
Abstract
In the present work, magnetic graphene oxide–based solid-phase extraction combined with dispersive liquid–liquid microextraction has been developed for the extraction and preconcentration of some widely used pesticide residues in juice and pulp of fruits before their determination with gas chromatography–flame ionization detection. In this method, an appropriate weight of a synthetic magnetic graphene oxide is added into fruit pulp and homogenized for a specified time. Then, the magnetic graphene oxide containing the adsorbed pesticides is separated from pulp using a magnet and the pesticides are desorbed using an appropriate volume of a water-miscible organic solvent. In the following, the eluent is mixed with a low water-soluble extraction solvent (at a microliter level) and rapidly dispersed into the fruit juice. The optimum conditions were found to be 150 mg of magnetic graphene oxide, 4.0 min for extraction time, 1.0 mL of acetonitrile as the eluent, 4.0 min for adsorption time, and 105 μL of chloroform as an extraction solvent. An excellent linearity was achieved in the range of 3.5–10,000 μg kg−1 (r2 ≥ 0.997). The limits of detection and quantification ranged from 1.0–6.0 μg kg−1 and 3.5–20 μg kg−1, respectively. A good repeatability was obtained with the relative standard deviation ≤ 9%. Furthermore, extraction recoveries and enrichment factors were in the range of 69–81% and 340–399, respectively.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Fruits and vegetables form the major component of food consumed by humans because of their high nutritional value and are therefore not surprising to know that many nutritional experts and other health professionals recommend the daily intake of them (Fernandes et al. 2012). However, at the same time, they can be a source of toxic substances such as pesticides (Knežević and Serdar 2009). Although the use of pesticides in the agricultural applications provides a wide range of beneficial effects, their misuse can result in unacceptable levels of their residues in the products such as fruits (Timofeeva et al. 2017). Thus, the determination of pesticide residues in fruits are essential for the sake of human health. Many techniques such as gas chromatography (GC) (Fernandes et al. 2018; Lu et al. 2012; Valles et al. 2012), high performance liquid chromatography (HPLC) (Ma et al. 2018; Romero-González et al. 2008; Wang et al. 2018b), and capillary electrophoresis (Bakar et al. 2012; Juan-García et al. 2010) were applied for the determination of pesticides at trace levels in real samples. Although the above-mentioned methods are sensitive instrumental techniques of analysis, another step known as sample preparation is still required. A perfect sample preparation method should be able to extract the analytes from the sample matrix and transfer them into a suitable phase for injection into instrumental system. Solid-phase extraction (SPE) (Moreno et al. 2018; Zhao et al. 2018) is one of the most common sample preparation methods. Up to now, different types of sorbents such as C18, polystyrene–divinylbenzene-based polymers, etc. have been utilized in SPE (Augusto et al. 2013). In recent years, magnetic sorbents such as graphene oxide are widely used as the adsorbent in SPE to overcome the difficulties associated with non-magnetic sorbents and the procedure is termed as magnetic solid-phase extraction (MSPE) (Safariková and Safarik 1999). Unlike traditional SPE, in MSPE, there is no need to pack the sorbent into a cartridge. MSPE overcomes phase separation difficulty through the application of an external magnetic field and is easier and faster than traditional SPE since time-consuming operations such as centrifugation or filtration used in traditional SPE are not used in MSPE (Xie et al. 2019; Li et al. 2018; Ghazaghi et al. 2017).
In recent years, for minimization of organic solvent consumption, microextraction techniques such as solid-phase microextraction (Dominguez et al. 2018; Fernandes et al. 2018; Salemi et al. 2018) and liquid phase microextraction (LPME) (Hashemi et al. 2017; Zohrabi et al. 2016; Wu et al. 2015; Zhang et al. 2008; Afshar Mogaddam et al. 2019) were developed. Compared with traditional methods, the above-mentioned microextraction techniques are more attractive due to their powerful clean-up ability, high enrichment capability, and low consumption of harmful organic solvents. One version of LPME is dispersive liquid–liquid microextraction (DLLME) that has been developed by Rezaee et al. in 2006 (Rezaee et al. 2006). It has a wide range of applications in trace or ultra-trace analysis of pesticide residues and inorganic analytes due to utilize the low volumes of solvents (Ahmad et al. 2015; Cunha et al. 2009; Boonchiangma et al. 2012; Anthemidis and Ioannou 2009; Wang et al. 2018a; Xu et al. 2016). DLLME not only isolates or pre-concentrates the analytes from the samples but also simultaneously reduces or even eliminates the interferences originally present in the sample, and facilitates their determinations at low levels. In addition, it also has the advantages of less solvent consumption, simple operation, and high enrichment factor. Therefore, it is regarded as an effective approach to pretreatment various analytes (Ding et al. 2018; Fan et al. 2014; Ho et al. 2013; Rezaee et al. 2013; Gao and Ma 2011; Amirkhizi et al. 2018; Li et al. 2017; Li et al. 2011; Wen et al. 2011; Ma et al. 2012). In most publications, when the samples are solid, such as fruits, their juices are extracted and the extraction procedure is carried out on the juices. However, a portion of the analytes may remain in the fruit pulps and discarded, resulting in a negative error in the determination of analytes in the samples.
In the present work, for the first time, a combination of MGO-based SPE and DLLME is used as an efficient sample pretreatment method for the extraction of some widely used pesticides (diazinon, chlorpyrifos, phosalone, and ametryn) from fruits including apple, cucumber, pear, and peach samples before their quantification by gas chromatography–flame ionization detection (GC–FID). These pesticides are selected since several previous studies in Iran claimed their presence in the fruits and their by-products (Pirsaheb et al. 2013; Farajzadeh et al. 2016; Pirsaheb et al. 2015). In the proposed method, a synthesized MGO is added into the fruit pulp and homogenized for a certain time. Then, MGO containing adsorbed pesticides are separated from pulp in the presence of an external magnetic field and the pesticides are desorbed using an appropriate volume of a water-miscible organic solvent which is used as a dispersive solvent in the following DLLME method. The main advantage of this method is that, despite most of the previous studies, the refuse of the fruit samples is not discarded and the proposed method is performed on both the juice and the pulp of the fruit which prevents a negative error in the determination of analytes in the samples. In addition, this method offers high enrichment factors (EFs) that can guarantee the determination of the target pesticides at trace levels in fruit samples.
Materials and Methods
Reagents and Solutions
Diazinon, chlorpyrifos, ametryn, and phosalone were purchased from Dr. Ehrenstorfer (Agsburg, Germany). Graphite powder (< 20 μm) was purchased from Sigma–Aldrich (St. Louis, USA). Reagents including FeCl2·4H2O, FeCl3·6H2O, KMnO4, BaCl2, NaNO3, NaCl, H2SO4, H2O2, NH4OH (30%, w/w), and HCl (37%, w/w) were purchased from Merck (Darmstadt, Germany). Acetone, methanol, dimethyl sulfoxide (DMSO), acetonitrile (ACN), chloroform, carbon tetrachloride, 1,2-dichloroethane (1,2-DCE), and 1,1,2-trichloroethane (1,1,2-TCE) were also purchased from Merck. Deionized water was obtained from Ghazi Company (Tabriz, Iran). A cube (5 cm × 2 cm × 2 cm) neodymium magnet (1 T) was purchased from ENES Magnesy Pawel Zientek (Warszawa, Poland). A stock solution of the studied pesticides was prepared by dissolving appropriate amounts of the analytes in ACN at a concentration of 1000 mg L−1 of each pesticide. A mixture standard solution of the analytes (1000 mg L−1 of each pesticide) in chloroform (extraction solvent) was prepared and directly injected into the separation system each day (three times) in order to evaluate the instrumental system quality and to calculate EFs and extraction recoveries (ERs) of the analytes.
Samples
Sixteen samples of fresh fruits including apple, cucumber, pear, and peach samples (four samples of each) were collected from a local supermarket (Tabriz, Iran). Seven grams of each fruit was homogenized in a food processor. Then, all of the homogenized fruit was placed into a mortar and spiked with each pesticide at a concentration of 200 μg kg−1 and gently blended for 4.0 min using a pestle. The obtained mixture was transferred into a beaker and stored in a refrigerator at a temperature of 4.0 °C for 48 h. In the following, the homogenous samples were squeezed by a manual fruit juice extractor and centrifuged at a rate of 5000 rpm for 3.0 min and its pulp and juice were separated. The obtained fruit juice was diluted to 10 mL with deionized water before analysis. Also, other samples (pesticide-free samples) of the apple, cucumber, pear, and peach were supplied from small farms (Tabriz, East Azerbaijan Province, Iran) in which no pesticide was used. They were used in the matrix effect study. The pesticide-free sample of apple was used as a blank in the optimization step and method validation.
Apparatus
Analysis of the selected analytes was carried out using an Agilent 7890A gas chromatograph (Agilent Technologies, CA, USA) equipped with a split/splitless inlet operated at 300 °C in a splitless/split mode (sampling time 1.0 min and split ratio of 1:10) and an FID. Nitrogen (99.999%, Gulf Cryo, United Arabic Emirates) was used as the carrier gas (at a constant flow of 1.0 mL min−1) and makeup gas (25 mL min−1). Chromatographic separation was achieved on an HP–5 capillary column (30 m × 0.32 mm i.d. with a 0.25-μm stationary film thickness) (Hewlett–Packard, Santa Clara, USA). The column oven temperature was initially held at 60 °C for 2.0 min, then raised to 300 °C at a rate of 15 °C min−1, and held at 300 °C for 4.0 min. The Chem Station software was used for data acquisition and processing. A 1.0-μL microsyringe (zero dead volume, Hamilton, Switzerland) was used for the injection of the samples into GC. Injection volume was 1.0 μL. The FID temperature was maintained at 300 °C. Hydrogen gas was generated with a hydrogen generator (GLAIND–2200, Dani, Italy) for FID at a flow rate of 40 mL min−1. Air flow rate for FID was 40 mL min−1. The magnetic property of the prepared MGO was characterized by Lake Shore 7304 vibrating sample magnetometer (VSM) (Lakeshore, USA).
Synthesis of MGO
Graphene oxides (GO) were synthesized by Hummer’s method (Hummers Jr and Offeman 1958) with a little modification. Briefly, the equal mass of graphite powder and NaNO3 (2.0 g, each substance) were added into 90 mL concentrated H2SO4 under continuous stirring in an ice batch. After 30 min, 5.0 g of KMnO4 was slowly added into the mentioned suspension at room temperature (25 °C). The reaction was continued for another 8.0 h. At last, residual \( {\mathrm{MnO}}_4^{-} \) ions were removed by adding 10 mL H2O2 (30%, w/w, analytical reagent). Then, final product was washed using HCl solution (1.0 mol L−1) until the \( {\mathrm{SO}}_4^{2-} \) ions could not be detectable by Ba2+ ions. The obtained solid was further washed with H2O for several times and dried at room temperature under vacuum condition. The MGO was obtained by chemical co-precipitation method. Typically, 0.5 g of the produced GO were dispersed into 500 mL water under vigorously stirring. Then, 4.6 g FeCl3·6H2O and 0.85 g FeCl2·4H2O were slowly added to the GO suspension at room temperature under N2 stream. After that, NH4OH (30%, w/w) was quickly added to precipitate Fe2+/Fe3+ ions for generating the magnetite particles (Fe3O4) at pH = 10.0. After remaining the mixture for 60 min reaction at 70 °C, the obtained MGO was separated in the presence of a magnet and washed three times with deionized water and ethanol, respectively, and finally dried at 80 °C under vacuum.
The room-temperature magnetization hysteresis curve is measured by VSM to study the magnetic properties of the prepared MGO. The obtained S-like magnetization hysteresis loop indicates that the prepared MGO is a superparamagnetic and the maximum saturation magnetization of MGO is 25.17 emu g−1, which was adequate to fulfill the separation of MGO from sample solutions using an external magnet.
Two-Step Enrichment Procedure of MGO-Based SPE Combined with DLLME
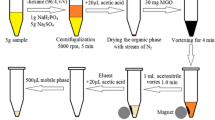
The pulp obtained from 7.0 g of blank apple or sample (see “Samples”) was placed into a mortar (50 mL capacity) and 150 mg of the sorbent (MGO) was added and gently blended for 4.0 min using a pestle. By this action, the analytes were adsorbed on the surface of MGO particles. Afterward, the mixture of pulp and sorbent was left to dry on a watch glass which placed in an autoclave (at 60 °C for 5 min). Then an external magnet was placed at the bottom of the watch glass and the analyte-loaded sorbent particles were separated due to the magnetic property of the MGO. In the next step, the pulp was easily discarded by inverting the watch glass in the presence of magnet and the isolated MGO particles were taken into a 15-mL glass test tube. Subsequently, 1.0 mL ACN as an elution solvent, was used to elute the analytes from the sorbent under sonication for 4.0 min. The obtained supernatant was removed and mixed with 105 μL chloroform (as an extraction solvent in the following DLLME). The obtained mixture was rapidly injected into the diluted juice (see “Samples”) placed into a 15-mL conical test tube using a 5.0-mL glass syringe. A cloudy solution resulted from dispersion of the tiny droplets of chloroform into the juice. The mixture was then centrifuged for 5 min at 5000 rpm, which led to the settling down of the dispersed droplets of the extractant at the bottom of the tube. After centrifuging, 10 ± 0.5 μL of the settled organic phase was obtained. Finally, an aliquot (1.0 μL) of the settled organic phase was removed and injected into the separation system for quantitative analysis. The proposed microextraction procedure is schematically shown in Fig. 1.

Microextraction and preconcentration procedure
Calculation of EF and ER
EF and ER are used to evaluate the extraction efficiency in different experimental conditions and they were calculated using the following equations:
where Csed, C0, Vsed, and M are the concentration of the analyte in the sedimented phase, the initial concentration of the analyte in the fruit sample, the volume of the sedimented phase (μL), and the weight of the fruit sample (g), respectively. nsed and n0 are the extracted and initial amounts of the analyte, respectively.
Results
Optimization of MSPE–DLLME Conditions
To improve the extraction efficiency and achieve the highest possible sensitivity, various parameters affecting the extraction process including weight of the sorbent, extraction time, type and volume of the elution solvent, desorption time, type and volume of the extraction solvent, and ionic strength were investigated and optimized. The experiments were conducted by variation of one parameter at a time, while the remaining parameters were kept constant.
Optimization of MSPE Conditions
Optimization of Sorbent Weight
The weight of sorbent directly affects the extraction efficiency of the analytes in the proposed method. In order to obtain the optimal weight of the sorbent, different weights of MGO varying in the range of 50–200 mg, were examined. The results (Fig. 2) show that 150 mg of the sorbent is enough for the quantitative sorption of the analytes. Therefore, 150 mg of the sorbent was selected as the most desirable weight of the sorbent for the extraction of analytes in the further experiments.

Selection of MGO weight. Conditions: sample, 7.0 g fruit spiked with the analytes at a concentration of 200 μg kg−1; extraction time, 5.0 min; elution solvent, ACN (1.0 mL); desorption time, 5.0 min; extraction solvent, 105 μL chloroform; ionic strength, 10%, w/v, NaCl; and centrifuge rate and time, 5000 rpm and 5.0 min, respectively. The error bars indicate the minimum and maximum of three determinations
Effect of Extraction Time
The time needed for quantitative adsorption of the analytes on the sorbent surface to reach the adsorption equilibrium is an effective parameter in the adsorption process, which depends on the physical and chemical properties of the sorbent. In order to obtain the best extraction time, five different extraction periods (1.0, 2.0, 3.0, 4.0, and 5.0 min) were tested under the gentle blending of MGO with pulp in a mortar using a pestle. Based on the obtained results, the highest analytical signals were achieved approximately after 4.0 min for analytes. Therefore, 4.0 min was chosen as the optimum extraction time for the following experiments.
Selection of Elution Solvent Type and Volume
The selection of a suitable solvent for desorbing the analytes from the sorbent surface is an important step in the proposed method that can affect the extraction efficiency. Therefore, several organic solvents including methanol, DMSO, acetone, and ACN were examined. The results in Fig. 3a demonstrate that the highest peak areas are obtained using ACN. Hence, ACN was chosen as a desorption solvent for the further experiments.

(a) Effect of elution solvent. Conditions: the same as those used in Fig. 2, except 150 mg MGO and 4.0 min were used as the sorbent weight and extraction time, respectively. (b) Effect of elution solvent volume. Conditions: The same as those used in Fig. 3a, except ACN was used as the elution solvent
To evaluate the effect of ACN volume, different volumes of ACN (0.5, 1.0, 1.5, and 2.0 mL) were studied. The obtained results (Fig. 3b) show that, the highest extraction efficiency is achievable when 1.0 mL ACN is used as the volume of the elution solvent. It was concluded that, at volumes less than 1.0 mL, ACN could not elute the analytes from the sorbent effectively and also the cloudy state did not form well in DLLME step, so the extraction efficiency was low. Also, at volumes higher than 1.0 mL, the extraction efficiency decreased, because of increasing solubility of the analytes in the aqueous phase in DLLME step. Therefore, l.0 mL was selected as the volume of the desorption solvent in the further experiments.
Desorption Time
Moreover, desorption time as the time needed for quantitative desorption of the analytes is another factor that affects the recovery of the analytes. The effect of desorption time was studied in the range of 1.0–5.0 min under sonication. The obtained results showed that the maximum efficiency was obtained at 4.0 min, and after that, it remained almost constant. Therefore, 4.0 min was selected as the optimum desorption time in the further experiments.
Reusability of the MGO
Reusability is an important factor when evaluating the efficiency of sorbents. In order to examine the reusability of the sorbent, after each use, the MGO was washed twice with ACN (each time by 2 mL along with vortexing for 3 min). The obtained results indicated that the MGO could be reused up to 7 successive extraction processes without a significant change in the extraction recovery of the analytes which indicated that MGO is a good sorbent for use with excellent reusability.
Optimization of DLLME Conditions
Selection of Extraction Solvent Type
Selection of an appropriate extraction solvent in DLLME should be based on the extraction capability of the solvent for the target analytes. It should also be water immiscible with density considerably different from that of water. Based on the above considerations, chloroform, carbon tetrachloride, 1,2-DCE, and 1,1,2-TCE were examined. To keep the sedimented extraction solvent volume constant (10 ± 0.5 μL), different volumes of the extraction solvents were accordingly used as follows: 70 μL of carbon tetrachloride, 65 μL of 1,1,2-TCE, 80 μL of 1,2-DCE, and 105 μL of chloroform. According to the results in Fig. 4, the highest extraction efficiency is achieved using chloroform. Thus, chloroform was chosen for the subsequent experiments.

Effect of extraction solvent. Conditions: the same as those used in Fig. 3b, except 1 mL was used as ACN volume
Optimization of Extraction Solvent Volume
The volume of the extraction solvent can directly affect the volume of the sedimented phase, extraction efficiency, and EF. For this purpose, different volumes of chloroform in the range of 85 to 145 μL with intervals of 20 μL were studied for DLLME performance. Based on the obtained results (data not shown), by increasing the volume of chloroform up to 105 μL, the analytical signals were increased and then declined with a further volume of the extraction solvent. In fact, the EF is decreased at high volumes of the extraction solvent, due to increasing the sedimented phase volume. Therefore, 105 μL of chloroform was chosen as the extraction solvent volume for more experiments.
Ionic Strength
One of the most exploited parameters during the optimization of a microextraction procedure is ionic strength due to the so-called salting-out effect. On the other hand, as the ionic strength of a solution increases along with the favorable salting-out effect, a second phenomenon can occur (increasing in the viscosity of the solution) which often hinders the process. Therefore, ionic strength should be optimized. For this purpose, different concentrations of NaCl in the range of 0–20%, w/v, with intervals of 5.0% were evaluated. The results showed that the analytical signals of the target analytes initially increased as the salt concentration increased up to 10%, w/v, and then gradually decreased with increasing NaCl concentration. Therefore, 10%, w/v, NaCl was added to the aqueous samples for the further studies.
Analytical Performance of the Proposed Method
After optimization of the whole procedure, quantitative parameters such as linearity, limits of detection (LOD) and quantification (LOQ), EF, ER, and repeatability were evaluated in the blank sample of apple in order to validate the proposed approach. All results are summarized in Table 1. The LODs and LOQs for the target analytes, calculated as the signal to noise ratios of 3.0 and 10 were from 1.0 to 6.0 and 3.5 to 20 μg kg−1, respectively. The ERs (at a concentration of 200 μg kg−1 of each analyte) were ranged from 69 to 81%. The EFs ranged between 340 and 399 depending on the analyte. The precision of the method, estimated from the relative standard deviation (RSD%), was determined by analyzing six replicates of the fruit sample containing the target analytes at three concentrations (25, 50, and 200 μg kg−1) in the same day (intra-day) and five consecutive days (inter-day). The RSD% varied from 2–8 to 4–9% for intra- and inter-day precisions, respectively.
Comparison of the Proposed Method with Current Methods
The present method was compared with other methods reported in the literature (Qu et al. 2010; Frıas et al. 2003; Ibrahim et al. 2010; Ravelo-Pérez et al. 2008; Wang et al. 2011) in terms of LOD, LOQ, intra-day RSD%, and EF for the determination of the analytes. The results are summarized in Table 2. The obtained LODs (1.0–6.0 μg kg−1) and LOQs (3.5–20 μg kg−1) in the presented method are comparable with other mentioned methods while in the mentioned methods a highly sensitive detection system such as mass spectrometry, nitrogen phosphorus detection, and electron capture detection which are inherently more sensitive than FID, have been used. The repeatability of the developed method is satisfactory and the obtained RSDs (4–8%) are comparable or better than the others. Also, this method has wider LRs (3.5–10,000 μg kg−1) compared to the others (0.01–1680 μg kg−1). In addition, the extraction time of the proposed method (20 min) is shorter than others, except SPME–GC–MS method. Moreover, the proposed procedure is characterized by low consumption of the organic solvent. It is environmentally friendly and produces low waste, except SPME and single-drop microextraction-based methods.
Application to Real Samples
To illustrate the applicability of the proposed method, it was applied to determine the selected analytes in sixteen samples of four fruit samples including apple, cucumber, peach, and pear (four samples of each). According to the obtained results, all of the evaluated samples were free of the target analytes. Figure 5 shows the typical GC–FID chromatograms for standard solution of the analytes (direct injection), pesticide-free apple, cucumber, peach, and pear samples spiked with the analytes (25 μg kg−1 of each analyte), and unspiked apple, cucumber, peach, and pear (one sample of each) in which the developed method was performed prior to chromatographic separation. To evaluate the anti-interference ability of the developed method (matrix effect study) in the apple, cucumber, peach, and pear samples, the added–found method was used. For this purpose, the apple, cucumber, peach, and pear samples were spiked with the analytes at three concentration levels (25, 100, and 200 μg kg−1 of each analyte) and the proposed method was applied on them for triplicates. The results obtained for the analytes in the samples in comparison with those obtained for their blank sample spiked at the same concentration were expressed as relative recoveries. According to the obtained results in Table 3, the mean relative recoveries of the studied pesticides in apple and cucumber samples ranged from 85 to 99 and 84 to 103%, respectively. In addition, mean relative recoveries of the analytes were obtained between 81–107 and 85–100% for peach and pear samples. In all cases RSD% values were less than 5%. These results indicate that the matrices of the samples have no significant effect on the proposed method according to SANCO guidelines which indicated that the mean relative recoveries in the range of 70 to 120% are acceptable for new methods (European Commission 2011). Therefore, the developed method could be used as an applicable and adequate method for the analysis of the selected pesticides in the studied samples.

Typical GC–FID chromatograms of (a) standard solution prepared in chloroform (100 mg L−1, each pesticide), (b) apple sample spiked with the selected analytes at a concentration of 25 μg kg−1, (c) apple sample, (d) cucumber sample spiked with the selected analytes at a concentration of 25 μg kg−1, (e) cucumber sample, (f) peach sample spiked with the selected analytes at a concentration of 25 μg kg−1, (g) peach sample, (h) pear sample spiked with the selected analytes at a concentration of 25 μg kg−1, and (i) pear sample after performing the proposed method, except chromatogram (a) in which direct injection was used. Peak identification: (1) diazinon; (2) ametryn; (3) chlorpyrifos; and (4) phosalone
Conclusions
In this study, a combination of MGO-based SPE and DLLME was introduced as an efficient method for the extraction, clean-up, and enrichment of some widely used pesticide residues in various fruit samples. The method was combined with GC–FID for quantitative analysis of the pesticides. To the best of our knowledge, this is the first report on the application of the present method for the extraction of the selected pesticides in fruit samples. The main advantage of the proposed method is that, despite most of the previous studies, the refuse of fruit samples is not discarded and the proposed method is performed on both juice and pulp of the fruit which prevents a negative error in the determination of the analytes in the samples. Also, the developed method not only preconcentrates the analytes, but also the other compounds present in the sample matrix are removed. Simplicity, short extraction time and being environmentally friendly as well as good analytical characters such as high EF, low LOD, and proper repeatability are other advantages of the proposed method. Also, successful applicability of the proposed method was confirmed via the simple chromatograms without interference peaks, indicating the superior clean-up and preconcentration of the proposed sample preparation method.
References
Afshar Mogaddam MR, Mohebbi A, Pazhohan A, Khodadadeian F, Farajzadeh MA (2019) Headspace mode of liquid phase microextraction: A review. Trends Anal Chem 110:8–14
Ahmad W, Al–Sibaai AA, Bashammakh AS, Alwael H, El–Shahawi MS (2015) Recent advances in dispersive liquid–liquid microextraction for pesticide analysis. Trends Anal Chem 72:181–192
Amirkhizi B, Nemati M, Arefhosseini SR, Hamedi Shahraki S (2018) Application of the ultrasonic–assisted extraction and dispersive liquid–liquid microextraction for the analysis of AFB1 in egg. Food Anal Methods 11:913–920
Anthemidis AN, Ioannou KIG (2009) Recent developments in homogeneous and dispersive liquid–liquid extraction for inorganic elements determination. Talanta 80:413–421
Augusto F, Hantao LW, Mogollón NGS, Braga SCGN (2013) New materials and trends in sorbents for solid–phase extraction. Trends Anal Chem 43:14–23
Bakar NBA, Makahleh A, Saad B (2012) In–vial liquid–liquid microextraction–capillary electrophoresis method for the determination of phenolic acids in vegetable oils. Anal Chim Acta 742:59–66
Boonchiangma S, Ngeontae W, Srijaranai S (2012) Determination of six pyrethroid insecticides in fruit juice samples using dispersive liquid–liquid microextraction combined with high performance liquid chromatography. Talanta 88:209–215
Cunha SC, Fernandes JO, Oliveira MBPP (2009) Fast analysis of multiple pesticide residues in apple juice using dispersive liquid–liquid microextraction and multidimensional gas chromatography–mass spectrometry. J Chromatogr A 1216:8835–8844
Ding M, Liu W, Peng J, Liu X, Tang Y (2018) Simultaneous determination of seven preservatives in food by dispersive liquid–liquid microextraction coupled with gas chromatography–mass spectrometry. Food Chem 269:187–192
Dominguez I, Arrebola FJ, Gavara R, Vidal JLM, Frenich AG (2018) Automated and simultaneous determination of priority substances and polychlorinated biphenyls in wastewater using headspace solid phase microextraction and high resolution mass spectrometry. Anal Chim Acta 1002:39–49
European Commission (2011) Method validation and quality control procedures for pesticide residues analysis in food and feed. Document No SANCO/12495/2011. Retrieved from http://ec.europa.eu/food/plant/protection/pesticides/docs/ qualcontrol_en.pdf. Accessed on 25 February 2018.
Fan Y, Hu S, Liu S (2014) Salting-out assisted liquid–liquid extraction coupled to dispersive liquid–liquid microextraction for the determination of chlorophenols in wine by high-performance liquid chromatography. J Sep Sci 37:3662–3668
Farajzadeh MA, Mohebbi A, Feriduni B (2016) Development of a simple and efficient pretreatment technique named pH–dependent continuous homogenous liquid–liquid extraction. Anal Methods 8:5676–5683
Fernandes VC, Domingues VF, de Freitas V, Delerue-Matos C, Mateus N (2012) Strawberries from integrated pest management and organic farming: Phenolic composition and antioxidant properties. Food Chem 134:1926–1931
Fernandes VC, Freitas M, Pacheco JG, Oliveira JM, Domingues VF, Delerue-Matos C (2018) Magnetic dispersive micro solid–phase extraction and gas chromatography determination of organophosphorus pesticides in strawberries. J Chromatogr A 1566:1–12
Frıas S, Rodrıguez M, Conde J, Pérez-Trujillo J (2003) Optimisation of a solid–phase microextraction procedure for the determination of triazines in water with gas chromatography–mass spectrometry detection. J Chromatogr A 1007:127–135
Gao Z, Ma X (2011) Speciation analysis of mercury in water samples using dispersive liquid–liquid microextraction combined with high–performance liquid chromatography. Anal Chim Acta 702:50–55
Ghazaghi M, Zavvar Mousavi H, Shirkhanloo H, Rashidi A (2017) Stirring–controlled solidified floating solid–liquid drop microextraction as a new solid phase–enhanced liquid–phase microextraction method by exploiting magnetic carbon nanotube–nickel hybrid. Anal Chim Acta 951:78–88
Hashemi B, Zohrabi P, Kim K-H, Shamsipur M, Deep A, Hong J (2017) Recent advances in liquid–phase microextraction techniques for the analysis of environmental pollutants. Trends Anal Chem 97:83–95
Ho YM, Tsoi YK, Leung KSY (2013) Ionic-liquid-based dispersive liquid–liquid microextraction for high-throughput multiple food contaminant screening. J Sep Sci 36:3791–3798
Hummers WS Jr, Offeman RE (1958) Preparation of graphitic oxide. J Am Chem Soc 80:1339–1339
Juan-García A, Font G, Juan C, Picó Y (2010) Pressurised liquid extraction and capillary electrophoresis–mass spectrometry for the analysis of pesticide residues in fruits from Valencian markets, Spain. Food Chem 120:1242–1249
Ibrahim WAW, Farhani H, Sanagi MM, Aboul-Enein H (2010) Solid phase microextraction using new sol–gel hybrid polydimethylsiloxane–2–hydroxymethyl–18–crown–6–coated fiber for determination of organophosphorous pesticides. J Chromatogr A 1217:4890–4897
Knežević Z, Serdar M (2009) Screening of fresh fruit and vegetables for pesticide residues on Croatian market. Food Control 20:419–422
Li X, Ma X, Huang R, Xie X, Guo L, Zhang M (2018) Synthesis of a molecularly imprinted polymer on mSiO2@Fe3O4 for the selective adsorption of atrazine. J Sep Sci 41:2837–2845
Li D, Ma X, Wang R, Yu Y (2017) Determination of trace bisphenol A in environmental water by high–performance liquid chromatography using magnetic reduced graphene oxide based solid–phase extraction coupled with dispersive liquid–liquid microextraction. Anal Bioanal Chem 409:1165–1172
Li J, Lu W, Ma J, Chen L (2011) Determination of mercury (II) in water samples using dispersive liquid–liquid microextraction and back extraction along with capillary zone electrophoresis. Microchim Acta 175:301–308
Lu D, Qiu X, Feng C, Lin Y, Xiong L, Wen Y, Wang D, Wang G (2012) Simultaneous determination of 45 pesticides in fruit and vegetable using an improved QuEChERS method and on–line gel permeation chromatography–gas chromatography/mass spectrometer. J Chromatogr B 895:17–24
Ma J, Wu G, Li S, Tan W, Wang X, Li J, Chen L (2018) Magnetic solid–phase extraction of heterocyclic pesticides in environmental water samples using metal–organic frameworks coupled to high performance liquid chromatography determination. J Chromatogr A 1553:57–66
Ma J, Lu W, Chen L (2012) Recent advances in dispersive liquid–liquid microextraction for organic compounds analysis in environmental water: a review. Curr Anal Chem 8:78–90
Moreno V, Llorent-Martínez EJ, Zougagh M, Ríos A (2018) Synthesis of hybrid magnetic carbon nanotubes–C18–modified nano SiO2 under supercritical carbon dioxide media and their analytical potential for solid–phase extraction of pesticides. J Supercrit Fluids 137:66–73
Pirsaheb M, Fattahi N, Shamsipur M (2013) Determination of organophosphorous pesticides in summer crops using ultrasound–assisted solvent extraction followed by dispersive liquid–liquid microextraction based on the solidification of floating organic drop. Food Control 34:378–385
Pirsaheb M, Fattahi N, Pourhaghighat S, Shamsipur M, Sharafi K (2015) Simultaneous determination of imidacloprid and diazinon in apple and pear samples using sonication and dispersive liquid–liquid microextraction. LWT Food Sci Technol 60:825–831
Qu LJ, Zhang H, Zhu JH, Yang GS, Aboul-Enein HY (2010) Rapid determination of organophosphorous pesticides in leeks by gas chromatography–triple quadrupole mass spectrometry. Food Chem 122:327–332
Ravelo-Pérez LM, Hernández-Borges J, Rodríguez-Delgado MÁ (2008) Multi–walled carbon nanotubes as efficient solid–phase extraction materials of organophosphorus pesticides from apple, grape, orange and pineapple fruit juices. J. Chromatogr A 1211:33–42
Rezaee M, Assadi Y, Hosseini MRM, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A 1116:1–9
Rezaee M, Mashayekhi HA, Saleh A, Abdollahzadeh Y, Naeeni MH, Fattahi N (2013) Determination of abamectin in citrus fruits using SPE combined with dispersive liquid–liquid microextraction and HPLC–UV detection. J Sep Sci 36:2629–2634
Romero-González R, Frenich AG, Vidal JM (2008) Multiresidue method for fast determination of pesticides in fruit juices by ultra performance liquid chromatography coupled to tandem mass spectrometry. Talanta 76:211–225
Safariková M, Safarik I (1999) Magnetic solid–phase extraction. J Magn Magn Mater 194:108–112
Salemi A, Khaleghifar N, Mirikaram N (2018) Optimization and comparison of membrane–protected micro–solid–phase extraction coupled with dispersive liquid–liquid microextraction for organochlorine pesticides using three different sorbents. Microchem J 144:215–220
Timofeeva I, Shishov A, Kanashina D, Dzema D, Bulatov A (2017) On–line in–syringe sugaring–out liquid–liquid extraction coupled with HPLC–MS/MS for the determination of pesticides in fruit and berry juices. Talanta 167:761–767
Valles NB, Retamal M, Mezcua M, Fernández-Alba A (2012) A sensitive and selective method for the determination of selected pesticides in fruit by gas chromatography/mass spectrometry with negative chemical ionization. J Chromatogr A 1264:110–116
Wang C, Qu G, Wang T, Deng F, Liang D (2018a) Removal of tetracycline antibiotics from wastewater by pulsed corona discharge plasma coupled with natural soil particles. Chem Eng J 346:159–170
Wang K, Xie X, Zhang Y, Huang Y, Zhou S, Zhang W, Lin Y, Fan H (2018b) Combination of microwave–assisted extraction and ultrasonic–assisted dispersive liquid–liquid microextraction for separation and enrichment of pyrethroids residues in Litchi fruit prior to HPLC determination. Food Chem 240:1233–1242
Wang Y, Wang Z, Zhang H, Shi Y, Ren R, Zhang H, Yu Y (2011) Application of pneumatic nebulization single–drop microextraction for the determination of organophosphorous pesticides by gas chromatography–mass spectrometry. J Sep Sci 34:1880–1885
Wen Y, Li J, Zhang W, Chen L (2011) Dispersive liquid–liquid microextraction coupled with capillary electrophoresis for simultaneous determination of sulfonamides with the aid of experimental design. Electrophoresis 32:2131–2138
Wu L, Hu M, Li Z, Song Y, Zhang H, Yu A, Ma Q, Wang Z (2015) Dynamic microwave–assisted extraction online coupled with single drop microextraction of organophosphorus pesticides in tea samples. J Chromatogr A 1407:42–51
Xie X, Ma X, Guo L, Fan Y, Zeng G, Zhang M, Li J (2019) Novel magnetic multi–templates molecularly imprinted polymer for selective and rapid removal and detection of alkylphenols in water. Chem Eng J 357:56–65
Xu S, Liu L, Wang Y, Zhou D, Kuang M, Fang D, Yang W, Wei S, Xiao A, Ma L (2016) Simultaneous determination of six synthetic phenolic antioxidants in edible oils using dispersive liquid–liquid microextraction followed by high-performance liquid chromatography with diode array detection. J Sep Sci 39:3205–3211
Zhang M, Huang J, Wei C, Yu B, Yang X, Chen X (2008) Mixed liquids for single–drop microextraction of organochlorine pesticides in vegetables. Talanta 74:599–604
Zhao P, Wang Z, Li K, Guo X, Zhao L (2018) Multi–residue enantiomeric analysis of 18 chiral pesticides in water, soil and river sediment using magnetic solid–phase extraction based on amino modified multiwalled carbon nanotubes and chiral liquid chromatography coupled with tandem mass spectrometry. J Chromatogr A 1568:8–21
Zohrabi P, Shamsipur M, Hashemi M, Hashemi B (2016) Liquid–phase microextraction of organophosphorus pesticides using supramolecular solvent as a carrier for ferrofluid. Talanta 160:340–346
Acknowledgments
The authors are grateful to Research Council of the University of Tabriz for financial support.
Funding
Behruz Feriduni has received a research grant from the University of Tabriz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Behruz Feriduni declares that he has no conflict of interest. Ali Mohebbi declares that he has no conflict of interest. Mir Ali Farajzadeh declares that he has no conflict of interest. Maryam Namvar declares that she has no conflict of interest.
Ethical Approval
This article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Feriduni, B., Mohebbi, A., Farajzadeh, M.A. et al. Magnetic graphene oxide–based solid-phase extraction combined with dispersive liquid–liquid microextraction for the simultaneous preconcentration of four typical pesticide residues in fruit juice and pulp. Food Anal. Methods 12, 2742–2752 (2019). https://doi.org/10.1007/s12161-019-01635-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-019-01635-z