
Abstract
Seaweeds are known for their nutritional benefits; however, they contain one of the highest levels of arsenic content. Arsenic species are found in various oxidation states in the environment, where inorganic arsenic is more toxic than organic ones. This study describes the optimization of extraction conditions for the determination of arsenic species, namely, arsenate [As(V)], arsenite, [As(III)], arsenobetaine (AsB), dimethylarsenic acid (DMA), and monomethylarsenic acid (MMA) in Kappaphycus alvarezii (K. alvarezii), a carrageenan-producing red seaweed, using high-performance liquid chromatography-inductively coupled plasma-mass spectrometry (HPLC-ICP-MS). A series of nitric acid solutions (0 to 2.0%) as extraction solutions and various extraction times (0 to 240 min) were investigated with heating at 90 °C. The proposed method was validated for matrix effects, linearity and working range, the limit of detection (LOD), the limit of quantification (LOQ), repeatability, within-lab reproducibility, and trueness using certified reference material (CRM, NMIJ 7405-a) and spiking with arsenic species. Among the nitric acid concentration tested, the concentration of 0.2% nitric acid gave recovery values of 86.8 to 94.2%, which complied with the European guidelines 2002/657/EC. Arsenic species were recovered at the maximum level from K. alvarezii at the extraction time of 60 min. The proposed method measured the As(V) concentration in CRM as 9.7 ± 0.3 mg kg−1 and therefore was compliant with the certified level of 10.1 ± 0.5 mg kg−1. The measurement of trueness for the fortified seaweed was within the acceptable limits, with recoveries between 87.8 and 107.4% for all five arsenic species. The proposed method demonstrated accuracy by complying with the provisions stated in the guidelines for validation performance characteristics. The efficacy of this method was further validated for the determination of arsenic species in K. alvarezii varieties.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The seaweed industry is a lucrative business in countries such as the Philippines, Indonesia, and Malaysia, where their coastlines provide a conducive environment for seaweed farming of various species types, namely Kappaphycus spp., Euchema denticulatum, and Gracilaria manilaensis (Phang et al. 2019). The global seaweed industry is worth more than USD 11.5 billion in 2020 (Seaweed Market: Global Industry Trends, Share, Size, Growth, Opportunity and Forecast (2021–2026) 2020). The Kappaphycus spp. is mainly cultivated in Sabah, Malaysia, and it represents one of the essential sources of carrageenan (Mohammad et al. 2019). Carrageenan is a family of sulfated linear polysaccharides with a fundamental structure of alternate units of D-galactose and 3,6-anhydro-galactose (3,6-AG) joined by α-1,3 and β-1,4-glycosidic linkages (Manuhara et al. 2016). The distinct thickening, gelling, and stabilizing properties of carrageenan are widely valued and utilized in the food industry for multiple applications. However, there has been a growing concern regarding its effects on human health as arsenic species have been detected in seaweed (Hussain et al. 2021). In addition, seaweed is known to be susceptible to the accumulation of arsenic from the environment (Klumpp and Peterson 1979; Klumpp 1980; Díaz et al. 2012; Chen and Zhang 2019).
Arsenic (As) is a ubiquitous metalloid that exists in the environment both from natural sources and anthropogenic activities (Park et al. 2019; Herath et al. 2020). The toxicity of arsenic is based on its chemical form or “species,” in which the inorganic forms of arsenic [arsenate, As(V) and arsenite, As(III)] are more toxic than the organic forms of arsenic [arsenobetaine (AsB), arsenocholine (AsC), dimethylarsenic acid (DMA), and monomethylarsenic acid (MMA)], with As(III) being more toxic than As(V) (Reis and Duarte 2019).
Anionic carboxyl and sulfate in the cell wall polysaccharides of seaweed such as carrageenan create perfect binding sites for heavy metals (Wang et al. 2015; Yong et al. 2017). Thus, to extract the arsenic species, these chemical bonds or binding sites need to be fragmented to ease the release of arsenic into the extracting medium or solution. Furthermore, since these elements are volatile, the extraction steps need to be performed carefully to avoid modifying their forms and concentrations (Rahman et al. 2012; Wolle and Conklin 2018b). Another vital factor to remember when considering an extraction procedure is that various arsenic species have different physical and chemical properties (Benramdane et al. 1999). This characteristic is essential when selecting a suitable extraction solution as the extraction efficiency depends on the solubilities of the arsenic compounds in the chosen solution and the ability of the solution to come into contact with the arsenic compounds (Kuehnelt et al. 2001; Reis and Duarte 2018).
The main goal for many emerging studies on arsenic speciation is the development of a rapid technique with a simplified sample treatment process, low reagent and consumables consumption, minimal use of expensive supporting equipment, as well as minimal contamination and wastage (Kazi et al. 2009; Park et al. 2019; Pétursdóttir and Gunnlaugsdóttir 2019; Bullen et al. 2020). The studies on arsenic speciation in seaweed are an ongoing process that is tedious and labor-intensive as some of the extraction processes are matrix-dependent, especially when the taxonomic differences between seaweed (red, green, or brown) are crucial in obtaining better extraction percentages and trustworthy results (Reis and Duarte 2018). Hence, sample extraction optimization processes are necessary to fulfill these requirements and to ensure that the arsenic species are efficiently solubilized without degradation or conversion for accurate quantification (Park et al. 2019). Numerous studies have experimented with different types of extraction techniques such as sonication, heating, microwave-assisted extraction, solid-phase extraction (SPE), and enzymatic hydrolysis (B'Hymer and Caruso 2004; García-Salgado et al. 2012; Narukawa et al. 2012; Khan et al. 2015; Wolle and Conklin 2018a; Huang et al. 2020; Narukawa et al. 2020). In addition, some experiments require the use of supporting equipment, specific enzymes, and consumables such as anion exchange cartridges, which are costly. Thus, identifying factors influencing arsenic species extraction, such as the type of extraction solution, incubation temperature and time, and species behavior, will ensure the achievement of higher extraction efficiency.
The extraction solution commonly used in the extraction process is either water, methanol (MeOH), a combination of MeOH and water, dilute nitric acid (HNO3), or dilute hydrochloric acid (HCl). Extraction using water is considered soft extraction, which is suitable for extracting polar natured arsenic species (Wolle and Conklin 2018a). The MeOH:water extraction solvent is widely recognized as a suitable extractor for organic arsenic. However, it decreases the extraction efficiency of inorganic arsenic and increases the solvent loading effect on inductively coupled plasma-mass spectrometry (ICP-MS), thus requiring increased power usage (Amaral et al. 2013). It has been shown that acidification increases the extraction efficiency by preserving the arsenic species as well as decomposing the sample matrix efficiently (Park et al. 2019). However, for samples that are extracted using HCI, the chloride will form argon chloride (40Ar35Cl) in the plasma in the ICP-MS, and this ion shares the same m/z ratio with arsenic (75As) (Wilschefski and Baxter 2019). Nitric acid has been reported as a strong oxidizing acid and produces less spectral interference in ICP analyses (Liu et al. 2020) besides generating better peak separation and faster analysis time (Park et al. 2019). In the notable works of arsenic species extraction from seaweed, the diluted HNO3 solutions were studied between 1 and 2.95% (Narukawa et al. 2012; Khan et al. 2015; Matsumoto-Tanibuchi et al. 2019; Park et al. 2019; Pétursdóttir and Gunnlaugsdóttir 2019; Kim et al. 2020; Lorenc et al. 2020). The challenge with these existing methods is that their nitric acid concentrations do not allow for efficient extraction of As species from carrageenan-producing seaweed due to their gelation properties. In addition to that, these studies were conducted on brown seaweed, which contains alginate with solubility and arsenic-binding capacity different from carrageenan (Alba and Kontogiorgos 2019). Therefore, a study to optimize the concentration of nitric acid in the extraction of As species from Kappaphycus spp. is necessary.
The extraction time required is dependent on the interaction of the extraction solution with the sample matrix and the rate of conversion of arsenic species (Ellwood and Maher 2003). Previous literature has stated extraction times that ranged from 30 min to overnight and even 24 h in some cases (Nan et al. 2018; Huang et al. 2020; Narukawa et al. 2020). The discrepancies in the duration between the improved and adopted methods may lead to delays when reporting arsenic species content of seaweed, especially for food safety monitoring.
Method validation is an essential protocol used to assess a method and to confirm the accuracy and reliability of the results, especially when the method is modified or optimized differently from its original method. This requirement is mandatory for the development of an accredited laboratory framework for regulatory purposes (Murphy et al. 2013). Various guidelines, protocols, and journals have proposed recommendations for the assessment of the performance characteristics in method validation. In addition, statistical procedures have been proven to provide valuable and realistic measurement indicators of study outcomes (Sanagi et al. 2010).
This study aims to develop a rapid and economical extraction method using an optimized heating protocol for the identification of five arsenic species (As(V), As(III), AsB, MMA, and DMA) in carrageenan-producing seaweed, K. alvarezii samples analyzed using HPLC-ICP-MS. Varied acid concentrations and extraction times were evaluated to identify the optimum conditions for arsenic species extraction. The optimized method was further validated for its matrix effect, LOD, LOQ, linearity and working range, repeatability, within-lab reproducibility, and trueness using fortified blank seaweed samples and certified reference material (CRM) to determine its fitness for purpose. The validated method was subsequently used to determine the arsenic species content of four varieties of K. alvarezii.
Materials and Methods
Instrumentation
Arsenic species from seaweed samples were determined using ICP-MS (NexION® 300XX, PerkinElmer Inc., CT, USA). The sample introduction system consisted of a Meinhard® modified type A concentric baffled cyclonic quartz spray chamber. The equipment is equipped with a unique triple-cone interface and quadrupole ion deflector. Potential polyatomic interferences such as 40Ar35Cl+ produced as a result of the reaction between argon gas and chlorine were eliminated using dynamic reaction cell (DRC) technology, and the As species were detected as 75As16O+ (m/z of 91) using oxygen reaction gas. The ICP-MS analytical conditions are shown in Table 1a.
The chromatographic separation of the arsenic species was achieved by the coupling of ICP-MS with HPLC (FLEXAR™ HPLC, PerkinElmer Inc., CT, USA) instrumentation consisting of a binary pump, autosampler, vacuum degasser, and column oven. An ODS C18 MG analytical column (Capcell Pak C18 MG; 100 Å, 5 µm, 250 mm × 4.5 mm, Shiseido Ltd., Tokyo, Japan) was used for separation and connected to the switching valve using PEEK tubing prior to ICP-MS detection. The analytical conditions for the HPLC setup are shown in Table 1b.
For data acquisition, peak signals for speciated arsenic compounds were obtained at m/z 75 (As) and displayed as mass intensity–time plots. The arsenic species concentrations were computed using peak area intensities and compared with established standard concentrations.
Reagents and Standards
Ultrapure water (Water Purification Systems, Younglin, Anyang, Korea) was used to prepare the standards and reagents. For speciation analyses, As(III) and As(V) solutions at concentrations of 1000 mg L−1, respectively, were procured from Perkin Elmer (CT, USA). Dimethylarsinic acid (DMA) was obtained as cacodylic acid (98%), and arsenobetaine (AsB) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Monosodium acid methane arsonate sesquihydrate (MMA) was purchased from Chem Service (West Chester, PA, USA). DMA, AsB, and MMA stock solutions were prepared separately by dissolving their salts in ultrapure water accordingly. All prepared solutions were stored in the dark at 5 °C to inhibit decomposition. The Hijiki seaweed certified reference material (CRM) (NMIJ CRM 7405-a) was obtained from the National Metrology Institute of Japan (Ibaraki, Japan) and used for method validation. Concentrated 65% nitric acid (HNO3) was obtained from Merck (Suprapur grade, Darmstadt, Germany) and used for the extraction process after dilution.
For the mobile phase, 1-octanesulfonic acid sodium salt (OSA), malonic acid, tetramethylammonium hydroxide solution (TMAH), and methanol (HPLC grade) were obtained from Sigma-Aldrich (St. Louis, MO, USA) and Merck (Darmstadt, Germany), respectively. Ammonium hydroxide (Fisher Scientific, Pittsburgh, PA) was used to adjust the pH of the mobile phase.
Sample Preparation
Seaweed varieties belonging to the K. alvarezii species were harvested from Sempurna, Sabah, Malaysia, and sun-dried in the harvesting area before being transported to the laboratory. These varieties included K. alvarezii var brown tambalang, K. alvarezii var tangan-tangan, K. alvarezii var buaya, and K. alvarezii var durian. The samples were rinsed with distilled and deionized water to remove salt and epiphytes. The washed and drained seaweeds were then frozen at − 20 °C for 24 h before placing them in a freeze-dryer (Benchtop PRO 9L, Virtis SP Scientific, Warminster, PA, USA). The freeze-dried seaweeds were blended using a heavy-duty blender (Waring, Albuquerque, NW, USA) prior to milling in an ultra-centrifugal mill (Retsch, Haan, Germany) to achieve a particle size of 0.12 mm. The freeze-dried seaweed powders were stored in air-tight amber glass bottles (arsenic is light sensitive) at room temperature until further analyses.
Moisture Content Determination
The moisture content of each seaweed sample was determined by drying approximately 5 g of pulverized seaweed (in triplicates) in an oven (Memmert UF110, Schwabach, Germany) at 105 °C until a constant weight was achieved following the recommended guidelines from the Association of Official Analytical Chemists (AOAC) (2005).
Contamination Removal
For the removal of contamination, all glassware used for arsenic species analyses were washed with detergent and rinsed with ultrapure water. Then, they were soaked overnight in an acid bath containing 10% HNO3 solution and rinsed three times with ultrapure water before drying them in an oven (Memmert UF750, Schwabach, Germany). A reagent blank (without sample) and ultrapure water were analyzed to check for contamination.
Experimental Design for Arsenic Species Extraction Optimization
Single-factor experiments were employed for the optimization of the extraction method in this study. The independent variables consisting of acid concentration and extraction time were evaluated. The four varieties of K. alvarezii were pooled for these experiments. The arsenic species recovery level for each of the independent variables ranged between 80 and 110% as required by the Official Journal of the European Communities, 2002/657/EC (European Parliament and the Council of the European Union 2002).
Effect of Nitric Acid (HNO3) Concentration
Approximately 0.5 g of freeze-dried K. alvarezii powder was weighed into a 15-ml polytetrafluoroethylene (PTFE) centrifuge tube and spiked with 0.2 mg kg−1 of arsenic species prior to the addition of 10 ml of dilute HNO3 as an extraction solution. Different concentrations of HNO3 solution were applied: 0% (ultrapure water only), 0.05%, 0.10%, 0.20%, 0.50%, 1.00%, 1.50%, and 2.00%. The tubes were heated for 1 h at 90 °C in a water bath (Memmert WNB 7, Schwabach, Germany). After cooling the tubes to room temperature, they were centrifuged (Kubota 5500, Tokyo, Japan) at 2200 × g for 30 min, and each of their supernatants was collected and diluted five times or less with the mobile phase solution. The dilution factor varied depending on the total arsenic concentration. The diluted supernatant was filtered using a 0.45-µm PTFE membrane filter (Agilent, Santa Clara, CA, USA) prior to HPLC-ICP-MS analysis. The optimal HNO3 acid concentration was selected based on the recovery of the arsenic species that was spiked into the freeze-dried K. alvarezii powder. The optimization process was performed in triplicates for each variable by the addition of standard arsenic to sample and reagent blanks. The sample blank was prepared by boiling seaweed with 5% HNO3, while the reagent blank was HNO3 at each concentration being studied.
Effect of Extraction Time
The freeze-dried K. alvarezii powder was extracted using the optimal HNO3 concentration determined in the above experiment, and the extraction times were varied: 0, 15, 30, 45, 60, 90, 120, 150, 180, and 240 min. The optimum extraction time was then selected according to the recovery of arsenic species, as mentioned previously.
Statistical Analyses
The results were displayed as means ± standard deviations (SD) of a non-equal replicate of assays and analyzed using SPSS V.23. A one-way analysis of variance (ANOVA) was performed together with Scheffe’s test to determine the significant differences between means at the 5% significance level.
Method Validation
Method validation was performed to prove that the method employed was fit for its purpose, especially for future enforcement and monitoring activities. A single laboratory validation (SLV) was employed according to the method validation guidelines, journals, and International Organization for Standardization (ISO) standards (ISO 110951996; European Parliament and the Council of the European Union 2002; Thompson et al. 2002; ISO 5765–62003; ISO 5725–12003; International Organization of Vine and Wine (OIV) 2005; Association of Official Analytical Chemists (AOAC) International 2007; Magnusson 2014; Sanagi et al. 2010; Association of Official Analytical Chemists (AOAC) 2019). The tested validation parameters consisted of matrix effect, LOD, LOQ, linearity and working range, precision (repeatability and within-lab reproducibility), and trueness (spike recovery and CRM). To reflect the characteristics of the matrix of different varieties of K. alvarezii, each variety was used in equal amounts and homogenized for a representative sample.
Matrix Effect
The matrix effect was evaluated to identify any significant differences between calibrations using the standard solution and in the K. alvarezii (matrix). A set of six-point calibrations ranging from 0.015 to 0.075 mg kg−1 for As(V), As(III), AsB, DMA, and MMA were prepared using standard solutions and spiked into K. alvarezii powder. Both sets of calibrations were analyzed on the same day. An ordinary least squares (OLS) regression, Snedecor’s F test, and Student’s t-test parameters were calculated for both calibration curves. The F test was performed by calculating the residual variances, S2res using the formula shown below:
where S2res1 was the residual variance from the calibration in the standard solution and S2res2 was the residual variance from the calibration in K. alvarezii. The calculated F (F cal) values were then compared against F critical (F crit) values at a 95% confidence interval to identify any differences in the residual variances. A hypothesis test was performed, whereby Ho indicated that there was no difference in the residual variance (F cal < F crit), while H1 indicated a difference in the residual variance (F cal > F crit).
Next, the t value (t cal) was calculated using the Student’s t-test to determine the differences between both calibration slopes (standard solution and K. alvarezii). The t cal calculation was dependent on the residual variances. Therefore, if the residual variances from the F test were not different, the following formula was used:
where the slopes of regression (b) were represented by b1 (calibration using the standard solution) and b2 (calibration using K. alvarezii). The total levels of calibration (n) were represented by n1 (standard solution) and n2 (K. alvarezii). The residual variances (S2res) were represented by S2res1 (calibration using the standard solution) and S2res2 (calibration using K. alvarezii). The standard solution calibration values of the ith calibration level (xi) were represented by xi1 (standard solution) and xi2 (K. alvarezii). The means of all the standard calibration values of n calibration levels (\(\overline{x)}\) were represented by \({\overline{x}}_{1}\) (standard solution) and \({\overline{x}}_{2}\)(K. alvarezii).
However, if the residual variances from the F test were different, a different approach was used to calculate t cal based on the formula shown below:
where the standard deviations of the regression line slopes (S2b) were represented by S2b1 (standard solution) and S2b2 (K. alvarezii).
The hypothesis testing was performed by comparing the slopes for both calibrations using the Student’s t-test, where Ho indicated that the slopes were not different and Ha indicated there were differences in the slopes for both the standard solution and K. alvarezii calibrations. Two approaches were used to calculate the t crit value. In the first formula, t cal was compared to the t crit value at (n1 + n2)-4 degrees of freedom at a 95% confidence limit.
In the second formula for t cal, t crit was calculated based on the following formula:
where t1 and t2 were the theoretical t values at a 95% confidence limit with n1-2 and n2-2 degrees of freedom, respectively.
When t cal was less than or equal to the value of t crit, the null hypothesis, Ho, was accepted, and the extraction method could proceed using the standard solution calibration for routine analysis. However, if the t cal was more than t crit, the alternate hypothesis, Ha, was accepted, and it can be concluded that the slopes were different. This observation indicated that a matrix effect was present, whereby the calibration for this method should be prepared in K. alvarezii powder.
Limit of Detection (LOD)
The LOD was determined using linear regression (International Organization of Vine and Wine (OIV) 2005), whereby the instrument response was assumed to be linearly associated with the standard concentration at a lower concentration range. The equation used is as follows:
Six diluted standard solutions ranging between 0.005 and 0.035 mg kg−1 were homogenously prepared and analyzed in three different batches. The data obtained were used to calculate the parameters of the calibration functions. In addition to obtaining LOD, a predetermined value of the LOQ was also calculated. The formulas for LOD and predetermined LOQ calculations are shown below:
where sa represents the standard deviation of the intercept point, and b represents the slope of the regression line. The predetermined LOQ was confirmed in the following section.
Limit of Quantification (LOQ)
The predetermined LOQ value obtained in this study was further verified to determine if the LOQ value obtained was achievable. In total, ten independent samples (K. alvarezii powder) were spiked based on the predetermined LOQ value. For the acceptance of the predetermined value, the measured value must fulfill the two conditions shown below:
if \(\frac{\left|LOQ-\overline{\mathrm{\rm X} }\right|}{\frac{\sigma }{\sqrt{n}}}\) < 10, then the predetermined LOQ was considered valid,
if 5σ < LOQ, then the predetermined LOQ was accepted,
where \(\overline{x}\) was the mean value of the ten measurements, σ represented the standard deviation of the ten measurements, and n was the number of datasets.
Linearity and Working Range
ICP-MS was employed as it provides a wide linear dynamic range. A calibration curve was constructed, starting with the lowest LOQ level to the highest expected level. Six calibration concentrations were prepared and analyzed in three replicates with concentrations ranging from 0.025 to 0.250 mg kg−1 for As(III), As(V), MMA, and DMA, respectively, and 0.05 to 0.50 mg kg−1 for AsB.
The parameters of the calibration functions consisting of linearity, regression, and intercept significance were estimated based on the ISO 11095 recommendations (ISO 110951996), whereby the regression model was defined using OLS. For estimating the parameters, two types of charts were constructed. The first chart represented the values measured against the true values of the arsenic species standards. The second chart was a y-residual plot, in which the residual values were plotted against the arsenic species standard concentrations.
The confirmation of linearity was performed based on the lack-of-fit test and regression model test using ANOVA. The linearity was ascertained based on two hypotheses; regression model acceptance (if F cal > F crit) and linear model acceptance (if F cal < F crit). The significant test was performed using Snedecor’s F tests. Subsequently, the Student’s t-test was used to decide whether an intercept was significantly different from zero if the calibration was found to be linear:
where t cal represented the calculated t value, a was the intercept, and Sa was the standard deviation of the intercept. If t cal was lower than or equal to the critical value t distribution for n degrees of freedom at the 95% confidence limit, the intercept was not significantly different from zero and passed through the origin (zero).
Precision
To evaluate the precision for single laboratory validation (SLV), particularly for in-house methods, repeatability and within-lab reproducibility, or sometimes known as intermediate precision, were employed in this study (Bratinova et al. 2009). For repeatability, independent test results were acquired on identical test items analyzed by a single analyst using the same method and instrumentation in the same laboratory within a short period of time. On the other hand, for within-lab reproducibility, the same conditions as in repeatability were employed, except that more than one analyst performed the analysis over a longer duration.
For repeatability, blank K. alvarezii powder samples were spiked with three concentration levels of arsenic species standard, ranging from low (preferably at the LOQ level), middle to high. The spiked concentrations were 0.025 mg kg−1, 0.075 mg kg−1, and 0.250 mg kg−1 for As(III), As(V), MMA, and DMA, respectively, and 0.05 mg kg−1, 0.15 mg kg−1, and 0.50 mg kg−1 for AsB. Each concentration was analyzed in ten different batches by a single analyst with a set of duplicates for each concentration.
The same spiking concentrations used for the repeatability analysis were applied to the within-lab reproducibility test, in which three different analysts analyzed four different batches by running duplicates for each batch within a 3-month interval. All these concentrations were read against standard solution calibration that was within the working range.
Relative standard deviation was calculated for each concentration for both test conditions, namely RSDr for repeatability and RSDR for within-lab reproducibility. To evaluate the acceptance, the Horwitz equation was used to calculate the predicted relative standard deviation (RSD) (Association of Official Analytical Chemists (AOAC) 2019) as shown below:
where C represented the concentration expressed as a mass fraction of the concentration expressed in the powers of 10 (1 ppm = 10−6).
To fulfill the acceptance criteria for repeatability and within-lab reproducibility, the calculated RSDr and RSDR should be lower than the predicted RSDr and RSDR obtained from the Horwitz equation.
Precision limits of r and R were derived from the repeatability and within-lab reproducibility data, where r and R represented the absolute difference between the two test results obtained under repeatability and within-lab reproducibility conditions with a 95% confidence limit (2√2 = 2.8). They were acquired from the standard deviation and calculated according to the formula shown below:
where Sr and SR represented the standard deviations of the total differences of duplicate results for different batches for repeatability and within-lab reproducibility, respectively. These precision limits would be recommended for the evaluation of future test results for internal quality control.
Trueness
Trueness is an agreement that is established between the mean value derived from a broad series of test measurements and a conventional true value or an accepted reference value. In this study, trueness was estimated using a CRM Hijiki seaweed (NMIJ CRM 7405-a) and a series of spiked K. alvarezii powder. However, the CRM was only certified as reference material for As(V). Thus, a series of K. alvarezii powder spiked with other arsenic species were required. The procedure for trueness measurement was executed as stated in the Eurachem guidelines (Magnusson 2014) and the acceptance of the recovery percentage (80 to 110%) was based on the guidelines by the Official Journal of the European Communities, 2002/657/EC (European Parliament and the Council of the European Union 2002).
At least five replicates of NMIJ CRM 7405-a with a duplicate of each replicate were analyzed to evaluate the recovery percentage against the certified value. Estimation of trueness was obtained by spiking three levels of arsenic species standard into blank K. alvarezii powder: 0.025 mg kg−1, 0.075 mg kg−1, and 0.250 mg kg−1 for As(III), As(V), MMA, and DMA; 0.05 mg kg−1, 0.15 mg kg−1, and 0.50 mg kg−1 for AsB. The samples were analyzed in ten replicates with a duplicate of each replicate. The mean value of the duplicate was computed for each replicate and the total replicates to obtain the recovery percentage. The recoveries of arsenic species in CRM and spiked K. alvarezii powder that were within the accepted range were used to determine the accuracy of the validated method.
Proficiency Test
The extraction method was tested with the Food Analysis Performance Assessment Scheme (FAPAS) proficiency test of a metallic contaminant in seaweed, involving 52 participants from around the world, from October to November 2020. Dried seaweed powder was analyzed for quantification of seven heavy metals, including inorganic arsenic (As(V) and As(III)), which were present at natural levels. The analysis was carried out twice in triplicates. The final measurements were calculated as means and evaluated with Ζ-score (satisfactory if |z|≤ 2).
Application of the Optimized and Validated Method on K. alvarezii
The proposed method was subsequently employed to determine the arsenic species levels in K. alvarezii var brown tambalang, K. alvarezii var tangan-tangan, K. alvarezii var buaya, and K alvarezii var durian.
Results and Discussion
Effect of Nitric Acid Concentration on the Extraction Efficiency
The selection of a suitable extraction solution was performed using increasing concentrations of HNO3 solution for 1 h at 90 °C. K. alvarezii powder was spiked with 0.2 mg kg−1 of arsenic species to compare its extraction efficiency and the results are shown in Fig. 1.

Effect of nitric acid concentration on recoveries of five arsenic species from K. alvarezii (N = 3) spiked with 0.2 mg kg−1 of each arsenic species. The extraction was performed at 90 °C for 1 h. Results are shown in percentages (mean recovery ± SD). The error bars represent the standard deviations of all points within the measurement region. Acceptable recovery is between 80 and 110% for concentrations above 10 µg kg.−1 according to the Official Journal of the European Communities (2002/657/EC)
The results revealed significant differences (p < 0.05) in the recoveries of the arsenic species using the eight acid concentrations investigated. These results indicated that the acid solution with a concentration of 0.2% HNO3 produced recoveries ranging between 86.8 and 94.2%. This finding was in agreement with the recommended recovery percentage of 80 to 110% proposed by the Official Journal of the European Communities, 2002/657/EC (European Parliament and the Council of the European Union 2002). The recovery percentage using 0.2% HNO3 solution was also found to be higher than the recovery obtained using other acid concentrations. This HNO3 concentration is five times lower than that recommended by other researchers, and it was able to overcome the problem of sample congealment, which can occur during the extraction of As species from K.alvarezii.
The addition of nitric acid into the extraction process is known to improve the extraction efficiencies of arsenic species not only in marine animals but also in marine plants (Hsieh and Jiang 2012). The low concentration of nitric acid has been shown to disintegrate the polysaccharides in marine and terrestrial plants (Sadee et al. 2016). To determine the natural forms of the arsenic species, it is crucial to preserve As(III) and As(V) during the extraction process, as they are a redox couple and readily interconvert if the oxidative or reductive properties of the extraction solution change prior to analysis. The stability of the As(III) and As(V) could only be achieved using a narrow range of HNO3 concentrations (within 0 to 1.00%) (Amaral et al. 2013). By considering the sample complexity and arsenic species conversion, the extraction process with 0.2% HNO3 solution revealed satisfactory extraction efficiency.
It should also be noted that when the HNO3 concentration was increased from 0.50 to 2.00%, a decline in the extraction efficiency was detected (61.7 to 79.9%). Narukawa et al. (2012) suggested that nitric acid was vital in destroying the seaweed body tissue. However, arsenic species are likely to decompose in harsh conditions, and it becomes challenging to determine the natural content of arsenic species in the seaweed matrix. The extraction efficiencies gradually improved (53.2 to 84.3%) from extraction with ultrapure water to that with 0.05 to 0.10% HNO3 solution. These two HNO3 concentrations were added to observe whether the extraction behavior did come closer to that of ultrapure water, as suggested by Narukawa et al. (2014).
A low recovery (less than 5%) was observed at 0% HNO3 solution (ultrapure water) for all five arsenic species. During the extraction with 0% HNO3 solution, the liquid became viscous after the heating process, and only a small amount of supernatant was able to be decanted for injection into HPLC-ICP-MS. The viscosity of the extract could be due to the carrageenan in the seaweed samples. As mentioned earlier, one of the challenges in arsenic species extraction is the chemical conversion of the arsenic species. It has also been previously stated that noninvasive extraction method such as using water does not alter the arsenic species, especially the conversion of As(III) to As(V) (Narukawa et al. 2014; Wolle and Conklin 2018b). However, it was duly observed that the drawback of water as an extractant was the production of a highly viscous solution that significantly decreased the recoveries (Hirata and Toshimitsu 2005; Park et al. 2019) of the arsenic species. Raab et al. (2005) stated that water extraction efficiency depends on the type of seaweed or algae. It was observed that Sea Lettuce and Black Moss gave low recoveries (30% and less than 5%, respectively), while Hijiki showed a recovery of between 77 and 84%. In this study, K. alvarezii was added to the list of seaweeds that has a low recovery of arsenic species when water was used as an extractant.
In summary, for optimization of the following extraction parameters, 0.2% HNO3 solution was selected as the most appropriate extractant.
Effect of Extraction Time
Extraction times were evaluated to achieve optimum extraction efficiency, rapid extraction process, and high sample throughput. The 0 min was a non-heating process, where the sample was extracted after the weighing step. Figure 2a–d shows the effect of extraction time on arsenic species concentration in K. alvarezii. Arsenobetaine was not recovered from K. alvarezii, thus the results are only presented for As(V), As(III), MMA, and DMA.

Effect of the extraction time (in min) on concentration (mg kg.−1) of a As(V), b As(III), c MMA, and d DMA from K. alvarezii (N = 3) extracted using 0.2% of HNO3 at 90 °C. Results are shown in concentration (mean recovery ± SD). The error bars represent the standard deviations of all points within the measurement region
There was a gradual increase in the concentrations of arsenic species from K. alvarezii with an increase in the extraction time, but they declined after adequate interaction of the sample with the extractant. The highest concentration was achieved at 60 min for As(V), As(III), and DMA, while for MMA, 120 min was necessary to achieve the maximum concentration. MMA might have required a longer time to weaken its covalent bond with one or more carbon atoms containing functional groups in K. alvarezii to be released into the diluted acid (Huang et al. 2020). A noticeable decline in the levels of As(V) (from 0.431 to 0.306 mg kg−1) and As(III) (from 0.047 to 0.010 mg kg−1) extracted was observed at 240 min and 120 min, respectively. There was a possibility of absorption of the analyte by the seaweed matrix at the longer extraction time (Amaral et al. 2013), in addition to a change in the stability of the arsenic species (Herath et al. 2020).
Many studies have stated that the length of the extraction time did not affect extraction efficiency (Tukai et al. 2002; Güngör and Elik 2007; Kazi et al. 2009; Narukawa et al. 2017), and any additional extraction time would create a plateau (Güngör and Elik 2007; Narukawa et al. 2017). Therefore, a shorter extraction time was preferred to preserve the arsenic species, mainly to prevent the conversion of As(V) to As(III) (Amaral et al. 2013). However, this study has illustrated that extending the extraction time gives more information about the extraction efficiency against time. As a compromise between prevention of the conversion of As(V) to As(III) and maintaining the yield of the arsenic species, the optimum extraction time of 60 min was selected as it was also proven to be an economical and cost-effective way to extract all the arsenic species.
Matrix Effect
The experiment in this section was used to determine if the sample matrix influenced the analyte response. Table 2 shows the Snedecor’s F and Student’s t-test for the arsenic species matrix effect. The slopes of the calibration using standard solutions and spiked K. alvarezii did not differ statistically, whereby the calculated value of the t-test (t cal) was lower than the t critical value (t cal < t crit) for all arsenic species at a 95% confidence level. These results indicated that there was no effect of the seaweed matrix on the arsenic species studied. Therefore, calibration using standard solutions can be utilized for arsenic species determination as this approach would provide practicality and simplicity for routine analysis compared to enriched matrix calibration (Trindade et al. 2020).
Limit of Detection and Quantification
The linear ordinary least-squares regression analysis parameters were calculated by analyzing three batches at six different concentration levels. The concentration level and the response of each concentration level were used to acquire the two coefficients of the calibration curve. Additionally, a lack-of-fit test was used to prove that the curve was linear. The standard deviation of the intercept point (sa) and the slope of the regression line (b) were also calculated from the above parameters.
The standard deviation of the intercept point (sa), the slope of the regression line (b), LOD, and LOQ values of the arsenic species are shown in Table 3. The LOD for the arsenic species demonstrated a low concentration, which ranged from 0.008 to 0.010 mg kg−1, thus reflecting the lowest detection by the HPLC-ICP-MS for this particular arsenic species.
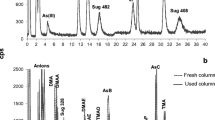
The predetermined value of LOQ observed in this study differed slightly from the confirmed LOQ. The predetermined LOQ was estimated by multiplying the standard deviation of the intercept (sa) with the slope of the regression line (b) by ten. This value was used as a guide to proceed with the experiments for the LOQ confirmation using a real sample; K. alvarezii. For LOQ confirmation, the predetermined value might have differed as it needed to fulfill two conditions for the confirmation. The confirmed LOQ in Table 3 shows there was a slight difference with the predetermined LOQ value, and it was adjusted to a more uniform value to ease the preparation of the mixed reference material for routine analysis (0.025 mg kg−1 for As(V), As(III), MMA and DMA; 0.050 mg kg−1 for AsB were used). Only AsB showed a higher level of LOQ compared to the rest of the arsenic species because, at this concentration, the response (intensity count) of AsB was equal to the rest of the arsenic species. Although the LOQ of AsB was doubled compared to that of the other arsenic species, it did not suppress the rest of the peaks observed in the HPLC-ICP-MS chromatogram in Fig. 3.

HPLC-ICP-MS chromatogram of arsenic species in K. alvarezii spiked at LOQ levels; 0.025 mg kg−1 for As(V), As(III), MMA, and DMA; 0.050 mg kg.−1 for AsB. The extraction of arsenic species from K. alvarezii was performed in 0.2% HNO3 at 90 °C for 1 h
For the monitoring of food regulation and safety standards for seaweed in countries like Malaysia, Australia, and New Zealand, inorganic arsenic [As(V) and As(III)] is monitored at the level of 1.0 mg kg−1, while in France, the safety limit is 3.0 mg kg−1 (Food Regulations 2019; Australia New Zealand Food Standards Code, schedule 19, maximum levels of contaminants and natural toxicants 2021; Edible seaweed and microalgae, regulatory status in France and Europe 2020). The results of this study show that the LOQ values for As(V) and As(III) were 40 times lower than the regulatory limit. Since the arsenic species in seaweed is one of the primary concerns of the food industries, achieving a low LOQ is vital to monitor the contamination rate.
The linear regression approach for LOD and LOQ determinations can also be used for other contaminants parameters such as pesticides and drug residues. However, it is most suitable for analytical methods that do not have background noises (International Organization of Vine and Wine (OIV) 2005). This procedure was adopted in this study as it uses a low concentration range that is close to zero for the calibration curve. Several studies (García-Salgado et al. 2012; Hsieh and Jiang 2012; Khan et al. 2015; Zmozinski et al. 2015) have adopted this approach as it takes into consideration errors in the concentrations and the instrument responses (del Río Bocio et al. 2003). Studies employing the classical approach of signal-to-noise ratio or blank determination exhibited slightly higher LOD and LOQ values (Hirata and Toshimitsu 2005; Han et al. 2008; Choi et al. 2011), as previously shown by Sanagi et al. (2009). Since arsenic species are found in trace amounts in food matrices, low LOD and LOQ values are desirable for quantification of the level of contamination. The concentrations of the arsenic species in this study are shown on a dry weight basis, in which the moisture content obtained for the K. alvarezii was 67%.
The LOD and LOQ obtained in this study were compared with that of other studies for As(V), As(III), DMA, MMA, and AsB in various types of seaweeds (Table 4). They were found to be comparable with those of studies using the heating extraction method with HNO3 and isocratic condition for HPLC-ICP-MS (Kim et al. 2020; Peng et al. 2020) but better than that reported by Matsumoto-Tanibuchi et al. (2019). Khan et al. (2015) utilized sonication with a mixture of MeOH and HNO3 as extraction solvent, and the extract was eluted using the anion-exchange cartridge. However, a comparison of detectability levels for As species quantification was not possible since this study provided its information on a unit as ppb, and it can be interpreted either in solution or in seaweed sample. Pétursdóttir and Gunnlaugsdóttir (2019) obtained lower concentrations of LOD and LOQ; however, hydride generation is only feasible for inorganic arsenic. Some studies obtained their LOD and LOQ in solution instead of in the actual matrix (Choi et al. 2011; Khan et al. 2015; Pétursdóttir and Gunnlaugsdóttir 2019), in which the values of the LOD or LOQ would appear to be lower. The LOD and LOQ obtained in the solution are only a representation of the instrumentation detection limit (IDL) (Belter et al. 2014; Kumar et al. 2021). The usage of the actual sample in LOQ determination in the method validation gives an accurate representation of the sample treatment from extraction to quantification.
Linearity and Working Range
The results from the linearity experiments using six concentration levels were analyzed for regression and lack of fit using the simple OLS regression method. Additionally, a significance test was performed to confirm if the y-intercept passed through the origin. The statistical data was transformed into a line fit plot and a y-residual plot.
The concentrations investigated in this study were correlated to their responses using ANOVA (Table 5). The results indicated that the alternate hypothesis was rejected for all arsenic species, and the hypothesis for regression was accepted. For the lack-of-fit test, F cal was lower than the corresponding F crit, and thus, the linearity was accepted for the arsenic species. If the calibration line displayed a significant curvature, the null hypothesis of linearity would be rejected. If this were to occur, more studies are required to find more suitable models (Sanagi et al. 2010).
A visual examination of y-residual plots indicated possible outliers for all the arsenic species and revealed no other apparent deficiencies. Therefore, these plots can be employed to reveal any heteroscedastic data formation. Furthermore, the concentrations investigated in this study (0.025 to 0.250 mg kg−1 for As(III), As(V), MMA, and DMA; 0.05 to 0.50 mg kg−1 for AsB) illustrated a homogeneous scatter, thus indicating that the distribution of the data in this experiment was relatively homoscedastic (Sanagi et al. 2010) due to the narrow study range selected as shown in Fig. 4a–e.

y-residual plots of residual (yi–íi) versus concentrations (mg kg.−1) of the five arsenic species; a As(V), b As(III), c AsB, d DMA, and e MMA and dashed lines are ± t(0.05,p − 2)Sres
Once the calibration was found to be linear, a final test was performed to determine if the linear calibration curve passed through the origin. The Student’s t-test in Table 6 showed that all the calibration curves for the arsenic species passed through the origin. Therefore, a more straightforward calibration approach could be utilized in routine analyses of all arsenic species. If a significant nonzero intercept was obtained, the accuracy of the method must be established (Moosavi and Ghassabian 2018).
According to Araujo (2009) and Moosavi and Ghassabian (2018), most studies (Hirata and Toshimitsu 2005; Choi et al. 2011; Khan et al. 2015; Park et al. 2019) commonly failed to indicate the statistical methods used to evaluate the linear relationship. The FDA guidance for the validation of analytical procedures proposed the use of appropriate statistical methods for the evaluation of linearity (International Conference on Harmonization (ICH) 2005). It was also emphasized that the “r-test” was unlikely to be tested for linearity as it cannot be concluded that r = 0.999 is more linear than r = 0.997. Hence, R2 was not exerted as an acceptance of linearity, although the data was recorded in this study.
Linearity in the validation procedure encapsulated the capability of the method in producing test results that were directly proportional to the analyte in the matrix over various analyte concentrations of interest (working concentration range). The linearity of the proposed working range for all five arsenic species investigated in this study has been statistically proven and can be used for routine analyses.
Repeatability and Within-Lab Reproducibility
The values of 3.4 to 12.9% and 6.6 to 19.0% for the relative standard deviation for repeatability (RSDr) and relative standard deviation for within-lab reproducibility (RSDR), respectively, were found to be lower than the predicted RSDr and RSDR, thus indicating that the method was capable of producing repeatable results (Christensen and Granby 2001). These results also indicated the errors from repeatability were minor since it was analyzed in a singular condition as compared to reproducibility, which produced more substantial errors due to multiple sources. The precision data are summarized in Table 7.
The precision data illustrated the similarity of the results obtained by the proposed method. This method was applied in various experimental conditions to include the random errors that could occur during extreme measurement conditions (Magnusson 2014). The data collected from the precision analyses are essential for the measurement of uncertainty. Additionally, the precision limits (r and R) derived from the standard deviations of repeatability and within-lab reproducibility are useful as acceptance criteria for internal quality control in routine analyses (ISO 5765–62003).
Trueness
In this study, the value of As(V) derived from NMIJ CRM 7405-a was 9.7 ± 0.3 mg kg−1 with a mean recovery (N = 6) of 96.0 ± 2.5% (SD). This result was in good agreement with the certified value of 10.1 ± 0.5 mg kg−1. CRM 7405-a has been used in previous studies to validate the accuracy of the method for seaweed and has shown recoveries of above 90% (Khan et al. 2015; Matsumoto-Tanibuchi et al. 2019; Park et al. 2019). Undeniably, there is an urgent need for the development of certified reference material with more arsenic species to enhance the applicability of the analytical methods selected (Wolle and Conklin 2018a).
Since no CRMs for As(III), AsB, MMA, and DMA in seaweed were available commercially, the targeted analytes were spiked into a blank seaweed at three concentration levels, ranging from low, middle, to high. Although the trueness of As(V) was determined through CRM, the analyte was still included for spiking. The recoveries of As(V), As(III), AsB, MMA, and DMA ranged between 87.8 and 107.4% and complied with the recommended values of 80 to 110% proposed by the Commission Decision 657/2002/EC (European Parliament and the Council of the European Union 2002) as shown in Table 7.
CRM analysis and recovery study were used to determine the trueness of this method for the determination of arsenic species, and it was found that the proposed method is suitable and efficient. Similar to the precision study that indicates the random errors of the method, the determination of trueness estimates the magnitude to which the systematic errors can affect this method (Araujo 2009).
Based on the results of the validation parameters analyzed, the extraction method with HPLC-ICP-MS used in this study was found to be efficient for the determination of the arsenic species in carrageenan-producing seaweed samples.
Proficiency Test
The concentration of inorganic arsenic (As(V) and As(III)) in the dried seaweed sample from the proficiency test provider was 56.3 ± 2.3 mg kg−1. The assigned value with standard deviation from the consensus of the results submitted by the 52 participants was 52.0 ± 4.59 mg kg−1. Therefore, the obtained result was satisfactory because the Ζ-score calculated was 0.9 corresponding to |z|≤ 2. Besides the assessment of the performance of the laboratory for the testing of this method, it also provides a comparison of performance with that of other laboratories internationally.
Method Validation Using Seaweed Samples
The identification of arsenic species in the actual seaweed sample (Kappaphycus spp.) was performed using the validated method. Chromatograms depicting the identity of four arsenic species peaks consisting of As(V), As(III), MMA, and DMA based on their retention times relative to their standards in K. alvarezii var brown tambalang, K. alvarezii var tangan-tangan, K. alvarezii var buaya, and K. alvarezii var durian are shown in Fig. 5a–d. In all four seaweed varieties, a peak was observed to elute before DMA. The unidentified peak was detected and reported as arsenosugar by several studies (Raab et al. 2005; Han et al. 2008; Choi et al. 2011; Yu et al. 2015; Kroukamp et al. 2019). This compound is possibly a component of the metabolism of arsenic in marine organisms (Hirata and Toshimitsu 2005). The nonavailability of this compound commercially poses a challenge in establishing its presence in seaweeds (García-Salgado et al. 2012).

HPLC-ICP-MS chromatograms for arsenic species and an unidentified peak in the extracts from four varieties of four varieties K. alvarezii: a K. alvarezii var brown tambalang, b K. alvarezii var tangan-tangan, c K. alvarezii var buaya, and d K. var durian
The mean concentration (± standard deviation) of each arsenic in the K. alvarezii varieties is presented in Table 8. As(V), As(III), DMA, and MMA were present in all the samples. Whereas, AsB was not detected in any of the K. alvarezii varieties. The occurrence of AsB is likely to be linked with epiphytes that adhere to the seaweed (Wolle and Conklin 2018a). MMA was predominant in both K. alvarezii var brown tambalang and K. alvarezii var tangan-tangan. DMA and As(V) were prevalent in K. alvarezii var buaya and K. alvarezii var durian, respectively. The variation of arsenic species levels in K. alvarezii varieties is due to the degree of reduction and methylation that occur in the seaweed (Farías et al. 2007). It is reported that arsenate is readily and actively taken up by seaweeds from seawater, where it is reduced to As(III), methylated to MMA and DMA, and then excreted (Ma et al. 2018). Different seaweeds may have different affinity for heavy metals due to different reasons, including their cell wall structures with different amounts and compositions of polysaccharides (Yong et al. 2017). In this study, it is suggested that the capacity for the absorption, retention, and excretion of As differs among the varieties in the same Kappaphycus spp. Comparing the results to the recommended values for inorganic As in food regulation and safety standards in Malaysia, Australia, and New Zealand for seaweed (Food Regulations 2019; Australia New Zealand Food Standards Code, schedule 19, maximum levels of contaminants and natural toxicants 2021), it can be seen that Kappaphycus spp. analyzed were safe, and their contribution of inorganic arsenic [A(V) + As(III)] is within the permissible specified limits.
Conclusions
The extraction conditions of the proposed method in this study were optimized and validated for the determination of As(V), As(III), AsB, DMA, and MMA in carrageenan-producing seaweed using HPLC-ICP-MS. The method featured a short extraction time of 1 h at 90 °C and the use of a low concentration of the nitric acid solution (0.2% HNO3). It was deemed to be rapid and economical without affecting the integrity of the arsenic species. Validation of the method was performed according to the guidelines proposed by the Official Journal of the European Communities, 2002/657/EC (European Parliament and the Council of the European Union 2002). There was no matrix effect observed throughout this study. The proposed method achieved a low LOQ of 0.025 mg kg−1 for As(V), As(III), MMA, and DMA, respectively, and 0.050 mg kg−1 for AsB, thereby fulfilling the Malaysian regulatory limit of 1.0 mg kg−1 for inorganic arsenic (Food Regulations 2019). The random and systematic errors in this method were addressed through the evaluation of accuracy based on precision and trueness studies. The RSD values for repeatability and within-lab reproducibility were below 20% and close to the predicted RSD. The measurement of trueness with recovery percentages for CRM and the spiked samples above 90% was within the range of 80 to 110% as indicated by the Official Journal of the European Communities, 2002/657/EC (European Parliament and the Council of the European Union 2002). The validation parameters were also statistically evaluated to demonstrate that the method is fit for its purpose. The proposed method can also be utilized for the determination of arsenic species in similar matrices, such as other carrageenan-containing seaweeds.
Data Availability
Data is available on request from the authors.
References
Alba K, Kontogiorgos V (2019) Seaweed Polysaccharides (Agar, Alginate Carrageenan). In: Melton L, Shahidi F, Varelis P (eds) Encyclopedia of Food Chemistry. Academic Press, Oxford, pp 240–250. https://doi.org/10.1016/B978-0-08-100596-5.21587-4
Amaral CD, Nobrega JA, Nogueira AR (2013) Sample preparation for arsenic speciation in terrestrial plants–a review. Talanta 115:291–299. https://doi.org/10.1016/j.talanta.2013.04.072
Araujo P (2009) Key aspects of analytical method validation and linearity evaluation. J Chromatogr B 877:2224–2234. https://doi.org/10.1016/j.jchromb.2008.09.030
Association of Official Analytical Chemists (AOAC) (2005) Official method 930.04 moisture in plants. In: Horwitz W, Latimer G (eds), 18th edn. Association of Official Analytical Chemists, Maryland, pp 1–4
Association of Official Analytical Chemists (AOAC) (2019) Official methods of analysis of AOAC international-guidelines for dietary supplements and botanicals: Appendix K. In: Horwitz W, Latimer G (eds), 20th edn. Association of Official Analytical Chemists, Maryland, pp 1–32
Association of Official Analytical Chemists (AOAC) International (2007) Analytical Laboratory Accreditation Criteria Committee (ALACC) Guide : How to meet ISO 17025 requirements for method verification.
Australia New Zealand Food Standards Code - schedule 19 - maximum levels of contaminants and natural toxicants (2021). https://www.legislation.gov.au/Details/F2021C00628. Accessed 30 Jul 2021
B’Hymer C, Caruso JA (2004) Arsenic and its speciation analysis using high-performance liquid chromatography and inductively coupled plasma mass spectrometry. J Chromatogr A 1045:1–13. https://doi.org/10.1016/j.chroma.2004.06.016
Belter M, Sajnóg A, Barałkiewicz D (2014) Over a century of detection and quantification capabilities in analytical chemistry–historical overview and trends. Talanta 129:606–616. https://doi.org/10.1016/j.talanta.2014.05.018
Benramdane L, Bressolle F, Vallon JJ (1999) Arsenic speciation in humans and food products: a review. J Chromatogr Sci 37:330–344. https://doi.org/10.1093/chromsci/37.9.330
Bratinova S, Raffael B, Simoneau C (2009) Guidelines for performance criteria and validation procedures of analytical methods used in controls of food contact materials. JRC Scientific and Technical Reports, EUR 24105 EN, 1st edn.
Bullen JC, Torres-Huerta A, Salaün P, Watson JS, Majumdar S, Vilar R, Weiss DJ (2020) Portable and rapid arsenic speciation in synthetic and natural waters by an As(V)-selective chemisorbent, validated against anodic stripping voltammetry. Water Res 175:115650. https://doi.org/10.1016/j.watres.2020.115650
Chen L, Zhang L (2019) Arsenic speciation in Asiatic algae: case studies in Asiatic continent. In: Duarte AC, Reis V (eds) Comprehensive analytical chemistry, 1st edn. Elsevier B.V., Amsterdam, pp 199–245. https://doi.org/10.1016/bs.coac.2019.03.004
Choi H, Park SK, Kim DS, Kim M (2011) Determination of 6 arsenic species present in seaweed by solvent extraction, clean-up, and LC-ICP/MS. Food Sci Biotechnol 20:39–44. https://doi.org/10.1007/s10068-011-0006-9
Christensen HB, Granby K (2001) Method validation for strobilurin fungicides in cereals and fruit. Food Addit Contam 18:866–874. https://doi.org/10.1080/02652030121435
del Río Bocio FJ, Riu J, Boqué R, Rius FX (2003) Limits of detection in linear regression with errors in the concentration. J Chemom 17:413–421. https://doi.org/10.1002/cem.818
Díaz O, Tapia Y, Muñoz O, Montoro R, Velez D, Almela C (2012) Total and inorganic arsenic concentrations in different species of economically important algae harvested from coastal zones of Chile. Food Chem Toxicol 50:744–749. https://doi.org/10.1016/j.fct.2011.11.024
Edible seaweed and microalgae - Regulatory status in France and Europe (2020) Centre d'Étude et de Valorisation des Algues. https://www.ceva-algues.com/en/document/edible-algae-regulatory-update/. Accessed 30 Jun 2020
Ellwood MJ, Maher WA (2003) Measurement of arsenic species in marine sediments by high-performance liquid chromatography–inductively coupled plasma mass spectrometry. Anal Chim Acta 477:279–291. https://doi.org/10.1016/S0003-2670(02)01414-9
European Parliament and the Council of the European Union (2002) Commission Decision 2002/657/EC. Off J Eur Communities L221:8–36. https://op.europa.eu/de/publication-detail/-/publication/ed928116-a955-4a84-b10a-cf7a82bad858/language-en. Accessed 30 Mac 2020
Farías S, Smichowski P, Vélez D, Montoro R, Curtosi A, Vodopívez C (2007) Total and inorganic arsenic in Antarctic macroalgae. Chemosphere 69:1017–1024. https://doi.org/10.1016/j.chemosphere.2007.04.049
Food Regulations (2019) Food Act 1983 (Act 281); & Food Regulations 1985. August 2019 edn. International Law Book Services, Kuala Lumpur
García-Salgado S, Quijano M, Bonilla M (2012) Arsenic speciation in edible alga samples by microwave-assisted extraction and high performance liquid chromatography coupled to atomic fluorescence spectrometry. Anal Chim Acta 714:38–46. https://doi.org/10.1016/j.aca.2011.12.001
Güngör H, Elik A (2007) Comparison of ultrasound-assisted leaching with conventional and acid bomb digestion for determination of metals in sediment samples. Microchem J 86:65–70. https://doi.org/10.1016/j.microc.2006.10.006
Han C, Cao X, Yu JJ, Wang XR, Shen Y (2008) Arsenic speciation in Sargassum fusiforme by microwave-ssisted extraction and LC-ICP-MS. Chromatographia 69:587–591. https://doi.org/10.1365/s10337-008-0917-7
Herath I, Kumarathilaka P, Bundschuh J, Marchuk A, Rinklebe J (2020) A fast analytical protocol for simultaneous speciation of arsenic by ultra-high performance liquid chromatography (UHPLC) hyphenated to inductively coupled plasma mass spectrometry (ICP-MS) as a modern advancement in liquid chromatography approaches. Talanta 208:1–13. https://doi.org/10.1016/j.talanta.2019.120457
Hirata S, Toshimitsu H (2005) Determination of arsenic species and arsenosugars in marine samples by HPLC-ICP-MS. Anal Bioanal Chem 383:454–460. https://doi.org/10.1007/s00216-005-3413-z
Hsieh YJ, Jiang SJ (2012) Application of HPLC-ICP-MS and HPLC-ESI-MS procedures for arsenic speciation in seaweeds. J Agric Food Chem 60:2083–2089. https://doi.org/10.1021/jf204595d
Huang SX, Jiang Q, Ding YF, Wang FJ, Zhu C (2020) Arsenic contents and speciation at different growth stages of Sargassum fusiforme [harv] setchell (hijiki), an edible seaweed. Appl Ecol Environ Res 18:1941–1952. https://doi.org/10.15666/aeer/1801_19411952
Hussain MM, Wang J, Bibi I, Shahid M, Niazi NK, Iqbal J, Mian IA, Shaheen SM, Bashir S, Shah NS, Hina K, Rinklebe J (2021) Arsenic speciation and biotransformation pathways in the aquatic ecosystem: the significance of algae. J Hazard Mater 403:1–13. https://doi.org/10.1016/j.jhazmat.2020.124027
International Conference on Harmonization (ICH) (2005) ICH harmonised tripartite guideline validation of analytical procedures: text and methodology Q2 (R1)
International Organization of Vine and Wine (OIV) (2005) Resolution Oeno10/2005: a practical guide for the validation, quality control, and uncertainty assessment of an alternative oenological analysis method Compendium of international methods of wine and must analysis
ISO 5725–1 (2003) Accuracy (trueness and precision) of measurement methods and results—part 1: general principles and definitions. International Organization for Standardization, Geneva
ISO 5765–6 (2003) Accuracy (trueness and precision) of measurement methods and results—part 6: use in practice of accuracy values. International Organization for Standardization, Geneva
ISO 11095 (1996) Linear calibration using reference materials. International Organization for Standardization, Geneva
Kazi TG, Jamali MK, Arain MB, Afridi HI, Jalbani N, Sarfraz RA, Ansari R (2009) Evaluation of an ultrasonic acid digestion procedure for total heavy metals determination in environmental and biological samples. J Hazard Mater 161:1391–1398. https://doi.org/10.1016/j.jhazmat.2008.04.103
Khan N, Ryu KY, Choi JY, Nho EY, Habte G, Choi H, Kim MH, Park KS, Kim KS (2015) Determination of toxic heavy metals and speciation of arsenic in seaweeds from South Korea. Food Chem 169:464–470. https://doi.org/10.1016/j.foodchem.2014.08.020
Kim M-h, Kim J, Noh C-H, Choi S, Joo Y-S, Lee K-W (2020) Monitoring arsenic species content in seaweeds produced off the southern coast of Korea and its risk assessment. Environments 7:68
Klumpp DW (1980) Characteristics of arsenic accumulation by the seaweeds Fucus spiralis and Ascophyllum nodosum. Mar Biol 58:257–264. https://doi.org/10.1007/BF00390774
Klumpp DW, Peterson PJ (1979) Arsenic and other trace elements in the waters and organisms of an estuary in SW England. Environ Pollut 19:11–20. https://doi.org/10.1016/0013-9327(79)90109-5
Kroukamp EM, Godeto TW, Forbes PB (2019) Optimized extraction of inorganic arsenic species from a foliose lichen biomonitor. Environ Sci Pollut Res 26:29896–29907. https://doi.org/10.1007/s11356-019-06073-2
Kuehnelt D, Irgolic KJ, Goessler W (2001) Comparison of three methods for the extraction of arsenic compounds from the NRCC standard reference material DORM-2 and the brown alga Hijiki fuziforme. Appl Organomet Chem 15:445–456. https://doi.org/10.1002/aoc.189
Kumar Y, Tarafdar A, Badgujar PC (2021) Seaweed as a source of natural antioxidants: therapeutic activity and food applications. J Food Qual 2021:17. https://doi.org/10.1155/2021/5753391
Liu J, Shen Q, Wei F, Guo Z, Tian Y (2020) Differentiation of digestion method for heavy metals in river sediments based on organic matter gradients. In: IOP Conference Series: Earth and Environmental Science, Xiamen, 19–21 Nov 2019. vol 1. IOP Publishing, Bristol, pp 1–8. https://doi.org/10.1088/1755-1315/467/1/012128
Lorenc W, Kruszka D, Kachlicki P, Kozłowska J, Barałkiewicz D (2020) Arsenic species and their transformation pathways in marine plants. Usefulness of advanced hyphenated techniques HPLC/ICP-MS and UPLC/ESI-MS/MS in arsenic species analysis. Talanta 220:121384. https://doi.org/10.1016/j.talanta.2020.121384
Ma Z, Lin L, Wu M, Yu H, Shang T, Zhang T, Zhao M (2018) Total and inorganic arsenic contents in seaweeds: absorption, accumulation, transformation and toxicity. Aquaculture 497:49–55. https://doi.org/10.1016/j.aquaculture.2018.07.040
Magnusson B (2014) Eurachem Guide: The fitness for purpose of analytical methods: a laboratory guide to method validation and related topics 2nd edn. Eurachem
Manuhara GJ, Praseptiangga D, Riyanto RA (2016) Extraction and characterization of refined K-carrageenan of red algae [Kappaphycus Alvarezii (Doty ex PC Silva, 1996)] originated from Karimun Jawa Islands. Aquat Procedia 7:106–111. https://doi.org/10.1016/j.aqpro.2016.07.014
Matsumoto-Tanibuchi E, Sugimoto T, Kawaguchi T, Sakakibara N, Yamashita M (2019) Determination of inorganic arsenic in seaweed and seafood by LC-ICP-MS: method validation. J AOAC Int 102:612–618. https://doi.org/10.5740/jaoacint.18-0148
Mohammad SM, Mohd Razali SF, Mohamad Rozaiman NHN, Laizani AN, Zawawi N (2019) Application of seaweed (Kappaphycus alvarezii) in Malaysian food products. Int Food Res J 26:1677–1687
Moosavi SM, Ghassabian S (2018) Linearity of calibration curves for analytical methods: a review of criteria for assessment of method reliability. In: Stauffer TM (ed) Calibration and validation of analytical methods: a sampling of current approaches. IntechOpen, London, pp 109–127. https://doi.org/10.5772/intechopen.72932
Murphy CJ, MacNeil JD, Capar SG (2013) Best practices for single-laboratory validation of chemical methods for trace elements in foods. Part I—background and general considerations. J AOAC Int 96:190–203. https://doi.org/10.5740/jaoacint.12-204
Nan K, He M, Chen B, Chen Y, Hu B (2018) Arsenic speciation in tree moss by mass spectrometry based hyphenated techniques. Talanta 183:48–54. https://doi.org/10.1016/j.talanta.2018.02.055
Narukawa T, Chiba K, Sinaviwat S, Feldmann J (2017) A rapid monitoring method for inorganic arsenic in rice flour using reversed phase-high performance liquid chromatography-inductively coupled plasma mass spectrometry. J Chromatogr A 1479:129–136. https://doi.org/10.1016/j.chroma.2016.12.001
Narukawa T, Hioki A, Chiba K (2012) Aqueous extraction of water-soluble inorganic arsenic in marine algae for speciation analysis. Anal Sci 28:773–779. https://doi.org/10.2116/analsci.28.773
Narukawa T, Raber G, Itoh N, Inagaki K (2020) A new candidate reference material for inorganic arsenic and arsenosugars in hijiki seaweed: first results from an inter-laboratory study. Anal Sci 36:233–239. https://doi.org/10.2116/analsci.19P306
Narukawa T, Suzuki T, Inagaki K, Hioki A (2014) Extraction techniques for arsenic species in rice flour and their speciation by HPLC–ICP-MS. Talanta 130:213–220. https://doi.org/10.1016/j.talanta.2014.07.001
Park MK, Choi M, Kim L, Choi SD (2019) An improved rapid analytical method for the arsenic speciation analysis of marine environmental samples using high-performance liquid chromatography/inductively coupled plasma mass spectrometry. Environ Monit Assess 191. https://doi.org/10.1007/s10661-019-7675-x
Peng S, Yuling G, Xiaolin Z, Ruia Y (2020) Determination of four arsenic species in algae by high-performance liquid chromatography coupled with hydride generation atomic fluorescence spectrometry. Química Nova 43:1074–1077. https://doi.org/10.21577/0100-4042.20170574
Pétursdóttir ÁH, Gunnlaugsdóttir H (2019) Selective and fast screening method for inorganic arsenic in seaweed using hydride generation inductively coupled plasma mass spectrometry (HG-ICPMS). Microchem J 144:45–50. https://doi.org/10.1016/j.microc.2018.08.055
Phang SM, Yeong HY, Lim PE (2019) The seaweed resources of Malaysia. Bot Mar 62:265–273. https://doi.org/10.1515/bot-2018-0067
Raab A, Fecher P, Feldmann J (2005) Determination of arsenic in algae–results of an interlaboratory trial: determination of arsenic species in the water-soluble fraction. Microchim Acta 151:153–166. https://doi.org/10.1007/s00604-005-0395-7
Rahman MA, Hasegawa H, Lim RP (2012) Bioaccumulation, biotransformation and trophic transfer of arsenic in the aquatic food chain. Environ Res 116:118–135. https://doi.org/10.1016/j.envres.2012.03.014
Reis V, Duarte AC (2019) Occurrence, distribution, and significance of arsenic speciation. In: Duarte AC, Reis V (eds) Comprehensive analytical chemistry, 1st edn. Elsevier B.V., Amsterdam, pp 1–14. https://doi.org/10.1016/bs.coac.2019.03.006
Reis VAT, Duarte AC (2018) Analytical methodologies for arsenic speciation in macroalgae: a critical review. Trends Anal Chem 102:170–184. https://doi.org/10.1016/j.trac.2018.02.003
Sadee BA, Foulkes ME, Hill SJ (2016) An evaluation of extraction techniques for arsenic in staple diets (fish and rice) utilising both classical and enzymatic extraction methods. Food Addit Contam Part A 33:433–441. https://doi.org/10.1080/19440049.2015.1132479
Sanagi MM, Ling SL, Nasir Z, Hermawan D, Wan Ibrahim WA, Naim AA (2009) Comparison of signal-to-noise, blank determination, and linear regression methods for the estimation of detection and quantification limits for volatile organic compounds by gas chromatography. J AOAC Int 92:1833–1838. https://doi.org/10.1093/jaoac/92.6.1833
Sanagi MM, Nasir Z, Ling SL, Hermawan D, Ibrahim WAW, Naim AA (2010) A practical approach for linearity assessment of calibration curves under the International Union of Pure and Applied Chemistry (IUPAC) guidelines for an in-house validation of method of analysis. J AOAC Int 93:1322–1330. https://doi.org/10.1093/jaoac/93.4.1322
Seaweed Market: Global industry trends, share, size, growth, opportunity and forecast (2021–2026) (2020) https://www.imarcgroup.com/seaweed-market. Accessed 25 Jan 2021
Thompson M, Ellison SLR, Wood R (2002) Harmonized guidelines for single-laboratory validation of methods of analysis. Pure Appl Chem 74:835–855. https://doi.org/10.1351/pac200274050835
Trindade AC, Araújo SA, Amorim FAC, Silva DS, Alves JPS, Trindade JS, Aguiar RM, Bezerra MA (2020) Development of a method based on slurry sampling for determining Ca, Fe, and Zn in coffee samples by flame atomic absorption spectrometry. Food Anal Methods 13:203–211. https://doi.org/10.1007/s12161-019-01578-5
Tukai R, Maher WA, McNaught IJ, Ellwood MJ (2002) Measurement of arsenic species in marine macroalgae by microwave-assisted extraction and high performance liquid chromatography–inductively coupled plasma mass spectrometry. Anal Chim Acta 457:173–185. https://doi.org/10.1016/S0003-2670(02)00018-1
Wang Y, Wang S, Xu P, Liu C, Liu M, Wang Y, Wang C, Zhang C, Ge Y (2015) Review of arsenic speciation, toxicity and metabolism in microalgae. Rev Environ Sci Biotechnol 14:427–451. https://doi.org/10.1007/s11157-015-9371-9
Wilschefski SC, Baxter MR (2019) Inductively coupled plasma mass spectrometry: introduction to analytical aspects. Clin Biochem Rev 40:115–133. https://doi.org/10.33176/AACB-19-00024
Wolle MM, Conklin SD (2018a) Speciation analysis of arsenic in seafood and seaweed: part I—evaluation and optimization of methods. Anal Bioanal Chem 410:5675–5687. https://doi.org/10.1007/s00216-018-0906-0
Wolle MM, Conklin SD (2018b) Speciation analysis of arsenic in seafood and seaweed: part II-single laboratory validation of method. Anal Bioanal Chem 410:5689–5702. https://doi.org/10.1007/s00216-018-0910-4
Yong WTL, Chin JYY, Thien VY, Yasir S (2017) Heavy metal accumulation in field cultured and tissue cultured Kappaphycus alvarezii and Gracilaria changii. Int Food Res J 24:970–975
Yu LL, Wei C, Zeisler R, Tong J, Oflaz R, Bao H, Wang J (2015) An approach for identification and determination of arsenic species in the extract of kelp. Anal Bioanal Chem 407:3517–3524. https://doi.org/10.1007/s00216-015-8567-8
Zmozinski AV, Llorente-Mirandes T, López-Sánchez JF, da Silva MM (2015) Establishment of a method for determination of arsenic species in seafood by LC-ICP-MS. Food Chem 173:1073–1082. https://doi.org/10.1016/j.foodchem.2014.10.102
Acknowledgements
The authors would like to express their gratitude to the Food Safety and Quality Programme, Ministry of Health, Malaysia, for the financial support provided through funding, and Universiti Malaysia Sabah for providing the seaweed samples. The authors greatly appreciate the support provided by the Head of Laboratory and staff of Selangor Food Safety and Quality Laboratory, Ministry of Health, for the provision of laboratory facilities and technical support. The authors would also like to thank the Director-General of Health, Ministry of Health, for permission to publish this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
This article does not contain any studies performed using human participants or animals.
Consent to Participate
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Subramaniam, G., Bakar, J., Surugau, N. et al. Optimization of Extraction Conditions and Validation of the Method for Determination of Arsenic Species in Carrageenan-Producing Seaweed, Kappaphycus spp., Using HPLC-ICP-MS. Food Anal. Methods 15, 2858–2878 (2022). https://doi.org/10.1007/s12161-022-02334-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-022-02334-y