914–31. https://doi.org/10.1016/j.jacc.2007.09.008.
Maruo Y, Ueda Y, Murayama K, Takeda A. A case report of Leigh syndrome diagnosed by endomyocardial biopsy. Eur Heart J Case Rep. 2021;5(2):ytaa582. https://doi.org/10.1093/ehjcr/ytaa582.
Ware JS, Li J, Mazaika E, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374(3):233–41. https://doi.org/10.1056/NEJMoa1505517.
Carroll CJ, Brilhante V, Suomalainen A. Next-generation sequencing for mitochondrial disorders. Br J Pharmacol. 2014;171(8):1837–53. https://doi.org/10.1111/bph.12469.
Wilcox NS, Prenner SB, Cevasco M, et al. End stage mitochondrial cardiomyopathy and heart transplantation due to biallelic pathogenic C1QBP variants. Circ Genom Precis Med. 2022;15(2):e003559. https://doi.org/10.1161/CIRCGEN.121.003559.
Feichtinger RG, Olahova M, Kishita Y, et al. Biallelic C1QBP mutations cause severe neonatal-, childhood-, or later-onset cardiomyopathy associated with combined respiratory-chain deficiencies. Am J Hum Genet. 2017;101(4):525–38. https://doi.org/10.1016/j.ajhg.2017.08.015.
Wang J, Li H, Sun M, et al. Early onset of combined oxidative phosphorylation deficiency in two Chinese brothers caused by a homozygous (Leu275Phe) mutation in the C1QBP gene. Front Pediatr. 2020;8:583047. https://doi.org/10.3389/fped.2020.583047.
Webster G, Reynolds M, Arva NC, et al. Mitochondrial cardiomyopathy and ventricular arrhythmias associated with biallelic variants in C1QBP. Am J Med Genet A. 2021;185(8):2496–501. https://doi.org/10.1002/ajmg.a.62262.
Ng YS, Alston CL, Diodato D, et al. The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease. J Med Genet. 2016;53(11):768–75. https://doi.org/10.1136/jmedgenet-2016-103910.
•• Parikh S, Karaa A, Goldstein A, et al. Solid organ transplantation in primary mitochondrial disease: proceed with caution. Mol Genet Metab. 2016;118(3):178-184. https://doi.org/10.1016/j.ymgme.2016.04.009. Review of solid organ transplantation in primary mitochondrial disorders including long term poutcome in cardiac transplant.
van Rahden VA, Fernandez-Vizarra E, Alawi M, et al. Mutations in NDUFB11, encoding a complex I component of the mitochondrial respiratory chain, cause microphthalmia with linear skin defects syndrome. Am J Hum Genet. 2015;96(4):640–50. https://doi.org/10.1016/j.ajhg.2015.02.002.
Reinson K, Kovacs-Nagy R, Oiglane-Shlik E, et al. Diverse phenotype in patients with complex I deficiency due to mutations in NDUFB11. Eur J Med Genet. 2019;62(11):103572. https://doi.org/10.1016/j.ejmg.2018.11.006.
Lichtenstein DA, Crispin AW, Sendamarai AK, et al. A recurring mutation in the respiratory complex 1 protein NDUFB11 is responsible for a novel form of X-linked sideroblastic anemia. Blood. 2016;128(15):1913–7. https://doi.org/10.1182/blood-2016-05-719062.
Rea G, Homfray T, Till J, et al. Histiocytoid cardiomyopathy and microphthalmia with linear skin defects syndrome: phenotypes linked by truncating variants in NDUFB11. Cold Spring Harb Mol Case Stud. 2017;3(1):a001271. https://doi.org/10.1101/mcs.a001271.
Shehata BM, Cundiff CA, Lee K, et al. Exome sequencing of patients with histiocytoid cardiomyopathy reveals a de novo NDUFB11 mutation that plays a role in the pathogenesis of histiocytoid cardiomyopathy. Am J Med Genet A. 2015;167A(9):2114–21. https://doi.org/10.1002/ajmg.a.37138.
Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2016;18(8):891–975. https://doi.org/10.1002/ejhf.592.
Clarke SL, Bowron A, Gonzalez IL, et al. Barth syndrome. Orphanet J Rare Dis. 2013;8:23. https://doi.org/10.1186/1750-1172-8-23.
Raman SV, Hor KN, Mazur W, et al. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(2):153–61. https://doi.org/10.1016/S1474-4422(14)70318-7.
Yeoh T, Hayward C, Benson V, et al. A randomised, placebo-controlled trial of carvedilol in early familial dilated cardiomyopathy. Heart Lung Circ. 2011;20(9):566–73. https://doi.org/10.1016/j.hlc.2011.06.004.
• Chatfield KC, Sparagna GC, Chau S, et al. Elamipretide improves mitochondrial function in the failing human heart. JACC Basic Transl Sci. 2019;4(2):147–157. https://doi.org/10.1016/j.jacbts.2018.12.005. Potentially promising new therapy for mitochondrial cardiomyopathies.
Daubert MA, Yow E, Dunn G, Marchev S, Barnhart H, Douglas PS, O'Connor C, Goldstein S, Udelson JE, Sabbah HN. Novel mitochondria-targeting peptide in heart failure treatment: a randomized, placebo-controlled trial of elamipretide. Circ Heart Fail. 2017;10(12):e004389. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004389
Sabbah HN, Gupta RC, Singh-Gupta V, Zhang K, Lanfear DE. Abnormalities of mitochondrial dynamics in the failing heart: normalization following long-term therapy with elamipretide. Cardiovasc Drugs Ther. 2018;32(4):319–28. https://doi.org/10.1007/s10557-018-6805-y.
•• Parikh S, Goldstein A, Karaa A, Koenig MK, Anselm I, Brunel-Guitton C, Christodoulou J, Cohen BH, Dimmock D, Enns GM, Falk MJ, Feigenbaum A, Frye RE, Ganesh J, Griesemer D, Haas R, Horvath R, Korson M, Kruer MC, Mancuso M, McCormack S, Raboisson MJ, Reimschisel T, Salvarinova R, Saneto RP, Scaglia F, Shoffner J, Stacpoole PW, Sue CM, Tarnopolsky M, Van Karnebeek C, Wolfe LA, Cunningham ZZ, Rahman S, Chinnery PF. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;19(12). https://doi.org/10.1038/gim.2017.107. Consensus statement on management for mitochondrial disorders.
Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17(9):689–701. https://doi.org/10.1038/gim.2014.177.
Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226(2):274–86. https://doi.org/10.1002/path.3028.
Scott Binder M, Roda RH, Corse AM, Sidhu S, Stewart S, Barth AS. Prevalence of heart disease in patients with mitochondrial abnormalities on skeletal muscle biopsy. Ann Clin Transl Neurol. 2021;8(4):825–30. https://doi.org/10.1002/acn3.51327.
Author information
Authors and Affiliations
Contributions
A. T. and A. C. G. wrote and edited the manuscript.
Corresponding author
Ethics declarations
Ethics Statement
A retrospective chart review of the clinical course and laboratory test results was performed for patients presented. Informed consent was provided, and all patients were enrolled in the Children’s Hospital of Philadelphia (CHOP) Institutional Review Board (IRB) approved study #08–6177 (Marni J. Falk, PI) that allows for medical record reviews, medical photography, publication for educational purposes, and clinical cohort analyses.
Conflict of Interest
Dr Atif Towheed declares no conflict of interest. Dr Amy Goldstein is a consultant for Reneo Pharmaceuticals and on the Speakers Bureau for United Mitochondrial Disease Foundation and MitoAction.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Towheed, A., Goldstein, A.C. Genetics of Mitochondrial Cardiomyopathy. Curr Cardiovasc Risk Rep 17, 49–72 (2023). https://doi.org/10.1007/s12170-023-00715-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12170-023-00715-4