
Abstract
Lactobacillus sanfranciscensis, Lactobacillus brevis, and Lactobacillus curvatus are frequently isolated from cereal-based fermented foods and are the dominant lactic acid bacteria in Korean sourdough. Detection of the individual species and their enumeration during fermentation are of great importance for maintaining the quality of bakery products. Here, we developed a species-specific quantitative polymerase chain reaction (qPCR) method to monitor the population of these three bacterial species during cocultivation in sourdough. Target genes, including those encoding nucleoside hydrolase, hypothetical protein, and glyoxalase, were selected by MegaBLAST for the detection of L. sanfranciscensis, L. brevis, and L. curvatus, respectively. The specificities of PCR primer sets were verified with qPCR; constant cycle threshold (Ct) values were obtained from mixed genomic DNAs (gDNAs) of target and non-target strains. The qPCR results were unaffected by the sourdough matrix. The cell numbers derived from qPCR were 10–65% higher than those obtained using a conventional plate-counting method. The qPCR standard curves for the three Lactobacillus species were established, and their populations were successfully enumerated during sourdough propagation. This method would enable quantification of three Lactobacillus species during cocultivation in sourdough and provide useful information on microbial commensalism that is essential to obtain high-quality bread.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sourdough has been used in bread production worldwide due to the improved rheological, organoleptic, and nutritional properties compared to the products made by straight dough processes (Hansen and Schieberle 2005; Pétel et al. 2017). The difference between two bakery products is based on metabolic compounds produced by lactic acid bacteria (LAB) at dough stage (Gänzle and Ripari 2016). The beneficial contributions during sourdough fermentation from LAB include production of exopolysaccharides that improved volume, texture, and shelf life (Lin and Gänzle 2014) and formation of anti-microbial and volatile compounds (Choi et al. 2012; Leroy and De Vuyst 2004). Traditional sourdoughs have geographical and cultural identities all over the world because of their natural ingredients as well as fermentation methodologies, and thus these differences result in LAB microflora in sourdough (Dertli et al. 2016). In our previous study, we identified Lactobacillus sanfranciscensis, L. curvatus, and L. brevis as prevalent species in Korean sourdough that represented 56, 27, and 9% of the total isolates, respectively (Lee et al. 2015). They are also frequently detected in other types of sourdough originated from various substrates (De Vuyst et al. 2014; Liu et al. 2018). Growth of Lactobacillus spp. may positively or negatively affect to each other due to their metabolites such as organic acids and bacteriocins and their enzymatic activities including proteases and amylases (Zhang et al. 2019a). Nowadays, fermentation industries often employ defined-starter cultures consisted of couple of strains to produce high-quality products (Bintsis 2018; Özcan et al. 2020). As the same in the bakery industry, maintenance of the microbial ecosystem in the starter culture could improve and maintain the quality of the products, and thus their population changes during fermentation should be analyzed and monitored at species level (Gaglio et al. 2020). However, there are no methods to specifically and simultaneously quantify these species in sourdough.
Compositions of complex microflora in sourdough result in difficulties in enumeration of particular species by conventional plate counting (Moser et al. 2017; Yazar and Tavman 2012). To guarantee quality of the sourdough, controlling and monitoring microbiota for reproducibility throughout the fermentation progress has been considered necessary (Lin and Gänzle 2014; Park et al. 2019). For this, culture-independent methods like real-time quantitative polymerase chain reaction (qPCR) have been developed for enumerating individual species from complex DNA sample by quantifying the number of copies for target gene (Kim et al. 2015). This qPCR method can be potential alternative of conventional plate counting due to its rapidness and sensitivity for the enumeration of certain bacteria (He et al. 2017). However, the qPCR has often used universal primers targeting 16S rRNA-encoding gene, and it has difficulty in discrimination in species levels of Lactobacillus genus due to their similarities in nucleotide sequences (Pontonio et al. 2017; You and Kim 2020; Kim et al. 2020, b). In addition, monitoring population changes of individual species in the food samples fermented by multiple Lactobacillus species is much more difficult. Besides, the current metagenomic method based on 16S rRNA gene amplicon sequencing often does not reveal microbial diversity past the genus level and exhibits only moderately accurate profile (Hillmann et al. 2018). Therefore, a reliable and rapid method is needed to monitor dynamics of multiple Lactobacillus species during sourdough fermentation (You and Kim 2020).
In the present study, we developed a species-specific real-time quantitative polymerase chain reaction (qPCR) method for L. sanfranciscensis, L. brevis, and L. curvatus to monitor their population during cocultivation in sourdough. To target the species-specific genes of L. brevis and L. curvatus, we searched the unique nucleotide sequences present in the corresponding genomes using the Mega Basic Local Alignment Search Tool (MegaBLAST) analysis of genome sequences of LAB. Based on the results, we selected the genes for a glyoxalase and hypothetical protein as targets for the species-specific detection of L. brevis and L. curvatus, respectively. For L. sanfranciscensis, we used our previously developed species-specific primer set targeting the nucleoside hydrolase gene of the bacterium (Lee et al. 2015). The specificities of the PCR primer sets were verified by colony PCR and qPCR against different species of LAB. The standard curves for qPCR of the three Lactobacillus species were successfully established and applied to monitor the changes in their populations during sourdough propagation.
Materials and Methods
LAB and Culture Conditions
Table 1 lists the microorganisms used in this study. L. sanfranciscensis, L. brevis, and L. curvatus were isolated from the Korean sourdough inoculated with traditional nuruk, a preculture of natural microorganisms on a disc prepared from cooked rice and wheat bran. The culture stocks stored at −80 °C were thawed and incubated in MRS medium (Difco, MI, USA) at 30 °C with shaking at 250 rpm under anaerobic conditions. Batch fermentations of mixed cultures were carried out in 500-mL baffled flasks containing 100 mL of MRS broth at 30 °C and 250 rpm. The initial pH of the MRS medium was adjusted to pH 6.5 using 2 N sodium hydroxide (NaOH).
Design of Strain-Specific qPCR Primer Sets
The complete genome sequence data of L. brevis KB290 and L. curvatus CRL705 were retrieved from National Center for Biotechnology Information (NCBI: http://www.ncbi.nlm.nih.gov/genome/). To select each qPCR target, their unknown, hypothetical, and unannotated genes from each genome sequence were screened in priority, and genetic uniqueness was examined with BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). As per the comparative genomics, qPCR target genes that lacked any significant similarity in nucleotide sequence among other LAB were selected, and the final target nucleotide sequences were chosen after repetitive assessment for specificity. Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) was used to confirm the specificities of primers toward qPCR amplicon. All primer sets were synthesized by Bioneer Co. (Daejeon, Korea).
Genomic DNA (gDNA) Preparation
For the extraction of gDNA from pure cultures in MRS media, the cells were cultivated until the density reached 108-109 cells. Cells were centrifuged at 13,000 × g for 2 min and the supernatant was discarded. To extract total DNA from sourdough samples, 10 g of sourdough was dissolved in a sterile 0.85% sodium chloride (NaCl) solution and the final volume was made up to 100 mL. After complete resuspension, 50 mL of the suspension was centrifuged at 200 × g for 5 min to remove large particles, and the cells were harvested by centrifugation for 15 min at 13,000 × g. Cell pellets were washed twice with 1 mL of sterile phosphate-buffered saline (PBS: 8.0 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4 in 1 L at pH 7.4) and the chromosomal DNA was prepared using the genomic DNA prep kit (SolGent, Seoul, Korea). The quantity and purity of gDNA were measured with NanoVue Plus (GE Healthcare Bio-Sciences Co., Piscataway, NJ, USA).
Conditions for PCR and qPCR
PCR was carried out in a 20 μL reaction mixture containing 10 pM primer set, 100 ng of extracted gDNA, 8 μL of double distilled water, and 10 μL of the AccuPower™ PCR PreMix (Bioneer Co., Daejeon, Korea). GeneAmp PCR System 2400 (Applied Biosystems, Foster city, CA, USA) was used, and the amplification program was as follows: denaturation step at 94 °C for 5 min; 40 cycles of denaturation at 94 °C for 1 min, annealing at 40 °C for 1 min, and extension at 72 °C for 1 min; and a final extension step at 72 °C for 10 min (Venturi et al. 2012). DNA electrophoresis was carried out for 1 h at 90 V using Vari-Gel™ Horizontal Mini System (Denville Scientific, MA, US) filled with Tris-acetate-ethylenediaminetetraacetic acid (EDTA) buffer (TAE: 40 mM Trizma base, 20 mM acetic acid, and 1 mM EDTA at pH 8.3).
The qPCR was carried out in a final volume of 25 μL in a reaction mixture comprising 12.5 μL of SYBR® Premix Ex Taq (Takara Bio. Inc., Shiga, Japan), 8.5 μL of sterile distilled water, 2 μL of template DNA (< 500 ng), and 0.4 μM of each primer. CFX ManagerTM real-time thermal cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA) was used, and annealing temperatures ranging from 58 to 63 °C were tested to determine optimum qPCR conditions yielding the lowest Ct value, a high final fluorescence value, and a melting curve with a single peak. All samples were analyzed in duplicates. Thermal cycling, fluorescence data collection, and data analysis were carried out using CFX ManagerTM Software v 3.0 (Bio-Rad Laboratories). The Ct value was defined as the PCR cycle at which the fluorescence signal exceeded the background level.
Enumeration of Three LAB by qPCR
For enumeration of L. sanfranciscensis, L. brevis, and L. curvatus in sourdough samples, their Ct values were interpolated on a linear regression model generated from the gDNA extracted from reference strain cultures in MRS media. Serial dilutions ranging from 103 to 109 copies/μL of each gDNA were prepared, and standard curves were obtained by plotting the Ct values against the target gene copy number/μL via qPCR. Each standard curve was derived from qPCR data in triplicates. The reliability criteria were determined by the square of correlation coefficient (R2) and amplification efficiency. R2 value is indicative of how well the data fit to the first-order equation, suggestive of the agreement between actual Ct values in triplicates and the linear range of the assay. Efficiency of qPCR (E) was calculated as follows: E = 10(−1/b) − 1, where b is the slope of the linear fit. Quantification limit was defined as the lowest concentration of gDNA at which linearity was maintained. Intra-assay repeatability was evaluated on triplicates of one of the gDNA samples analyzed in a single qPCR experiment. The coefficient of variation (CV) based on Ct values was obtained at different concentrations of gDNA not only in the same qPCR run but also in three independent qPCR runs, estimating intra-assay and inter-assay reproducibility. Copy numbers (or cell numbers) were calculated for all standards using the following formula: Number of copies/μL = (6.022 × 1023 × gDNA concentrations)/number of bases pairs × 660 Da, where 6.022 × 1023 is Avogadro’s number (molecules/mole) and 660 Da is the average weight of a single base pair (Dhanasekaran et al. 2010).
Sourdough Fermentation and Propagation
L. sanfranciscensis, L. brevis, and L. curvatus were precultured in MRS medium at 30 °C and 250 rpm under anaerobic conditions. Cells were recovered by centrifugation at 10,000 × g for 2 min and suspended in sterile 0.85% NaCl. To prepare 500 g of sourdough, wheat flour (225 g), rye (25 g), tap water (250 mL), and a mixture (5 mL) of three microbial suspensions were continuously mixed for 5 min on a mixer (Chopin & Co., Boulogne, Seine, France). Sourdoughs were fermented at 23 °C for 5 h and stored at 4 °C for 19 h in the back-slopping process during the 7 days of long propagation.
Statistical Analysis
Statistical analysis of the qPCR results was performed using SPSS 12.0 (SPSS Inc, Chicago, IL, USA). Data were considered statistically significant when p < 0.05.
Results
Species-Specific Primers
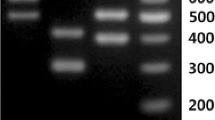
To develop species-specific primer sets for LAB, we retrieved the complete genome sequence data and screened candidate unique genes in their genomes. The genes that showed high identity or similarity with other species in nucleotide sequences were excluded by consecutively running MegaBLAST. The selection criteria were optimized for both highly similar sequences (MegaBLAST) and more dissimilar sequences (discontinuous MegaBLAST). Several candidate genes were used to match and finally the hypothetical gene (LVISKB_0189) of L. brevis and glyoxalase gene (EHE85023.1) of L. curvatus were selected as targets for qPCR. The corresponding primer sets to obtain final qPCR amplicons were designed for the amplification of each PCR amplicon below 170 bp, as primers producing around 150 bp-sized amplicons are ideal for real-time qPCR (Dhanasekaran et al. 2010). As shown in Fig. 1, the two primer sets BrevF/BrevR and CurvF/CurvR originating from the hypothetical gene of L. brevis and glyoxalase gene of L. curvatus, respectively, showed no sequence match with any other sequences uploaded in NCBI database. In addition, these primer sequences were commonly present in the gDNAs of 15 strains of L. brevis (Fig. 2a) and 12 strains of L. curvatus (Fig. 2b), respectively. The resulting primer sets are listed in Table 2. In addition, we used our previously developed species-specific primer set for L. sanfranciscensis targeting the nucleoside hydrolase gene of the bacterium (Lee et al. 2015).

Alignments of Lactobacillus brevis hypothetical protein gene a and L. curvatus glyoxalase gene b with nucleotide sequences from other species that showed sequence similarity based on discontinuous MegaBLAST (NCBI). The alignment was conducted based on MULTALIN (http://multalin.toulouse.inra.fr/multalin/), and intermediate sequences were omitted. Abbreviations: KB290, L. brevis; CRL, L. curvatus CRL270. Species-specific primers were designed based on the boxes

a Homologous region alignment of hypothetical protein gene (LVISKB_0189) from 12 strains of Lactobacillus brevis showing sequence differences (white column). The annealing sites for the designed primers are outlined. b Homologous region alignment of glyoxalase gene (EHE85023.1) from 12 strains of L. curvatus showing sequence differences (white column). The annealing sites for the designed primers are outlined
Assessment of Primer Specificity Using PCR Amplification
The specificity of each primer set was assessed with PCR using gDNAs prepared from pure cultures of seven different LAB such as L. sanfranciscensis, L. brevis, L. curvatus, L. plantarum, L. buchneri, L. sakei, and Pediococcus pentosaceus. The references of LAB were selected such that they were mainly detected in sourdough or nuruk. As shown in Fig. 3, single specific bands of expected sizes were obtained from each PCR reaction; the length of PCR products was 129 bp for L. sanfranciscensis, 162 bp for L. brevis, and 120 bp for L. curvatus, while we failed to detect any nonspecific amplification with gDNA samples of other bacteria. The results of DNA sequencing of PCR amplicons sub-cloned into the TA cloning vector also proved that each primer set amplified the right nucleotide regions located at the intended loci in their genomes (data not shown). Thus, the primer sets could be used to selectively detect the gDNA of L. sanfranciscensis, L. brevis, or L. curvatus.

Species-specific PCR detection. Lactobacillus brevis (left), L. sanfranciscensis (center), and L. curvatus (right) were analyzed with agarose gel electrophoresis of PCR amplicons a and real-time PCR amplification. Lane M, DNA marker; lane 1, L. brevis 118; lane 2, L. brevis 111; lane 3, L. sanfranciscensis 117; lane 4, L. sanfranciscensis 142; lane 5, L. curvatus 104; lane 6, L. curvatus 114; lane 7, L. sakei; lane 8, Pediococcus pentosaceus 224; lane 9, L. buchneri; lane 10, L. plantarum; [bp], nucleotide base pair
Assessment of Primer Specificity Using qPCR
Primer specificity is especially important when qPCR is associated with SYBR Green, as this intercalating dye is nonspecific and detects all double-stranded DNA fragments. The specificity of each primer set was assessed with qPCR amplification of gDNAs from 12 different bacterial species, mainly covering bacteria that are commonly found in sourdough. Table 3 show that the Ct values ranged from 10.62 to 13.53 under optimal conditions with the amount of DNA present. Non-target DNA showed Ct values over 27, which was different from the value reported for the negative control (blank). In addition, single peaks were observed on the amplicon melting curves for each primer set of L. sanfranciscensis, L. brevis, and L. curvatus at their melting temperatures (Tm) of 84.5, 87, and 78 °C, respectively (data not shown). These observations indicate the specificity of the primer sets against their target DNAs during the amplification reaction of real-time PCR.
Validation of qPCR Assays
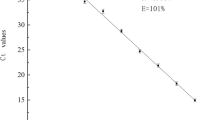
Standard curves for the quantification of L. sanfranciscensis, L. brevis, and L. curvatus were obtained from 10-fold serial dilutions of gDNAs of L. sanfranciscensis 101, L. brevis 111, and L. curvatus 114 isolated from nuruk (Fig. 4). A good linear correlation was obtained between the Ct values and number of genome copies for all standard curves, and the corresponding R2 values ranged from 0.995 to 0.999. These correlation coefficients demonstrated the reliability of the qPCR assays over a range of 5 log units. The slope of the standard curve of the gDNA for L. curvatus was −3.27, which was close to that of a PCR with an efficiency of approximately 100% (−3.30) (Chandelier et al. 2006). In contrast, the slopes of the gDNAs for L. sanfranciscensis and L. brevis were less than −3.30, indicative of an efficiency < 100%. The intra- and inter-assay CV values (mean over standard deviation ×100) were measured and the intra-assay CV value was < 1.95% for L. sanfranciscensis primer set, < 1.76% for L. brevis primer set, and < 1.45% for L. curvatus primer set. The inter-assay CV values were slightly higher than the intra-assay CV values.

Standard curves generated from the threshold cycle (Ct) values plotted against the estimated logarithm of each target gene concentration (copies/μL) of Lactobacillus sanfranciscensis a, L. brevis b, and L. curvatus c. Ten-fold serial dilutions of the DNA extracted from L. sanfranciscensis 101, L. brevis 111, and L. curvatus 114 were performed
Sourdough has a highly complex matrix composed of various nutrients and matrix. There is a possibility that the nutrients and matrix may affect the gDNA extraction yield and even interfere with qPCR assay. Therefore, the recovery yield of gDNA purified from the MRS medium or sourdough was compared to determine the effect of sourdough matrix. For this, an equal number of LAB were mixed with the MRS medium or dough, and gDNAs were extracted. The qPCR analysis results showed that there is no significant difference (p < 0.05) in cell number obtained by qPCR regardless of the strain, revealing no interfering effect of the sourdough matrix on DNA recovery (Fig. 5).

DNA extraction yield from MRS medium and sourdough samples
To assess the applicability of qPCR for the direct quantification of L. sanfranciscensis, L. brevis, and L. curvatus in sourdough samples, the cell number of these strains obtained by qPCR was compared with that obtained with the traditional MRS plate counting method. Figure 6 shows that the cell number enumerated by the plate counting method tended to be lower than that observed with the qPCR assay (Fig. 6). In particular, the number of L. sanfranciscensis and L. brevis determined with qPCR increased by 50 and 65%, respectively, as compared with that evaluated with the plate counting method, while the number of L. curvatus was almost constant. The limit of qPCR for cell enumeration is that it may result in false positives or high estimation of cell number, as all gDNAs extracted from cells could be amplified regardless of cell viability (Taylor et al. 2014). Therefore, while the increment is not much, the cell numbers obtained by qPCR method in this study may include both live and dead cells.

Comparison of quantification results using qPCR and plate cell counting
Quantification of Three LAB in Sourdough Samples
The qPCR method developed herein using species-specific primer sets was applied to monitor changes in microbial population during back-slopping of sourdough. L. sanfranciscensis, L. brevis, and L. curvatus were inoculated in the dough at different cell densities for sourdough fermentation. For back-slopping, a part of the dough was used as the inoculum for the second fermentation on the following day and so on. During this microbial propagation process of sourdough, any changes in the microbial population were measured with qPCR. As shown in Fig. 7, the number of L. curvatus continued to decline during the back-slopping process, while that of L. brevis was constant. In contrast, L. sanfranciscensis population increased and it became the most dominant LAB strain after the seventh cycle. This result reveals the applicability of the qPCR method developed to monitor the population sizes of L. sanfranciscensis, L. brevis, and L. curvatus during their growth in sourdough.

Persistence of Lactobacillus sanfranciscensis, L. brevis, and L. curvatus starters in sourdough during 7 days of long propagation. Symbols are population sizes of L. sanfranciscensis 101 ( ), L. brevis 111 (
), L. brevis 111 (  ), and L. curvatus 114 (
), and L. curvatus 114 ( ). Quantification was performed with the qPCR method
). Quantification was performed with the qPCR method
Discussion
Microbiota greatly contributes to the differences in the characteristics of similarly matured products during processing. These dynamics are largely influenced by the interactions between microorganisms (e.g., microbial co-operation and antagonism) and have a marked impact on the survival, growth, and activity of different microbial populations during processing. It is, therefore, important to reliably quantify physiologically active populations in terms of dynamic changes to understand the role of each microbial population (Achilleos and Berthier 2013). The sourdough has complex microbial composition and the interactions between microorganisms are key to their performance in food habitats (Liu et al. 2018; Sieuwerts et al. 2018). For understanding microbiota of sourdough, studies used molecular fingerprinting techniques: random amplification of polymorphic DNA (RAPD) (Vogelmann and Hertel 2011; Rizzello et al. 2011), rep-PCR (Liu et al. 2016), multiplex PCR (Zhang et al. 2019b), PCR- denaturing gradient gel electrophoresis (PCR-DGGE) (De Vuyst and Vancanneyt 2007; Settanni et al. 2006), and amplified fragment length polymorphism (AFLP) (Fusco et al. 2016). Recently, metagenomics allows high-resolution genomic analysis of unculturable microbes and correlation of the genomes with particular functions in the environment (Menezes et al. 2020). Those methods are easy to identify which microbes exist in sourdough (Meroth et al. 2003), but the accurate quantification of individual species is still challenging (Gao and Li 2018).
To measure the population of microorganism in sourdough, qPCR has been employed using universal primers targeting 16S rRNA-encoding gene (Pontonio et al. 2017). Generally, strains that show > 98% similarity in the 16 S rRNA gene sequence are regarded as the same species (Stackebrandt and Ebers 2006). Therefore, comparison of 16S rRNA gene sequence has limitation in discriminating species level such as L. acidophilus group (including L. acidophilus, L. helveticus, L. gallinarum, L. cripsatus) (You and Kim 2020) and L. casei group (including L. casei, L. paracasei, L. rhamnosus) (Kim et al. 2020b). To overcome this restriction, unique genes of the species other than 16S rRNA gene can be searched and used as target genes for qPCR; glutamate decarboxylase gene for L. reuteri in sourdough (Su et al. 2011), phenyalanine-tRNA synthetase gene for L. helveticus in cheese, and sometimes a hypothetical protein gene for L. lactis in kimchi (Kim et al. 2020a). In this study, we confirmed that genes for nucleoside hydrolase, hypothetical protein, and glyoxalase are unique for L. sanfranciscensis, L. brevis, and L. curvatus, respectively, and those genes can be used as targets for accurate quantification of the species during sourdough fermentation by using qPCR method. This method would enable quantification of three Lactobacillus species during cocultivation in sourdough and provide useful information on microbial commensalism that is essential to produce high-quality sourdough.
Conclusions
We developed a species-specific qPCR method for L. sanfranciscensis, L. brevis, and L. curvatus to monitor their growth during sourdough fermentation. The genes encoding nucleoside hydrolase, hypothetical protein, and glyoxalase were selected as targets for the species-specific detection. The specificity of PCR primer sets was verified against different species of LAB and different ratios of target and non-target DNAs. The qPCR methods developed estimated slightly higher number (< 65%) of LAB cell as compared with the traditional plate counting method, probably owing to the detection of both live and dead cells. Considering DNA recovery, no significant interfering effect of sourdough matrix was observed. The standard curves for qPCR of three species were established and successfully applied to monitor their growth during sourdough propagation. Thus, tools that rapidly quantify LAB populations during sourdough fermentation are warranted to facilitate optimization of the entire fermentation process for reproducible organoleptic qualities of products.
References
Achilleos C, Berthier F (2013) Quantitative PCR for the specific quantification of Lactococcus lactis and Lactobacillus paracasei and its interest for Lactococcus lactis in cheese samples. Food Microbiol 36(2):286–295
Bintsis T (2018) Lactic acid bacteria as starter cultures: an update in their metabolism and genetics. AIMS Microbiol 4(4):665–684
Chandelier A, Ivors K, Garbelotto M, Zini J, Laurent F, Cavelier M (2006) Validation of a real-time PCR method for the detection of Phytophthora ramorum. EPPO Bull 36:409–414
Choi H, Kim YW, Hwang I, Kim J, Yoon S (2012) Evaluation of Leuconostoc citreum HO12 and Weissella koreensis HO20 isolated from kimchi as a starter culture for whole wheat sourdough. Food Chem 134:2208–2216
De Vuyst L, Van Kerrebroeck S, Harth H, Huys G, Daniel HM, Weckx S (2014) Microbial ecology of sourdough fermentations: diverse or uniform? Food Microbiol 37:1–29
De Vuyst L, Vancanneyt M (2007) Biodiversity and identification of sourdough lactic acid bacteria. Food Microbiol 24(2):120–127
Dertli E, Mercan E, Arıcı M, Yılmaz MT, Sağdıç O (2016) Characterisation of lactic acid bacteria from Turkish sourdough and determination of their exopolysaccharide (EPS) production characteristics. LWT-Food Sci Technol 71:116–124
Dhanasekaran S, Doherty TM, Kenneth J (2010) Comparison of different standards for real-time PCR-based absolute quantification. J Immunol Methods 354:34–39
Fusco V, Quero GM, Chieffi D, Franz CM (2016) Identification of Lactobacillus brevis using a species-specific AFLP-derived marker. Int J Food Microbiol 232:90–94
Gaglio R, Alfonzo A, Barbera M, Franciosi E, Francesca N, Moschetti G, Settanni L (2020) Persistence of a mixed lactic acid bacterial starter culture during lysine fortification of sourdough breads by addition of pistachio powder. Food Microbiol 86:103349
Gänzle M, Ripari V (2016) Composition and function of sourdough microbiota: From ecological theory to bread quality. Int J Food Microbiol 239:19–25
Gao Y, Li H (2018) Quantifying and comparing bacterial growth dynamics in multiple metagenomic samples. Nat Methods 15(12):1041–1044
Hansen A, Schieberle P (2005) Generation of aroma compounds during sourdough fermentation: applied and fundamental aspects. Trends Food Sci Technol 16:85–94
He S, Hong X, Huang T, Zhang W, Zhou Y, Wu L, Yan X (2017) Rapid quantification of live/dead lactic acid bacteria in probiotic products using high-sensitivity flow cytometry. Methods Appl Fluoresc 5(2):024002
Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, Knight R, Knights D (2018) Evaluating the information content of shallow shotgun metagenomics. mSystem 3(6):e00069–e00018
Kim SA, Bae JH, Seong H, Han NS (2020a) Development of Leuconostoc lactis–specific quantitative PCR and its application for identification and enumeration in fermented foods. Food Anal Methods 1–8
Kim DH, Chon JW, Kim H, Kim HS, Choi D, Hwang DG, Seo KH (2015) Detection and enumeration of lactic acid bacteria, acetic acid bacteria and yeast in kefir grain and milk using quantitative real-time PCR. J Food Saf 35(1):102–107
Kim E, Yang SM, Cho EJ, Kim HY (2020b) Novel real-time PCR assay for Lactobacillus casei group species using comparative genomics. Food Microbiol 103485
Lee H, Baek H, Lim SB, Hur JS, Shim S, Shin SY, Han NS, Seo J (2015) Development of species-specific PCR primers and polyphasic characterization of Lactobacillus sanfranciscensis isolated from Korean sourdough. Int J Food Microbiol 200:80–86
Leroy F, De Vuyst L (2004) Lactic acid bacteria as functional starter cultures for the food fermentation industry. Trends Food Sci Technol 15:67–78
Lin XB, Gänzle MG (2014) Quantitative high-resolution melting PCR analysis for monitoring of fermentation microbiota in sourdough. Int J Food Microbiol 186:42–48
Liu T, Li Y, Chen J, Sadiq FA, Zhang G, Li Y, He G (2016) Prevalence and diversity of lactic acid bacteria in Chinese traditional sourdough revealed by culture dependent and pyrosequencing approaches. LWT-Food Sci Technol 68:91–97
Liu X, Zhou M, Jiaxin C, Luo Y, Ye F, Jiao S, Lü X (2018) Bacterial diversity in traditional sourdough from different regions in China. LWT-Food Sci Technol 96:251–259
Menezes LAA, Sardaro MS, Duarte RTD, Mazzon RR, Neviani E, Gatti M, Lindner JDD (2020) Sourdough bacterial dynamics revealed by metagenomic analysis in Brazil. Food Microbiol 85:103302
Meroth CB, Walter J, Hertel C, Brandt MJ, Hammes WP (2003) Monitoring the bacterial population dynamics in sourdough fermentation processes by using PCR-denaturing gradient gel electrophoresis. Appl Environ Microbiol 69(1):475–482
Moser A, Berthoud H, Eugster E, Meile L, Irmler S (2017) Detection and enumeration of Lactobacillus helveticus in dairy products. Int Dairy J 68:52–59
Özcan E, Seven M, Şirin B, Çakır T, Nikerel E, Teusink B, Toksoy Öner E (2020) Dynamic co-culture metabolic models reveal the fermentation dynamics, metabolic capacities and interplays of cheese starter cultures. Biotechnol Bioeng
Park DM, Bae JH, Kim M, Kim H, Kang S, Shim S, Lee D, Seo JH, Han NS (2019) Suitability of Lactobacillus plantarum SPC-SNU 72-2 as a probiotic starter for sourdough fermentation. J Microbiol Biotechnol 29(11):1729–1738
Pétel C, Onno B, Prost C (2017) Sourdough volatile compounds and their contribution to bread: a review. Trends Food Sci Technol 59:105–123
Pontonio E, Di Cagno R, Mahony J, Lanera A, De Angelis M, van Sinderen D, Gobbetti M (2017) Sourdough authentication: quantitative PCR to detect the lactic acid bacterial microbiota in breads. Sci Rep 7(1):1–13
Rizzello CG, Cassone A, Coda R, Gobbetti M (2011) Antifungal activity of sourdough fermented wheat germ used as an ingredient for bread making. Food Chem 127(3):952–959
Settanni L, Valmorri S, van Sinderen D, Suzzi G, Paparella A, Corsetti A (2006) Combination of multiplex PCR and PCR-denaturing gradient gel electrophoresis for monitoring common sourdough-associated Lactobacillus species. Appl Environ Microbiol 72(5):3793–3796
Sieuwerts S, Bron PA, Smid EJ (2018) Mutually stimulating interactions between lactic acid bacteria and Saccharomyces cerevisiae in sourdough fermentation. LWT-Food Sci Technol 90:201–206
Stackebrandt E, Ebers J (2006) Taxonomic parameters revisited: tarnished gold standards. Microbiol Today 33:152–155
Su MS, Schlicht S, Gänzle MG (2011) Contribution of glutamate decarboxylase in Lactobacillus reuteri to acid resistance and persistence in sourdough fermentation. Microb Cell Factories 10(S1):S8
Taylor MJ, Bentham RH, Ross KE (2014) Limitations of using propidium monoazide with qPCR to discriminate between live and dead Legionella in biofilm samples. Microbiol Insights 7:15–24
Venturi M, Guerrini S, Granchi L, Vincenzini M (2012) Typing of Lactobacillus sanfranciscensis isolates from traditional sourdoughs by combining conventional and multiplex RAPD-PCR profiles. Int J Food Microbiol 156:122–126
Vogelmann SA, Hertel C (2011) Impact of ecological factors on the stability of microbial associations in sourdough fermentation. Food Microbiol 28(3):583–589
Yazar G, Tavman Ş (2012) Functional and technological aspects of sourdough fermentation with Lactobacillus sanfranciscensis. Food Eng Rev 4(3):171–190
You I, Kim EB (2020) Genome-based species-specific primers for rapid identification of six species of Lactobacillus acidophilus group using multiplex PCR. PLoS One 15(3):e0230550
Zhang G, Tu J, Sadiq FA, Zhang W, Wang W (2019a) Prevalence, genetic diversity, and technological functions of the Lactobacillus sanfranciscensis in sourdough: a review. Compr Rev Food Sci Food Saf 18(4):1209–1226
Zhang B, Yang Z, Huang W, Omedi JO, Wang F, Zou Q, Zheng J (2019b) Isoflavone aglycones enrichment in soybean sourdough bread fermented by lactic acid bacteria strains isolated from traditional Qu starters: Effects on in vitro gastrointestinal digestion, nutritional, and baking properties. Cereal Chem 96(1):129–141
Acknowledgments
The present work was supported by the SPC Group and Traditional Culture Convergence Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Plannig (NRF-2017M3C1B5019292).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests
Shin Dal Kang, Sangmin Shim, Deukbuhm Lee, and Jin-Ho Seo are employed at the Research Institute of Food and Biotechnology, SPC Group, that sponsored this research.
Ethical approval
This article does not contain any studies with human participants or animals.
Informed consent
Not applicable
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Baek, HW., Kim, SA., Min, WK. et al. A Species-Specific qPCR Method for Enumeration of Lactobacillus sanfranciscensis, Lactobacillus brevis, and Lactobacillus curvatus During Cocultivation in Sourdough. Food Anal. Methods 14, 750–760 (2021). https://doi.org/10.1007/s12161-020-01920-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-020-01920-2