
Abstract
Magnetic solid phase extraction (MSPE) is a valuable tool for the extraction and preconcentration of analytes from environmental, food, and biological samples because of its convenience, promptness, and simplicity. In this study, a graphene oxide-based magnetic nanocomposite (MGO) was synthesized and characterized by various instrumental methods. An effective MSPE procedure coupled with high-performance liquid chromatography (MSPE-HPLC) was developed for the preconcentration and determination of the patulin (PAT) in apple juice samples. Critical experimental parameters of MSPE that could affect the extraction efficiencies, such as the amount of MGO, the pH, and ionic strength of the sample solution; the extraction time; and the desorption conditions, were investigated. After optimizing the conditions, the detection limit of this method provided was 2.3 μg/kg; the recoveries were in the range of 68.7–83.6 %, with relative standard deviations (RSDs) ranging between 1.8–8.7 %. It was confirmed that the graphene-based magnetic nanocomposite was a kind of effective MSPE material used for the PAT analyses in apple juice samples.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mycotoxins are secondary metabolites produced by certain fungi on growing plants and are toxic to humans and animals. Their occurrence, toxicology, and impact on stakeholders have been a subject of continuous concern (Berthiller et al. 2013; Marin et al. 2013). Among commonly known mycotoxins, patulin (4-hydroxy-4H-furo[3,2-c]pyran-2[6H]-one) (PAT) is produced by Aspergillus, Penicillium, and Byssochlamys mold species that are commonly found on apples. Apple products made from partially damaged or bruised apples prior to processing are more susceptible to PAT contamination. Infants and young children who consume higher amounts of apple products than adults do are at higher risk of PAT intoxication (Beretta et al. 2000; Brandon et al. 2012).
The International Agency for Research on Cancer has classified PAT as Group 3 (not carcinogenic). The occurrence of PAT as toxic contaminant in apple products is prevalent and warrants regular monitoring of PAT in apple products. Regulatory levels of PAT were established in China, India, Japan, and USA. The maximum limit for PAT for apple juice is 50 μg/kg. In the Europe, additional limits for solid apple products (25 μg/kg) and products designed for infants and young children (10 μg/kg) have been applied (Commission Regulation 2006).
Analytical methods involving various sample pretreatment and instrumental detection techniques employed for PAT determination are mostly based on chromatography (Turner et al. 2009; Li et al. 2013). High-performance liquid chromatography (HPLC) coupled with ultraviolet detection (HPLC-UV) is extensively used, presumably due to the natural polarity of PAT and its strong absorption of UV light (van Egmond et al. 2007), in regulatory control (Marin et al. 2011; Pique et al. 2013) and risk assessment (Murillo-Arbizu et al. 2009) purpose. Recently, HPLC coupled with tandem mass spectrometry (HPLC-MS/MS) has gained increasing use (Vaclavikova et al. 2015; Zhang et al. 2014; Beltran et al. 2014). Additional techniques such as fluorescence competitive immunoassay (de Champdore et al. 2007), micellar electrokinetic capillary chromatography (Victor-Ortega et al. 2013), and gas chromatography–mass spectrometry (GC-MS) have also being developed and employed for determining PAT in apple juice (Rodriguez-Carrasco et al. 2012; Kharandi et al. 2013).
HPLC-UV determination of PAT in apple juice usually encountered typical matrix interferences such as 5-hydroxymethylfurfural and phenolic compounds (Mochizuki et al. 2009). The presence of extra interfering peaks renders the determination more difficult. Additional pretreatments of sample are warranted include liquid–liquid extraction (LLE) (Beltran et al. 2014), dispersive liquid–liquid microextraction (DLLME) (Mohammadi et al. 2013; Victor-Ortega et al. 2013), matrix solid-phase dispersion (MSPD) (Wu et al. 2008), solid-phase extraction (SPE) (Gokmen et al. 2005; Li et al. 2007), and liquid-liquid extraction tandem solid-phase extraction (LLE-SPE) (Valle-Algarra et al. 2009). The selection of appropriate adsorbent is an important factor to obtain good recovery in SPE. Typical use of C18 cartridges (Valle-Algarra et al. 2009), Oasis HLB cartridges (Desmarchelier et al. 2011), silica gel cartridges (Barreira et al. 2010), and inorganic salt adsorbent cartridges (for elimination of interferences) (Iha et al. 2009) have been reported for the determination of PAT.
Magnetic solid-phase extraction (MSPE) is a new technique for the sorption of analytes from a large volume of sample solution using magnetic or magnetizable adsorbents. By taking advantages of the excellent magnetic responsibility and high dispersibility of the adsorbents, MSPE has been demonstrated as a simple, rapid, reliable, and green sample pretreatment technique for a variety of inorganic and organic compounds from various matrices (Zhao et al. 2011; Wierucka and Biziuk 2014; Yan et al. 2014). Nanomaterials (NMs) possessing unique physical and chemical properties play a critical role in sample preparation (Zhang et al. 2013). Toward this end, we have explored various types of NMs for chemical analysis (Lee et al. 2010; Gonavelli et al. 2013; Wu et al. 2013). Graphene and its derivatives such as graphene oxide (GO), a two-dimensional few-layer carbon nanosheet with hexagonal packed lattice structure, serves as a potential building platform for chemical functionalization. GO possesses high surface-to-volume ratio, tunable surface functional groups (such as abundant hydrophilic groups including hydroxyl, epoxide, and carboxylic groups making it highly dispersed in aqueous solution; large amounts of benzene-ring substructure offering π–π interaction and cation–π interactions with analytes containing ring and unsaturated bonds), and free of leachable impurities, making it an attractive sorbent for SPE. However, its small size would cause high pressure and adsorbent loss during elution, making it less practical to be directly used as SPE adsorbent (Sitko et al. 2013). The introduction of magnetic property into GO, termed magnetic GO (MGO), simultaneously offers high sorption capacity, and convenient magnetic controllable separation could overcome this problem. MGO have been used for pretreatment of trace amounts of polycyclic aromatic hydrocarbons (Han et al. 2012) and chlorophenols (Pan et al. 2014) in environmental water as well as heavy metals in biological samples (Sun et al. 2015). However, to the best of our knowledge, there is no report on pretreatment of PAT in apple juice by MSPE. We therefore conducted this research to study the use of MSPE with MGO adsorbents followed by HPLC detection for the determination of PAT in apple juice.
Experimental
Chemicals and Materials
Patulin standard was purchased from Sigma-Aldrich (USA). Graphite flakes (99 %) were purchased from Alfa Aesar (USA). Sodium nitrate (98.5 %) was purchased from Showa chemicals (Japan), potassium permanganate (99 %) from BDH Ltd (USA), hydrogen peroxide (30 %) from Sigma Aldrich, and H2SO4 (98 %) from Fluka (USA). Iron (III) chloride hexahydrate (FeCl3·6H2O, 99 %) was purchased from Riedel-de Haën AG (USA). Iron (II) sulfate heptahydrate (FeSO4·7H2O, 99 %) was purchased from Sigma-Aldrich (USA). The C18-SPE cartridges (500 mg, 6 mL) were purchased from Applied Separations (USA). HPLC-grade acetonitrile and methanol were purchased from Tedia Co. (USA). Ethyl acetate was purchased from Avantor Performance Materials Inc. (USA). Deionized (DI) water was obtained from a Milli-Q waters system (USA). All the other reagents were of analytical grade and purchased from Jing Ming Chemical Factory (Taiwan).
Synthesis of GO
The GO was synthesized according to our previous work (Gonavelli et al. 2013) with slight modifications. A beaker (500 mL) equipped with a magnetic stirring bar was charged with H2SO4 (72 mL) and cooled at 0–5 °C. Graphite flakes (1.5 g) were added slowly with vigorous stirring, followed by adding slowly NaNO3 (1.5 g) and KMnO4 (4.5 g). The temperature of the reaction mixture was maintained below 10 °C during the adding process. The mixture was allowed to warm to room temperature (RT) and stirred for 1 h. The RT mixture was charged with DI water (120 mL) and stirred for 30 min while the temperature was increased to 95 °C. The mixture was poured into the DI water (300 mL), followed by slow addition of H2O2 (10 mL). The solution was filtered and the residual material was subjected to wash with 5 % HCl and DI water as well as repeated centrifugation. The resultant GO material was dried in a freeze-dryer overnight.
Synthesis of MGO
The GO (0.1 g) was exfoliated in DI water (10 mL) by sonication for 40 min. Iron (III) chloride hexahydrate (FeCl3·6H2O, 0.54 g) and iron (II) sulfate heptahydrate (FeSO4·7H2O, 0.28g) were dissolved in DI water (40 mL) and added slowly into the GO suspension. An ammonia solution (0.5 mL) was quickly added to the solution and stirred for 1 h at 60 °C. The MGO product was collected by an external magnetic field and washed with DI water three times, and then dried in a freeze-dryer overnight. The dry MGO was ground to powder using a mortar before use.
Characterization of MGO
The physical and chemical properties of MGO were characterized by transmission electron microscopy (TEM), Fourier transform infrared (FT-IR), and superconducting quantum interference device (SQUID). The morphology and size were investigated using a JEM-2100 TEM (JEOL, Japan) at an accelerating voltage of 200 kV. Infrared analyses were conducted with a Vertex 80v FTIR (Bruker, German) spectrometer where KBr was used to prepare the sample tablets. Magnetic properties were measured using an MPMS5 SQUID (Quantum Design, USA) at 300 K over a range of applied fields from −10,000 to 10,000 Oe.
Sample Treatment and Clean-Up
The apple juice samples were purchased from local retail markets and stored at 4 °C. A schematic of the MSPE procedure for PAT determination using MGO is shown in Fig. 1. The procedure consists of seven steps. (1) The apple juice sample (5 g) was placed into a centrifuge tube (15 mL) followed by addition of ethyl acetate-hexane (96:4, v/v) (5 mL), NaH2PO4 (1 g), and anhydrous Na2SO4 (5 g). (2) The tube was shaken in an orbital shaker for 10 min, followed by centrifugation at 5000 rpm for 5 min. The upper layer organic phase (3 mL) was transferred to a centrifuge tube followed by addition of acetic acid (20 μL). (3) The solvent was evaporated to near dryness under a gentle N2 stream at 45 °C. The residue was dissolved in acidic water solution (pH = 6, 2 mL) followed by addition of MGO (30 mg). (4) The mixture solution was vortexed for 4 min on a slow-moving platform shaker. (5) The solvent was pipetted out when the PAT-adsorbed MGO adsorbents were magnetically assembled and immobilized by an external magnet. An aliquot of acetonitrile (1.0 mL) was added and vortexed for 1 min for PAT desorption. (6) The PAT-containing eluent was pipetted into another centrifuge tube when the PAT-free MGO adsorbents were magnetically assembled and immobilized by an external magnet. Addition of acetic acid (20 μL) was conducted. (7) The acidic eluent was evaporated to near dryness under a gentle N2 stream at 45 °C. The residue was dissolved in HPLC mobile phase (500 μL) and ready for HPLC analysis.

Schematic of MSPE procedure for the determination of PAT using MGO
HPLC-UV Analysis
The samples were analyzed using a liquid chromatographic system consisted of a quaternary pump P4000, autosampler AS3000, detector UV 6000LP (Thermo Fisher Scientific, USA). Separation was carried out on a reverse phase Agilent (USA) ODS column (I.D. 4.6 mm × 250 mm, 5.0 μm). The flow rate of the mobile phase was 1.0 mL/min with an injection volume of 20 μL. Eluent A was acetonitrile and eluent B was water containing 0.5 % acetic acid. A gradient elution was performed by changing the mobile phase composition as follows: 0–2 min, 1 % A; 2–25 min, 1–10 % A; 25–26 min, 10–99 % A; 26–35 min, 99 % A; 35–36 min, 99–100 % A; 36–45 min, 100 % A. The detection was performed at 275 nm during the 20-min run.
Results and Discussion
The MSPE method used in this study differs from traditional SPE method in that the MGO adsorbents is well dispersed in the sample solution and rapidly contact the analytes by vortexing. The non-polar PAT is associated with MGO surface via π–π and cation–π interactions. The hydrophilic and more polar components are left in the sample solution. The PAT-adsorbed MGO adsorbents could be easily isolated from the sample solution using an external magnet. Subsequent elution using solvent of high analyte solubility could effectively desorb the weakly bound PAT from MGO. The combined effects facilitate the sorption probability and selectivity of PAT by MGO. As the MGO adsorbents involved in this study is different from the SPE in previous studies (Gokmen et al. 2005; Li et al. 2007; Wu et al. 2008), we characterized the as-prepared MGO first to ensure its physiochemical property, followed by an optimization study of sorption/desorption parameters to gain insight into the MSPE process as well as analytical performance, and finally, a comparison of the proposed MSPE method with reported methods was carried out for reference.
Characterization of MGO
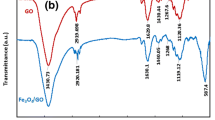
The morphology of the GO and MGO were characterized by transmission electron microscopy (TEM). The GO nanosheets exhibited irregular shapes and contained some wrinkles, which maintained a large surface area (Fig. 2a). The magnetic nanoparticles (NPs) uniformly distributed on GO nanosheets and were spherical in shape with a mean diameter of approximately 10–15 nm (Fig. 2b). A superconducting quantum interference device (SQUID) was used to investigate the magnetic properties of MGO, which exhibited good superparamagnetic property with a saturation magnetization value (Ms) ca.32 emu g−1 at 300 K and very low coercivity (Hc) ca. 7 Oe (Fig. 2c). The inset illustrated that MGO aggregated (within 1 min) onto the vial inner wall by an external magnetic field. The superparamagnetic behavior of as-prepared MGO was realized based on the S-like appearance of magnetization hysteresis curve and nearly zero magnetic remanence. Furthermore, the saturation magnetization value of 32 emu g−1 clearly indicated that the MGO can be quickly separated from the solution just by using an external magnet. The Fourier transform infrared (FT-IR) spectrum of GO (Fig. 2d upper curve) showed O–H stretching around 3350 cm−1, surface carboxylic groups C=O–O (1720 cm−1), aromatic C=C (1620 cm−1), carboxyl O=C–O (1360 cm−1), and alkoxy C–O (1050 cm−1). The FT-IR spectrum of MGO (Fig. 2d lower curve) revealed that aromatic alkene C=C (1570 cm−1), carboxyl O=C–O (1360 cm−1), and C–O (1200 cm−1) are the remaining functional groups, demonstrating that a number of oxygen functionalities were successfully reduced. The Fe–O bond (575 cm−1) and amine N–H broad band (3000–3300 cm−1 centered at 3200 cm−1) confirmed the magnetic functionalities of GO.

TEM images of a GO and b MGO. c Magnetization hysteresis curve of MGO at 300 K. d FT-IR spectra of GO and MGO
Optimization of adsorption and desorption conditions
The criteria for the optimization study are based on recovery from fortified samples by measuring the PAT amount eluted from the sorbent and comparing it to the fortified amount. The recovery value depends on both sorption efficiency and desorption efficiency, which reflects the retention and elution behavior, respectively. In order to gain insight into the MSPE process, we conducted the optimization study by either optimizing sorption while controlling desorption or vice versa. The experimental factors influencing sorption (PAT retention) include sorbent amount, sorption time, sample pH, and sample ionic strength; whereas eluent solvent and volume are the factors influencing desorption (PAT elution). Furthermore, the extent of reduced interference represented by elution profiles was also taken into consideration in the optimization study. The evaluation of optimal conditions is based on the triplicate analysis of matrix-matched blanks fortified with 50 μg/kg of PAT.
Effect of MGO Amount on Sorption Efficiency
The effect of MGO amount ranging from 5 to 50 mg on PAT sorption efficiency was investigated. Figure 3a shows that as the MGO amount increases, the PAT recovery gradually increases and levels off at 30 mg, indicating that 30-mg MGO is sufficient to sorb ~0.25-g PAT in apple juice. The PAT recovery shows minor decrease at 40 mg, presumably due to insufficient eluent volume (1 mL) for complete PAT elution when using excess amount of sorbent. Optimal recovery is obtained by using 30 mg of MGO amount.

a Effect of MGO amount on MSPE sorption of PAT (sorption time = 5 min, sample pH = 6, eluting solvent = acetonitrile, solvent volume = 1.0 mL). b Effect of sorption time on MSPE sorption of PAT (MGO amount = 30 mg, sample pH = 6, eluting solvent = acetonitrile, solvent volume = 1.0 mL). The error bars represent the standard deviation of three independent measurements.
Effect of Sorption Time on Sorption Efficiency
The effect of sorption time ranging from 1 to 10 min on PAT sorption efficiency was investigated. Figure 3b shows that as the sorption time increases, the PAT recovery gradually increases, levels off at 4 min, and remains constant thereafter. Optimal recovery is obtained by using 4 min of sorption time.
Effect of Sample pH and Ionic Strength on Sorption Efficiency
The sample pH is known to affect the stability of ionizable compounds. For example, the practice of keeping sample pH slightly acidic was used to avoid PAT degradation (Valle-Algarra et al. 2009). By adding appropriate volumes of 1.0 % HCl or 1.0 % ammonium hydroxide into the sample solution, the effect of sample pH ranging from 2 to 8 on PAT sorption efficiency was investigated. Figure 4a shows that as the pH increases, the PAT recovery slightly decreases until pH = 4, followed by apparent increase until pH = 7, and decreases again until pH = 8. The appearance of the data is presumably due to the difference in formation and solubility of PAT in samples with different pH. PAT is preferably soluble in the pH = 4 sample, rendering lowest sorption efficiency by MGO. The downward PAT recovery after sample pH = 7 is presumably due to PAT becoming unstable in alkaline condition. Moreover, the acidic environment generally tends to enhance sorption of acidic interfering species, which is observed in the elution profiles of samples with low pH. A compromise of optimal PAT recovery and reduced interference is obtained by using sample pH = 6.

a Effect of sample pH on MSPE sorption of PAT (MGO amount = 30 mg, sorption time = 5 min, eluting solvent = acetonitrile, solvent volume = 1.0 mL). b Effect of ionic strength on MSPE sorption of PAT (MGO amount = 30 mg, sorption time = 5 min, sample pH = 6, eluting solvent = acetonitrile, solvent volume = 1.0 mL). The error bars represent the standard deviation of three independent measurements.
By adding appropriate NaCl to adjust sample ionic strength, the effect of sample ionic strength ranging from 0 to 200 mM on PAT sorption efficiency was investigated. Figure 4b shows that as the ionic strength increases, the PAT recovery does not show apparent variation. The sample ionic strength is therefore not adjusted in the follow studies.
Effect of Eluting Solvent Strength and Volume of Desorption Efficiency
The strength and volume of the eluting solvent is known to influence the desorption efficiency in SPE. The effect of the eluting solvent strength (ethanol, ethyl acetate, methanol, ethyl acetate/acetonitrile 1:1, and acetonitrile) volume (0.5 to 3.0 mL) on PAT desorption efficiency was investigated.
Figure 5a shows that as the eluting solvent strength increases, the PAT recovery increases proportionally with the polarity index of the solvent (from ethanol to acetonitrile, 4.3, 4.4, 5.1, 5.6, and 5.8), demonstrating that acetonitrile is the most appropriate solvent.

a Effect of eluting solvent strength on MSPE desorption of PAT (MGO amount = 30 mg, sorption time = 5 min, sample pH = 6, eluting solvent volume = 1.0 mL). b Effect of eluting solvent volume on MSPE desorption of PAT (MGO amount = 30 mg, sorption time = 5 min, sample pH = 6, eluting solvent = acetonitrile).
The effect of the eluting solvent volume ranging from 0.5 to 3.0 mL on PAT desorption efficiency was investigated. Figure 5b shows that as the solvent volume increases, the PAT recovery rapidly increases and levels off at 1.0 mL, remains constant until 2.0 mL, and apparently decreases until 3.0 mL. A compromise of optimal PAT recovery and minimal eluting solvent volume is obtained by using 1.0 mL of eluting solvent volume.
Reusability of MGO
The regeneration and reusability potential of the MGO adsorbent was also investigated. The MGO could be reused for MSPE of PAT after eluting the adsorbed PAT. The MGO is stable and reused at least ten adsorption-elution cycles without significant loss of sorption capacity. The results indicate that the MGO adsorbent is stable and free of carryover of analytes during MSPE procedure, demonstrating good reusability.
Analytical Performance and Applications
After establishing the optical experimental conditions, a series of experiments were carried out to evaluate the interferences removal capability of the MSPE technique first. The evaluation was conducted by pretreating a pair of PAT-free (Fig. 6a, c, e) and PAT-fortified (0.1 mg/L, Fig. 6b, d, f) apple juice samples by MSPE as well as by popular LLE and C18 CPE. The presence of many peaks in the HPLC-UV chromatograms of PAT-free (Fig. 6a) and PAT-fortified (Fig. 6b) apple juice samples indicates the presence of a complex matrix effect. The matrix effect is significantly improved as shown by the chromatograms subjected to C18 SPE pretreatment (Fig. 6c, d). Further improvement in reducing matrix effect by MSPE pretreatment is apparent as smaller number of interfering peaks is observed (Fig. 6e, f). It can be concluded that well-separated and symmetric PAT peak could be obtained by using the optimized experimental conditions.

Typical HPLC-UV chromatograms of a PAT-free apple juice sample by LLE, b PAT-fortified (0.1 mg/L) apple juice sample by LLE; c PAT-free apple juice sample by C18 SPE, d PAT-fortified (0.1 mg/L) apple juice sample by C18 SPE; e PAT-free apple juice sample by MSPE, f PAT-fortified (0.1 mg/L) apple juice sample by MSPE
Several important parameters including linearity, correlation coefficient (R 2), limit of detection (LOD), and limit of quantification (LOQ) were detected to validate the method. Standard curve based on series standards from 40 to 2000 μg/L was constructed and used to determine the samples. The linear equation was (Y = 1421X + 7870), and the correlation coefficient (R 2) was 0.9998. Under the optimized conditions, the LOD and LOQ were calculated using a signal-to-noise ratio of 3 and 10, respectively. The LOD and LOQ of PAT for apple juice were 2.3 and 7.7 μg/kg, respectively. The recovery and repeatability were determined at three fortification levels: 50, 100, and 200 μg/kg. As listed in Table 1, the recoveries of PAT were between 68.7 and 76.9 % for clear apple juice and cloudy apple juice, and the RSDs were between 3.2 and 8.7 %.
The performance of the developed MSPE method was compared with other reported methods such as SPE, MSPD, and dispersive liquid–liquid microextraction (DLLME) from the viewpoint of adsorbent, linearity, LOD, recovery, and RSD. As listed in Table 2, The LOD of this method was slightly less sensitive than that of the HPLC and mass spectrometry reported in the previous literatures. The detection limit is below the current legal standard of the European Commission (EC). Hence, this developed method is practical for the detection of PAT in apple juice samples. In summary, the developed analytical procedure reported in their work was proved to be effective, convenient, and accurate for routine analyses.
References
Barreira MJ, Alvito PC, Almeida CMM (2010) Occurrence of patulin in apple-based-foods in Portugal. Food Chem 121:653–658
Beltran E, Ibanez M, Sancho JV, Hernandez F (2014) Determination of patulin in apple and derived products by UHPLC-MS/MS. Study of matrix effects with atmospheric pressure ionisation sources. Food Chem 142:400–407
Beretta B, Gaiaschi A, Galli CL, Restani P (2000) Patulin in apple-based foods: occurrence and safety evaluation. Food Addit Contam 17:399–406
Berthiller F, Crews C, Dall'Asta C, De Saeger S, Haesaert G, Karlovsky P, Oswald IP, Seefelder W, Speijers G, Stroka J (2013) Masked mycotoxins: a review. Mol Nutr Food Res 57:165–186
Brandon EFA, Baars AJ, Te Biesebeek JD, Oomen AG, Bakker MI, De Heer C (2012) Risk assessment of patulin intake from apple containing products by young children. World Mycotoxin J 5:391–403
Commission Regulation (2006) (EC) No. 1881/2006 of 19 December 2006. Official Journal of the European Union 364:5
de Champdore M, Bazzicalupo P, De Napoli L, Montesarchio D, Di Fabio G, Cocozza I, Parracino A, Rossi M, D'Auria S (2007) A new competitive fluorescence assay for the detection of patulin toxin. Anal Chem 79:751–757
Desmarchelier A, Mujahid C, Racault L, Perring L, Lancova K (2011) Analysis of patulin in pear- and apple-based foodstuffs by liquid chromatography electrospray ionization tandem mass spectrometry. J Agric Food Chem 59:7659–7665
Funes GJ, Resnik SL (2009) Determination of patulin in solid and semisolid apple and pear products marketed in Argentina. Food Control 20:277–280
Gokmen V, Acar J, Sarioglu K (2005) Liquid chromatographic method for the determination of patulin in apple juice using solid-phase extraction. Anal Chim Acta 543:64–69
Gonavelli G, Chang CC, Ling YC (2013) Facile synthesis of smart magnetic graphene for safe drinking water: heavy metal removal and disinfection control. Acs Sustain Chem Eng 1:462–472
Han Q, Wang Z, Xia J, Chen S, Zhang X, Ding M (2012) Facile and tunable fabrication of Fe3O4/graphene oxide nanocomposites and their application in the magnetic solid-phase extraction of polycyclic aromatic hydrocarbons from environmental water samples. Talanta 101:388–395
Iha MH, de Souza SVC, Sabino M (2009) Single-laboratory validation of a liquid chromatography method for the determination of patulin in apple juice. Food Control 20:569–574
Kharandi N, Babri M, Azad J (2013) A novel method for determination of patulin in apple juices by GC-MS. Food Chem 141:1619–1623
Lee PL, Chiu YK, Sun YC, Ling YC (2010) Synthesis of a hybrid material consisting of magnetic iron-oxide nanoparticles and carbon nanotubes as a gas adsorbent. Carbon 48:1397–1404
Li J, Wu R, Hu Q, Wang J (2007) Solid-phase extraction and HPLC determination of patulin in apple juice concentrate. Food Control 18:530–534
Li P, Zhang Z, Hu X, Zhang Q (2013) Advanced hyphenated chromatographic-mass spectrometry in mycotoxin determination: current status and prospects. Mass Spectrom Rev 32:420–452
Malysheva SV, Di Mavungu JD, Boonen J, De Spiegeleer B, Goryacheva IY, Vanhaecke L, De Saeger S (2012) Improved positive electrospray ionization of patulin by adduct formation: usefulness in liquid chromatography-tandem mass spectrometry multi-mycotoxin analysis. J Chromatogr A 1270:334–339
Marin S, Mateo EM, Sanchis V, Valle-Algarra FM, Ramos AJ, Jimenez M (2011) Patulin contamination in fruit derivatives, including baby food, from the Spanish market. Food Chem 124:563–568
Marin S, Ramos AJ, Cano-Sancho G, Sanchis V (2013) Mycotoxins: occurrence, toxicology, and exposure assessment. Food Chem Toxicol 60:218–237
Mochizuki N, Hoshino M, Suga K, Sugita-Konishi Y (2009) Identification of an interfering substrate in apple juice and improvement for determination of patulin with high-performance liquid chromatography analyses. J Food Prot 72:805–809
Mohammadi A, Tavakoli R, Kamankesh M, Rashedi H, Attaran A, Delavar M (2013) Enzyme-assisted extraction and ionic liquid-based dispersive liquid-liquid microextraction followed by high-performance liquid chromatography for determination of patulin in apple juice and method optimization using central composite design. Anal Chim Acta 804:104–110
Murillo-Arbizu M, Amezqueta S, Gonzalez-Penas E, de Cerain AL (2009) Occurrence of patulin and its dietary intake through apple juice consumption by the Spanish population. Food Chem 113:420–423
Pan S, Zhou L, Zhao Y, Chen X, Shen H, Cai M, Jin M (2014) Amine-functional magnetic polymer modified graphene oxide as magnetic solid-phase extraction materials combined with liquid chromatography-tandem mass spectrometry for chlorophenols analysis in environmental water. J Chromatogr A 1362:34–42
Pique E, Vargas-Murga L, Gomez-Catalan J, de Lapuente J, Llobet JM (2013) Occurrence of patulin in organic and conventional apple-based food marketed in Catalonia and exposure assessment. Food Chem Toxic 60:199–204
Rodriguez-Carrasco Y, Berrada H, Font G, Manes J (2012) Multi- mycotoxin analysis in wheat semolina using an acetonitrile-based extraction procedure and gas chromatography-tandem mass spectrometry. J Chromatogr A 1270:28–40
Sitko R, Zawisza B, Malicka E (2013) Graphene as a new sorbent in analytical chemistry. Trends Anal Chem 51:33–43
Sun J, Liang Q, Han Q, Zhang X, Ding M (2015) One-step synthesis of magnetic graphene oxide nanocomposite and its application in magnetic solid phase extraction of heavy metal ions from biological samples. Talanta 132:557–563
Turner NW, Subrahmanyam S, Piletsky SA (2009) Analytical methods for determination of mycotoxins: a review. Anal Chim Acta 632:168–180
Vaclavikova M, Dzuman Z, Lacina O, Fenclova M, Veprikova Z, Zachariasova M, Hajslova J (2015) Monitoring survey of patulin in a variety of fruit-based products using a sensitive UHPLC-MS/MS analytical procedure. Food Control 47:577–584
Valle-Algarra FM, Mateo EM, Gimeno-Adelantado JV, Mateo-Castro R, Jimenez M (2009) Optimization of clean-up procedure for patulin determination in apple juice and apple purees by liquid chromatography. Talanta 80:636–642
van Egmond HP, Schothorst RC, Jonker MA (2007) Regulations relating to mycotoxins in food. Anal Bioanal Chem 389:147–157
Victor-Ortega MD, Lara FJ, Garcia-Campana AM, del Olmo-Iruela M (2013) Evaluation of dispersive liquid-liquid microextraction for the determination of patulin in apple juices using micellar electrokinetic capillary chromatography. Food Control 31:353–358
Wierucka M, Biziuk M (2014) Application of magnetic nanoparticles for magnetic solid-phase extraction in preparing biological, environmental and food samples. Trends Anal Chem 59:50–58
Wu R, Dang Y, Niu L, Hu H (2008) Application of matrix solid-phase dispersion-HPLC method to determine patulin in apple and apple juice concentrate. J Food Compos Anal 21:582–586
Wu MC, Deokar AR, Liao JH, Shih PY, Ling YC (2013) Graphene-based photothermal agent for rapid and effective killing of bacteria. Acs Nano 7:1281–1290
Yan S, Qi T, Chen D, Li Z, Li X, Pan S (2014) Magnetic solid phase extraction based on magnetite/reduced graphene oxide nanoparticles for determination of trace isocarbophos residues in different matrices. J Chromatogr A 1347:30–38
Zhang B, Zheng X, Li H, Lin J (2013) Application of carbon-based nanomaterials in sample preparation: a review. Anal Chim Acta 784:1–17
Zhang K, Wong JW, Mai H, Trucksess MW (2014) Dopant-assisted atmospheric pressure photoionization of patulin in apple juice and apple-based food with liquid chromatography-tandem mass spectrometry. J Agric Food Chem 62:4112–4118
Zhao G, Song S, Wang C, Wu Q, Wang Z (2011) Determination of triazine herbicides in environmental water samples by high-performance liquid chromatography using graphene-coated magnetic nanoparticles as adsorbent. Anal Chim Acta 708:155–159
Acknowledgments
This study was funded by the Basic Science Research Foundation (grant number 2452015203) of Northwest A&F University and Science and technology basic work (no. 2013FY113400).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Yutang Wang declares that he has no conflict of interest. Yanjie Wen declares that she has no conflict of interest. Yong-Chien Ling declares that he has no conflict of interest.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Wang, Y., Wen, Y. & Ling, YC. Graphene Oxide-Based Magnetic Solid Phase Extraction Combined with High Performance Liquid Chromatography for Determination of Patulin in Apple Juice. Food Anal. Methods 10, 210–218 (2017). https://doi.org/10.1007/s12161-016-0570-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0570-y