
Abstract
An analytical method applying ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS) for the determination of aflatoxins M2, M1, G2, G1, B2, B1, deoxynivalenol, ochratoxin A, fumonisins B1 and B2, hydrolyzed fumonisins B1 and B2, zearalenone and sterigmatocystin in cereal-based porridge destined for infant consumption was developed and validated. The mycotoxins were extracted using an adequate solvent ratio (n-hexane/3% formic acid solution/acetonitrile) under rapid shaking and sonication, without any further cleanup steps. Recoveries ranged from 63.5 to 113.2% and were considered satisfactory, with relative standard deviations lower than 20%. The limits of quantification ranged from 0.14 to 6.73 μg kg−1. The validated method was then applied to the determination of mycotoxins in 84 samples of cereal-based porridge destined for infant consumption obtained in the metropolitan region of Rio de Janeiro, RJ. Aflatoxins B1 and G1, fumonisins B1 and B2, hydrolyzed fumonisins B1 and B2, deoxynivalenol, and zearalenone were found, respectively, in 7.1, 2.4, 47.6, 3.6, 65.5, and 60.7% of the analyzed samples. The maximum permissible limits set by the Brazilian legislation were exceeded for at least one mycotoxin in 21 (25%) of the analyzed samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Mycotoxins are toxic compounds naturally produced as secondary metabolites by many filamentous fungi. Found as food contaminants worldwide, mycotoxins have caused significant economic losses in agriculture, and pose a very serious public health issue (Chu 1991; Murphy et al. 2006; Zain 2011; Rocha et al. 2014). In addition to several deleterious effects on human and animal health, some mycotoxins are classified by the International Agency for Research on Cancer (IARC) as human carcinogens or as potentially carcinogenic to humans (IARC 1993; Peraica et al. 1999; IARC 2002).
Taking into account their toxicity and occurrence, aflatoxins B1, B2, G1, and G2; deoxynivalenol; fumonisins B1 and B2; ochratoxin A; and zearalenone have been considered the main mycotoxins found in cereals and cereal-based products (Lee and Ryu 2017). Other mycotoxins have also been found in these foods, such as sterigmatocystin (Mol et al. 2016) and hydrolyzed fumonisins (Dombrink-Kurtzman and Dvorak 1999). Aflatoxins M1 and M2 and aflatoxins B1 and B2 hydroxylated metabolites, respectively, have been frequently found in milk (Peraica et al. 1999; Prandini et al. 2009; Sartori et al. 2015a). However, aflatoxins M1 and M2 may also be produced by fungi in minor amounts (Bräse et al. 2009; Filazi and Sireli 2013) and have also been reported in food other than milk, including corn (Shotwell et al. 1976; Vesonder et al. 1991; Ren et al. 2007; Huang et al. 2010; Ezekiel et al. 2012; Sartori et al. 2015b).
The contamination of cereal-based food destined for infant consumption by mycotoxins has been reported in several countries (Lombaert et al. 2003; Araguás et al. 2005; Tam et al. 2006; Baydar et al. 2007; D’Arco et al. 2008; Gottschalk et al. 2009; Kabak 2009; Alvito et al. 2010; Kostelanská et al. 2010; Romagnoli et al. 2010; Beltran et al. 2011; Cano-Sancho et al. 2011; Mahnine et al. 2012; Ozden et al. 2012; Rubert et al. 2012b; Juan et al. 2014; Zhang et al. 2014). In Brazil, high rates of mycotoxin occurrence in various cereals have been reported, including cereals used for the preparation of products intended for infant consumption (Maziero and Bersot 2010). However, few studies have determined mycotoxins in these products in Brazil (Castro et al. 2004; Caldas and Silva 2007).
Compliance with legislations regarding mycotoxin control in food requires the use of reliable analytical methods. In this scenario, several analytical approaches have been developed for the determination of mycotoxins in food (Krska et al. 2008; Cigić and Prosen 2009; Turner et al. 2009; Köppen et al. 2010; Saeger 2011). In recent years, liquid chromatography-tandem mass spectrometry (LC-MS/MS) has been widely applied in the determination of mycotoxins in different matrices (Turner et al. 2015; Berthiller et al. 2016; Berthiller et al. 2017). In addition, the increasing applicability of certain high-resolution mass spectrometry detectors for the determination of mycotoxins in food is also noted (Zachariasova et al. 2010; Rubert et al. 2012a; Fang et al. 2013; Jia et al. 2014; Righetti et al. 2016).
The selectivity of these techniques has enabled the simultaneous analysis of different classes of mycotoxins in several food matrices with minimum sample treatment (Sulyok et al. 2007; Mol et al. 2008; Frenich et al. 2009; Lacina et al. 2012). However, with regard to the determination of mycotoxins in food destined for infant consumption, most studies have reported more sophisticated sample treatment procedures using cleanup steps (Kostelanská et al. 2010; Rubert et al. 2012b; Zhang et al. 2014).
In this context, the aim of the present study was to develop and validate an analytical method for the determination of mycotoxins in commercial cereal-based porridge food destined for infant consumption by ultra-high performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). Sample treatment involves a simultaneous extraction and cleanup (deffating) step, followed by concentration of the mycotoxin-containing extracts. The validated analytical method was then applied to the analysis of 84 samples of cereal-based porridge destined for infant consumption obtained from the metropolitan region of Rio de Janeiro, RJ.
Materials and Methods
Reagents and Chemicals
Acetonitrile and methanol (HPLC-grade) were purchased from J.T.Baker (Phillipsburg, NJ, USA). Ammonium formate (>99%) and formic acid (mass spectrometry grade) were purchased from Sigma-Aldrich (St. Louis, MO, USA). n-Hexane (purity >96%), ethyl acetate (for analysis), and potassium hydroxide (pellets for analysis) were obtained from Merck (Darmstadt, Germany). Ultrapure water was obtained from a Milli-Q Gradient water system (Millipore, Bedford, MA, USA).
Standard Solutions
Solid standards of the aflatoxins B1, B2, G1, G2, M1, M2; ochratoxin A; and sterigmatocystin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Stock solutions of ochratoxin A (40 μg mL−1) were prepared in toluene/acetic acid (99:1, v/v). Individual stock solutions of sterigmatocystin and of the aflatoxins were prepared in acetonitrile at 10 μg mL−1. The concentrations of these standard solutions were determined by UV spectrophotometry (Horwitz and Latimer 2005). At least each 12 months, the stability of these solutions was checked by UV spectrophotometry. Stock solutions of fumonisins B1 and B2 (50 μg mL−1) in acetonitrile/water (1:1, v/v) and deoxynivalenol in acetonitrile (100 μg mL−1) were purchased from Fluka/Sigma-Aldrich (St. Louis, MO, USA). Stock solutions of zearalenone in acetonitrile (100.7 μg mL−1) were purchased from Biopure (Tulln, Austria). The hydrolyzed fumonisins B1 and B2 were prepared in the laboratory by hydrolysis of fumonisins B1 and B2 following the procedure described by Dall'Asta et al. (2009). Thus, an aliquot (5 mL) of a standard solution containing fumonisins B1 and B2 (50 μg mL−1) in acetonitrile/water (1:1, v/v) was evaporated to dryness under a gentle nitrogen flow in a 40 °C water bath. The residues were dissolved in 5 mL of a 2 mol L−1 KOH solution and left to react for 12 h at room temperature. The hydrolyzed fumonisins were then extracted three times with 10 mL of ethyl acetate, combined, and evaporated to dryness under a gentle nitrogen flow in a 40 °C water bath, and the residues were subsequently dissolved in 5 mL of methanol. The absence of the native fumonisins in this solution was verified by UHPLC-MS/MS. Thus, total conversion to the hydrolyzed forms was assumed and the concentrations of hydrolyzed fumonisins B1 and B2 in methanol were calculated as 28.1 and 27.6 μg mL−1, respectively. Aliquots from stock solutions were combined, and the volume was adjusted with methanol/water (1:1, v/v) to obtain an intermediate standard solution (10 ng mL−1 for ochratoxin A and aflatoxins M1, M2, G2, G1, B2, and B1; 40 ng mL−1 for sterigmatocystin; 200 ng mL−1 for hydrolyzed fumonisins B1 and B2, deoxynivalenol, and zearalenone; and 400 ng mL−1 for fumonisins B1 and B2). These solutions were diluted with methanol/water (1:1, v/v) to prepare working solutions. Intermediate and working solutions were prepared weekly. All the standard solutions were stored at −18 °C.
UHPLC-MS/MS Analysis
Liquid chromatography was performed using an ACQUITY UPLC™ system (Waters). A BEH C18 column (100 mm × 2.1 mm i.d., 1.7 μm particle size) was used as the stationary phase. The column temperature was maintained at 35 °C. The mobile phase flow rate was set at 0.3 mL min−1. A 0.3% acid formic solution was used as the aqueous mobile phase of the elution gradient for the determination of fumonisins, hydrolyzed fumonisins, and sterigmatocystin. The elution gradient began with 60% methanol, was increased to 80% during 3 min, and was held at 80% for 1 min. The system was then re-equilibrated for 2 min with 60% methanol. The injection volume was of 5 μL. A 5 mmol L−1 formate ammonium solution was used as the aqueous mobile phase of the elution gradient used for the determination of aflatoxins M2, M1, B2, B1, G2, and G1; ochratoxin A; deoxynivalenol; and zearalenone. The elution gradient began with 10% methanol, was increased to 100% during 4 min, and was held at 100% for 1.5 min. The system was then re-equilibrated for 2 min with 10% methanol. The injection volume was of 7.5 μL.
Analyte detection was performed using a tandem quadrupole mass spectrometer (Waters, Quattro Premier™ XE) equipped with an electrospray ionization (ESI) source operated in both positive and negative ionization modes. The optimized source parameters were capillary voltage 3.5 kV, extractor voltage 3 V, rf lens 0.1 V, multiplier 750 V, desolvation temperature of 350 °C, and source temperature of 120 °C. Nitrogen was used as the cone and desolvation gas at flows of 50 and 750 L h−1, respectively. Argon was used as the collision gas at a pressure of 4 × 10−3 mbar. The two ion transitions selected (m/z) for each mycotoxin and the acquisition conditions are displayed in Table 1.
Sample Preparation
Each sample (3 g) was weighed in 50-mL centrifuge tubes, and 5 mL of n-hexane, 5 mL of a 3% formic acid solution, and 10 mL of acetonitrile were added. The tubes were then shaken for 2 min using a vortex (IKA Works) and subsequently sonicated for 10 min. After these steps, the tubes were centrifuged at 3000 rpm for 7 min (Hitachi HIMAC CF 7D2). A 5-mL aliquot of the extracts (acetonitrile/water) was then concentrated to dryness under a gentle nitrogen flow in a 50 °C water bath (Turbo-Vac LV). Finally, the residues were dissolved with 1 mL of methanol/water (1:1, v/v) and the solutions thus obtained were filtered through 0.22-μm PVDF membrane filters.
Method Validation
Single laboratory validation was performed by evaluating the following analytical performance parameters: selectivity, matrix effect, linearity, trueness, precision (repeatability and intermediate precision), limit of detection (LOD), and limit of quantification (LOQ).
The selectivity of the method was evaluated by analyzing matrix blank samples (corn-based, oat-based, rice-based, oat/rice-based, and multicereal-based) regarding the presence of interfering signals eluted at the same time as the analytes in all porridge samples. The mycotoxins were identified in the samples by comparing the analyte retention times to the standard solution retention times. Confirmation was performed by comparison of the signal intensity ratios of the quantifier and qualifier ion transitions of each analyte in the samples with those obtained using standard solutions considering the maximum permitted tolerances according to the European Union (EC 2002).
To investigate matrix effects, calibration curves for each compound in the matrix extract (matrix-matched calibration) and in methanol/water (1:1, v/v) were prepared at eight concentration levels, ranging from 0.25 to 5 ng mL−1 (aflatoxins M2, M1, G2, G1, B2, and B1, and ochratoxin A), 1 to 20 ng mL−1 (sterigmatocystin), 5 to 100 ng mL−1 (hydrolyzed fumonisins B1 and B2, deoxynivalenol, and zearalenone), and 10 to 200 ng mL−1 (fumonisins B1 and B2). The calibration curve slopes were compared by an analysis of covariance (ANCOVA), considering a significance level of 5% (García-Campaña et al. 1997). Before the ANCOVA analysis, the homogeneity of the residual variances of all the calibration curves was verified (p values >0.05) applying a modified Levene test (Brown and Forsythe 1974).
Linearity was assessed using calibration curves with the same concentration levels used when studying the matrix effect (Souza and Junqueira 2005). Outliers were successively investigated by the Jackknife standardized residuals test (Belsley et al. 1980). The homoscedasticity of the residuals for all the calibration curves was verified by a modified Levene test (Brown and Forsythe 1974). The independency of the residuals for all calibration curves was verified by a Durbin-Watson statistical test (Durbin and Watson 1951). The normality of the residuals for all the calibration curves was verified by Ryan-Joiner’s test (Ryan and Joiner 1976). The regression significance and the lack of fit were performed by an analysis of variance (ANOVA) (Draper and Smith 1998).
The trueness and repeatability of the method were evaluated by recovery studies using oat/rice-based porridge samples spiked with the mycotoxins at four concentration levels, with four replicates for each level. Intermediate precision was performed by the analysis of spiked samples with the same concentrations used in the second concentration level, analyzed within 3 days by three different analysts.
Porridge samples spiked with the compounds in the lowest concentration level used in the recovery studies were used to determine the limit of detection (LOD) and limit of quantification (LOQ), considering signal-to-noise ratios of 3 and 10, respectively.
Samples of Cereal-Based Porridge Destined for Infant Consumption
A total of 84 samples of commercial cereal-based porridge destined for infant consumption were purchased randomly from local supermarkets in the metropolitan region of Rio de Janeiro, RJ, Brazil, between 2012 and 2014, from 13 different companies, representing 37 different brands, classified as corn-based (n = 30), rice-based (n = 20), oat-based (n = 7), oat/rice-based (n = 10), wheat-based (n = 3), and multicereal-based (n = 14). The samples were stored in their original packaging at −20 °C until analysis.
Results and Discussion
Optimization of the UHPLC-MS/MS Conditions
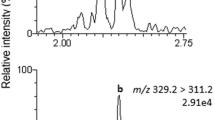
The mass spectrometry conditions were optimized by infusion of individual working standard solutions, between 500 and 1000 ng mL−1, using an ESI source operated in both positive and negative ionization modes. Initially, more intense precursor ions were selected and the cone voltage was optimized for each target compound with the mass spectrometer operating in the scan mode. Subsequently, collision energies were applied to obtain the product ions. Collision energies were optimized for each transition, and the two more intense product ions were selected as the quantification and confirmation transition ions. Ion transitions obtained in positive mode were selected for all mycotoxins, except for zearalenone. For this mycotoxin, the transitions obtained in positive ionization mode (m/z 319.3 > 301.2 and m/z 319.3 > 283.2) showed interfering peaks when the sample preparation method was applied to the cereal-based porridge samples. Thus, the ion transitions obtained in the negative mode were selected for zearalenone, providing satisfactory method sensitivity and selectivity (Fig. 1).

Chromatograms of a corn/oat-based porridge sample fortified with the zearalenone (10 μg kg−1) indicating a presence of matrix interferents (m/z 319.3 > 283.2) and b absence of matrix interferents (m/z 316.9 > 174.8)
The mobile phase composition was studied with the purpose of obtaining adequate chromatographic peak shapes and sensitivity for the mycotoxin analysis. The presence of carryover was evaluated by injecting a solution of target mycotoxins followed by solvent injections (methanol/water (1:1, v/v)).
Methanol was selected as the organic mobile phase because it provides higher sensitivity when compared to acetonitrile. Different compositions of the initial organic mobile phase (10, 25, 55, and 60%), with a linear gradient ending at 80 and 90% of the organic mobile phase, were evaluated. The efficiency of various additives in both mobile phases or only in the aqueous mobile phase (ammonium formate 5 mmol L−1, 0.1% of acid formic, 0.3% of acid formic, ammonium formate 5 mmol L−1/0.1% of acid formic, ammonium formate 5 mmol L−1/0.3% of acid formic), added to provide ionization and adequate peak shape for the analytes, was also studied.
A decrease in the concentration of the organic solvent in the initial mobile phase was found to increase sensitivity to the target compounds; adequate sensitivity was observed by using 10 and 25% of organic phase in the initial gradient; carryover was negligible (<1%) for ochratoxin A using these methods. For sterigmatocystin, a significant carryover (>3%) was observed. As reported previously (Plattner 1999; Tamura et al. 2012), the worst carryovers were observed for the fumonisins, mainly for fumonisin B2, since this compound appeared in several injection cycles after the injection of a standard solution or contaminated sample. In order to solve this problem, several solvents (methanol; 0.1% formic acid in methanol; 0.3% formic acid in methanol; 1% formic acid in methanol; methanol/water (1:1, v/v); 1% formic acid in water, acetonitrile, methanol (1:1:1:1, v/v/v)) were tested to be used as the injection syringe wash solvents. The tested washing solvents were not efficient in eliminating the carryover (fumonisins continued to appear in several injection cycles). This effect occurs due to the possible presence of reversible bonds of these substances with metal ions in the sample path of the chromatographic system, including the chromatographic columns (Tamura et al. 2014). Decrease of carryover was observed with increasing ratios of organic solvents in the initial mobile phase gradient, as previously reported (Tamura et al. 2011). In our study, absence of carryover was observed for fumonisins and sterigmatocystin using 60% of the organic solvent in the initial mobile phase gradient, 0.3% formic acid as the aqueous mobile phase, and a linear gradient ending in 80% of organic phase. As these conditions are inadequate for the determination of all the target compounds using one gradient, two methods were selected: the conditions described above were used for the determination of the fumonisins, hydrolyzed fumonisins, and sterigmatocystin, while 10% of organic solvent (methanol) in the initial gradient and a 5-mmol L−1 ammonium formate solution as the aqueous mobile phase were selected for aflatoxins M2, M1, B2, B1, G2, and G1; ochratoxin A; deoxynivalenol; and zearalenone.
After the selection of the mobile phase, different injection volumes (5, 7.5, and 10 μL) were evaluated with regard to chromatographic peak shape and method sensitivity. An injection volume of 5 μL was selected for fumonisins, hydrolyzed fumonisins, and sterigmatocystin, due to adequate sensitivity for all target substances. An injection volume of 7.5 μL was chosen for the determination of the other mycotoxins, since this is the highest volume that did not lead to chromatographic problems, mainly for deoxynivalenol.
Optimization of the Sample Treatment Method
Four methods frequently reported in the literature for the simultaneous determination of different classes of mycotoxins in foods were initially selected for evaluation (Sulyok et al. 2007; Mol et al. 2008; Tamura et al. 2011; Lacina et al. 2012). These methods were modified in order to reduce the analysis time and improve sensitivity. The experiments were performed in triplicate using oat/rice-based porridge samples fortified with target mycotoxins. Recoveries were determined using blank extracts fortified with mycotoxins at adequate concentrations, avoiding the influence of the matrix effect. The evaluated methods are described as follows. Method A, modified QuEChERS (quick, easy, cheap, effective, rugged, and safe) method (Tamura et al. 2011): 10 mL of water and 10 mL of acetonitrile were added to 3 g of the sample weighed in a 50-mL centrifuge tube. The mixture of salts (QuEChERS citrate) was then added, and the tube was immediately shaken for 1 min and centrifuged at 3000 rpm for 7 min. A 3-mL aliquot of the acetonitrile phase was evaporated to dryness under a gentle nitrogen flow at 45 °C. The residue was then dissolved with 1 mL of methanol/water (1:1, v/v). The solution thus obtained was filtered through a 0.22-μm filter before injection. Method B, modified QuEChERS method (Lacina et al. 2012): 10 mL of formic acid solution 2% and 10 mL of acetonitrile were added to 3 g of the sample weighed in a 50-mL centrifuge tube. A mixture of salts (4 g of magnesium sulfate and 1 g of sodium chloride) was then added, and the tube was immediately shaken for 1 min and centrifuged at 3000 rpm for 7 min. A 3-mL aliquot of the acetonitrile phase was evaporated to dryness under a gentle nitrogen flow at 45 °C. The residue was then dissolved with 1 mL of methanol/water (1:1, v/v). The solution thus obtained was filtered through a 0.22-μm filter before injection. Method C, solid-liquid extraction (Sulyok et al. 2007): 5 mL of formic acid solution 5% was added to 3 g of the sample weighed in a 50-mL centrifuge tube. Subsequently, 20 mL of acetonitrile was added and the tube was shaken for 2 min and sonicated for 10 min. The tube was then centrifuged at 3000 rpm for 7 min. A 5-mL aliquot of the extract was evaporated to dryness under a gentle nitrogen flow at 45 °C. The residue was then dissolved with 1 mL of methanol/water (1:1, v/v). The solution thus obtained was filtered through a 0.22-μm filter before injection. Sonication was used instead of shaking (60 min) used in the original methods. Method D, solid-liquid extraction (Mol et al. 2008): 5 mL of water was added to 3 g of the sample weighed in a 50-mL centrifuge tube. Subsequently, 15 mL of acetonitrile (1% formic acid) was added and the tube was shaken for 2 min and then sonicated for 10 min. The tube was then centrifuged at 3000 rpm for 7 min. A 5-mL aliquot of the extract was evaporated to dryness under a gentle nitrogen flow at 45 °C. The residue was then dissolved with 1 mL of methanol/water (1:1, v/v). The solution thus obtained was filtered through a 0.22-μm filter before injection. Sonication was used instead of shaking (60 min) used in the original methods.
The obtained results are displayed in Table 2. None of the evaluated methods obtained satisfactory results (70 to 120%) for all the evaluated mycotoxins, although satisfactory recoveries were obtained using method D for all mycotoxins except for hydrolyzed fumonisins B1 (49.9%) and B2 (29.8%). However, a better extraction efficiency of hydrolyzed fumonisins was observed in this method compared to method C (hydrolyzed fumonisins B1 (30.3%) and B2 (23.8%), demonstrating a possible relationship of extraction efficiency with the water/acetonitrile ratio used in these methods.
Taking into account these results, several experiments were conducted combining different extraction solvents and sonication times, in order to obtain an efficient method for the extraction of the target mycotoxins, including hydrolyzed fumonisins. Oat/rice-based porridge samples fortified with target mycotoxins were used in the experiments. A total of 13 combined experiments were performed in duplicate, described as follows: 5 mL of n-hexane was added (or not added) to 3 g of the sample weighed in a 50-mL centrifuge tube. Subsequently, 5 mL of water (or formic acid 3% solution) and 10 mL (or 15 mL) of acetonitrile were added. The tube was shaken for 2 min using a vortex and then sonicated for 10 min (or 30 min). The sample was then centrifuged at 3000 rpm for 7 min. A 5-mL aliquot of the acetonitrile/water phase was evaporated to dryness under a gentle nitrogen flow at 50 °C (Turbo-Vac LV). The residue was then dissolved with 1 mL of methanol/water (1:1, v/v). The solution thus obtained was filtered through a 0.22-μm filter. The conditions of each experiment and their results are displayed in Tables 3 and 4, respectively. The best results for target mycotoxins were obtained, including hydrolyzed fumonisins, using n-hexane (5 and 10 mL), a 3% formic acid solution, and 10 mL of acetonitrile in the extraction procedure. No significant difference was observed in the extraction efficiency for hydrolyzed fumonisins using 5 or 10 mL of n-hexane; since it uses less amount of solvent, the experiment condition using 5 mL of n-hexane was selected to investigate the influence of sonication time on the extraction of hydrolyzed fumonisins. However, the extraction efficiency of hydrolyzed fumonisins was not improved with increased extraction time (30 min). Thus, the procedure using 5 mL of n-hexane, a 3% formic acid solution, 10 mL of acetonitrile, and sonication for 10 min during the extraction step was selected for validation.
Method Validation
The developed analytical method was validated for the determination of 14 mycotoxins (aflatoxins M2, M1, G2, G1, B2, B1; deoxynivalenol; ochratoxin A; fumonisins B1 and B2; hydrolyzed fumonisins B1 and B2; zearalenone; and sterigmatocystin) in cereal-based porridge destined for infant consumption.
No interfering signals eluted at the same time as the analytes in all porridge samples (corn-based, oat-based, rice-based, oat/rice-based, and multicereal-based). Figure 2 displays a chromatogram of an oat/rice-based porridge sample fortified with target mycotoxins at the limit of quantification. The retention times and ion ratios, as well as the maximum permitted tolerances for ion ratios obtained for the target mycotoxins, are displayed in Table 1.

Chromatograms of an oat/rice-based porridge sample fortified with the target mycotoxins at the limit of quantification
The wide variety of porridge brands available in the market, as well as the absence of uncontaminated matrices for some mycotoxins, made it difficult to carry out matrix effect studies for all brands. Therefore, to evaluate the extension of the phenomena, matrix effects were studied using only the oat/rice-based porridge matrix.
Significant differences between the slopes of the calibration curves prepared in the solvent and in the matrix for several target mycotoxins were observed (p values <0.05), indicating that the matrix effect is significant for these compounds.
Sample dilutions (final extracts) and chromatographic gradient time effects were thus studied in order to reduce or eliminate matrix effects. The effect of sample dilution was assessed using four matrix proportions in the final extract (0.1, 0.25, 0.5, and 1 g mL−1), with the same mycotoxin concentrations in all extracts. The obtained results are displayed in Table 5. A decrease in the matrix effect for most mycotoxins was observed when increasing sample dilution. However, a significant matrix effect (>10%) for most mycotoxins was observed for all evaluated dilutions.
In an attempt to decrease the matrix effect, the gradient time was increased from 4 to 6 min. However, even though the matrix effect decreased up to 49% when increasing the gradient time for most of the assessed mycotoxins, a gradient time of 4 min was applied because of losses of chromatographic signal intensities (20 to 65%) and increases in the analysis time. Thus, matrix-matched calibration was used in routine analysis, while the standard addition method was used for quantifying mycotoxins when concentrations were higher than the maximum permissible concentration and when an uncontaminated matrix was not available to prepare matrix-matched calibrations (Mavungu et al. 2009; EC 2014).
In the linearity studies, the homoscedasticity, the independency of the residuals, and the normality of the residuals for all the calibration curves were confirmed (p values >0.05). A high regression significance (p values <0.001) and non-significant lack of fit (p values >0.05) were found, attesting to the linearity of the evaluated curves.
The trueness and repeatability results of the method are displayed in Table 6, where the repeatability and intermediate precision are expressed by the relative standard deviation (RSD %) and trueness by the recovery values. The recovery values ranged from 63.5 to 113.2%, with RSD lower than 20% for all studied mycotoxins under repeatability conditions. The RSD for the intermediate precision study was always lower than 12%. The results were satisfactory according the European Union criteria (EC 2002; EC 2006).
The values obtained for LOD and LOQ are also displayed in Table 6. The sensitivity of the method was considered suitable for the routine analysis of target mycotoxins in cereal-based porridge destined for infants, taking into account the maximum limit allowed for regulated mycotoxins (FAO 2004; Brasil 2011).
Sample Analyses
After validation, the developed method was used to determine target mycotoxins in samples of cereal-based porridge intended for infant consumption. The results are displayed in supplementary material (Online Resource 1).
The results were evaluated according to the normative resolution RDC No. 07/2011, of the current Brazilian legislation for food mycotoxin control (Brasil 2011). The permissible limits set by the Brazilian legislation were exceeded for at least one of the investigated mycotoxins in 21 (25%) of the samples.
Aflatoxin B1 was found in 6 (7.1%) of the analyzed samples at concentration levels between 0.07 and 2.06 μg kg−1. The incidence of aflatoxin B1 in the present study is low when compared to the 88, 50, and 25% values found by Baydar et al. (2007), Tam et al. (2006), and Alvito et al. (2010), respectively, when analyzing cereal-based foodstuffs for infant consumption.
Aflatoxin B1 exceeded the maximum limit allowed for the sum of aflatoxins B1, B2, G1, and G2 (1 μg kg−1) in cereal-based foodstuffs for infant consumption set by the Brazilian legislation in one sample (corn-based porridge). Aflatoxin G1 was detected in 2 (2.4%) of the samples.
Fumonisins B1 and B2 were both found in 40 (47.6%) of the analyzed samples. Fumonisins (B1 + B2) were found in concentration levels ranging 7 from 1500 μg kg−1, with an average of 283 μg kg−1; the concentration levels found in 15 (18%) of the analyzed samples exceeded the maximum permissible limit (200 μg kg−1) according to the Brazilian legislation.
These mycotoxins were found in all (100%) of the corn-based porridge samples. The high incidence and levels of fumonisins found in the corn-based porridge samples are in agreement with previous reports for corn-based foods destined for infant consumption in Brazil (Castro et al. 2004).
The hydrolyzed fumonisins B1 and B2 were found in 3 (3.6%) of the analyzed samples, indicating that fumonisins can be hydrolyzed during the industrial processing of these foods (Dombrink-Kurtzman and Dvorak 1999).
Deoxynivalenol was found in 55 (65.5%) of the analyzed samples at concentrations ranging from 2.0 to 332.5 μg kg−1, with an average of 68.1 μg kg−1. Concentrations exceeded the maximum limit (200 μg kg−1) set by the Brazilian legislation in 5 (6.0%) of the analyzed samples. The incidence of deoxynivalenol found herein was lower than that reported by Juan et al. (2014), of 76%, and higher than the 63, 40, 36, and 23.5% reported by Lombaert et al. (2003), Cano-Sancho et al. (2011), Romagnoli et al. (2010), and Zhang et al. (2014), respectively, in cereal-based foods for infant consumption.
Zearalenone was found in 51 (60.7%) of the analyzed samples at concentrations ranging between 0.64 and 60.8 μg kg−1, averaging 6.7 μg kg−1. The concentrations exceeded the maximum permissible levels allowed (20 μg kg−1) by the Brazilian legislation in four (4.8%) of the analyzed samples. The incidence found in this study was higher than the 33, 14, 6, and 2% reported, respectively, by Lombaert et al. (2003), Kostelanská et al. (2010), Rubert et al. (2012b), Romagnoli et al. (2010).
Figure 3 displays chromatograms of cereal-based porridge samples naturally contaminated by mycotoxins.

Chromatograms of cereal-based porridge samples naturally contaminated by mycotoxins
Conclusions
An UHPLC-MS/MS method for the determination of 14 mycotoxins in cereal-based porridge destined for infant consumption was developed and validated. The sample treatment method is very useful for routine analysis, as it involves a simple simultaneous extraction/cleanup step, followed by extract concentration. The validated method was applied for the determination of target mycotoxins in 84 samples of cereal-based porridge for infant consumption. Fumonisins can be considered the main issue regarding infant exposure by mycotoxins through porridge consumption. However, aflatoxin B1, deoxynivalenol, and zearalenone were also found in concentrations exceeding the maximum permitted limit stipulated by the Brazilian legislation. The simultaneous contamination of the porridge samples by different mycotoxin classes demonstrates the importance of the analytical method developed herein.
References
Alvito PC, Sizoo EA, Almeida CMM, van Egmond HP (2010) Occurrence of aflatoxins and ochratoxin A in baby foods in Portugal. Food Anal Methods 3:22–30
Araguás C, González-Peñas E, López de Cerain A (2005) Study on ochratoxin A in cereal-derived products from Spain. Food Chem 92:459–464
Baydar T, Erkekoglu P, Sipahi H, Sahin G (2007) Aflatoxin B1, M1 and ochratoxin A levels in infant formulae and baby foods marketed in Ankara, Turkey. J Food Drug Anal 15:89–92
Belsley DA, Kuh E, Welsch RE (1980) Regression diagnostics: identifying influential data and sources of collinearity. Wiley, New York 292 p
Beltran E, Ibanez M, Sancho JV, Cortes MA, Yusa V, Hernandez F (2011) UHPLC–MS/MS highly sensitive determination of aflatoxins, the aflatoxin metabolite M1 and ochratoxin A in baby food and milk. Food Chem 126:737–744
Berthiller F, Brera C, Crews C, Iha MH, Krska R, Lattanzio VMT, MacDonald S, Malone RJ, Maragos C, Solfrizzo M, Stroka J, Whitaker TB (2016) Developments in mycotoxin analysis: an update for 2014-2015. World Mycotoxin J 9:5–30
Berthiller F, Brera C, Iha MH, Krska R, Lattanzio VMT, MacDonald S, Malone RJ, Maragos C, Solfrizzo M, Stranska-Zachariasova M, Stroka J, Tittlemier SA (2017) Developments in mycotoxin analysis: an update for 2015-2016. World Mycotoxin J 10:5–29
Bräse S, Encinas A, Keck J, Nising CF (2009) Chemistry and biology of mycotoxins and related fungal metabolites. Chem Rev 109:3903–3990
Brasil (2011) Ministério da Saúde. Agência Nacional de Vigilância Sanitária. Resolução RDC n° 7, de 18 do fevereiro de 2011. Dispõe sobre limites máximos tolerados (LMT) para micotoxinas em alimentos. Diário Oficial da República Federativa do Brasil, Brasília 46:66–67
Brown BM, Forsythe AB (1974) Robust tests for the equality of variances. J Am Stat Assoc 69:364–367
Caldas ED, Silva ACS (2007) Mycotoxins in corn-based food products consumed in Brazil: an exposure assessment for fumonisins. J Agric Food Chem 55:7974–7980
Cano-Sancho G, Gauchi JP, Sanchis V, Marín S, Ramos AJ (2011) Quantitative dietary exposure assessment of the Catalonian population (Spain) to the mycotoxin deoxynivalenol. Food Additives and Contaminants: Part A 28:1098–1109
Castro MF, Shephard GS, Sewram V, Vicente E, Mendonça TA, Jordan AC (2004) Fumonisins in Brazilian corn based foods for infant consumption. Food Addit Contam 21:693–699
Chu FS (1991) Mycotoxins: food contamination, mechanism, carcinogenic potential and preventive measures. Mutat Res 259:291–306
Cigić IK, Prosen H (2009) An overview of conventional and emerging analytical methods for the determination of mycotoxins. Int J Mol Sci 10:62–115
D’Arco G, Fernández Franzón M, Font G, Damiani P, Mañes J (2008) Analysis of fumonisins B1, B2 and B3 in corn-based baby food by pressurized liquid extraction and liquid chromatography/tandem mass spectrometry. J Chromatogr A 1209:188–194
Dall'Asta C, Galaverna G, Mangia M, Sforza S, Dossena A, Marchelli R (2009) Free and bound fumonisins in gluten-free food products. Molecular Nutrition and Food Research 53:492–499
Dombrink-Kurtzman MA, Dvorak TJ (1999) Fumonisin content in masa and tortillas from Mexico. J Agric Food Chem 47:622–627
Draper N, Smith H (1998) Applied regression analysis. Wiley, New York 706 p
Durbin J, Watson GS (1951) Testing for serial correlation in least squares regression ii. Biometrika 38:159–178
EC European Commission (2002) Commission decision 2002/657/EC of 12 August 2002. Implementing Council Directive 96/23/EC concerning performance of analytical methods and the interpretation of results. Official Journal of the European Communities, Brussels, 2002, L 221/8
EC European Commission (2006) Commission regulation (EC) no. 401/2006. Laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off J Eur Union L 70:12–34
EC European Commission (2014) Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. SANCO/12571/2013
Ezekiel CN, Sulyok M, Warth B, Odebode AC, Krska R (2012) Natural occurrence of mycotoxins in peanut cake from Nigeria. Food Control 27:338–342
Fang LX, Xiong AZ, Wang R, Ji S, Yang L, Wang ZT (2013) A strategy for screening and identifying mycotoxins in herbal medicine using ultra-performance liquid chromatography with tandem quadrupole time-of-flight mass spectrometry. J Sep Sci 36:3115–3122
FAO Food and Agriculture Organization of the United States (2004) Worldwide regulations for mycotoxins in food and feed in 2003. FAO Food and Nutrition Paper 81, Rome
Filazi A, Sireli UT (2013) Occurrence of aflatoxins in food, aflatoxins—recent advances and future prospects, Prof. Razzaghi-Abyaneh M. (Ed.), ISBN: 978-953-51-0904-4, InTech, 2013. http://www.intechopen.com/books/aflatoxins-recent-advances-and-future-prospects/occurrence-of-aflatoxins-in-food. Acessed 20 April 2017
Frenich AG, Vidal JLM, González RR, Luiz MMA (2009) Simple and high-throughput method for the multimycotoxin analysis in cereals and related foods by ultra-high performance liquid chromatography/tandem mass spectrometry. Food Chem 117:705–712
García-Campaña A, Cuadros-Rodríguez L, Aybar-Muñoz J, Alés-Barrero F (1997) Chemometric protocol to validate an analytical method in the presence of corrigible constant and proportional systematic errors. J AOAC Int 80:657–664
Gottschalk C, Barthel J, Engelhardt G, Bauer J, Meyer K (2009) Simultaneous determination of type A, B and D trichothecenes and their occurrence in cereals and cereals products. Food Addit Contam 26:1273–1289
Horwitz W, Latimer GW (2005) Official methods of analysis of AOAC International, 18th edn. AOAC International, Gaithersburg
Huang B, Han Z, Cai Z, Wu Y, Ren Y (2010) Simultaneous determination of aflatoxins B1, B2, G1, G2, M1 and M2 in peanuts and their derivative products by ultra-high-performance liquid chromatography-tandem mass spectrometry. Anal Chim Acta 662:62–68
IARC International Agency for Research on Cancer (1993) Evaluation of carcinogen risks to humans. Some naturally occurring substances: foods items and constituents, heterocyclic aromatic amines and mycotoxins
IARC International Agency for Research on Cancer (2002) Monographs 56: 489–521. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 82: 301–366. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC, Lyon
Jia W, Chu X, Ling Y, Huang J, Chang J (2014) Multi-mycotoxin analysis in dairy products by liquid chromatography coupled to quadrupole orbitrap mass spectrometry. J Chromatogr A 1345:107–114
Juan C, Raiola A, Manes J, Ritieni A (2014) Presence of mycotoxin in commercial infant formulas and baby foods from Italian market. Food Control 39:227–236
Kabak B (2009) Ochratoxin A in cereal-derived products in Turkey: occurrence and exposure assessment. Food Chem Toxicol 47:348–352
Köppen R, Koch M, Siegel D, Merkel S, Maul R, Nehls I (2010) Applied microbiology and biotechnology determination of mycotoxins in foods: current state of analytical methods and limitations. Appl Microbiol Biotechnol 86:1595–1612
Kostelanská M, Sosnovcová I, Ondrej L, Hajslová J (2010) Determination of mycotoxins in infant and baby food using UPLC-MS/MS analytical method. Proceedings of 6 th ISC Modern Analytical Chemistry. Prague
Krska R, Schubert-Ullrich P, Molinelli A, Sulyok M, Macdonald S, Crews C (2008) Mycotoxin analysis: an update. Food Addit Contam 25:152–163
Lacina O, Zachariasova M, Urbanova J, Vaclavikova M, Cajka T, Hajslova J (2012) Critical assessment of extraction methods for the simultaneous determination of pesticide residues and mycotoxins in fruits, cereals, spices and oil seeds employing ultra-high performance liquid chromatography-tandem mass spectrometry. J Chromatogr A 1262:8–18
Lee HJ, Ryu D (2017) Worldwide occurrence of mycotoxins in cereals and cereal-derived food products: public health perspectives of their co-occurrence. Journal of Agricultural and Food Chemistry
Lombaert GA, Pellaers P, Roscoe V, Mankotia M, Neil R, Scott PM (2003) Mycotoxins in infant cereal foods from the Canadian retail market. Food Addit Contam 20:494–504
Mahnine N, Meca G, Fernández-Franzón M, Mañes J, Zinedine A (2012) Occurrence of fumonisins B1, B2 and B3 in breakfast and infant cereals from Morocco. Phytopathol Mediterr 51:193–197
Mavungu DDJ, Monbaliu S, Scippo ML, Maghuin-Rogister G, Schneider YJ, Larondelle Y, Callebaut A, Robbens J, Van Peteghem C, De Saeger S (2009) LC-MS/MS multi-analyte method for mycotoxin determination in food supplements. Food additives and contaminants. Part A Chem Anal Control Expo Risk Assess 26:885–895
Maziero MT, Bersot LS (2010) Micotoxinas em alimentos produzidos no Brasil. Revista Brasileira de Produtos Agroindustriais 12:89–99
Mol HG, Plaza-Bolaños P, Zomer P, de Rijk TC, Stolker AA, Mulder PP (2008) Toward a generic extraction method for simultaneous determination of pesticides, mycotoxins, plant toxins, and veterinary drugs in feed and food matrixes. Anal Chem 80:9450–9459
Mol HGJ, MacDonald SJ, Anagnostopoulos C, Spanjer M, Bertuzzi T, Pietri A (2016) European survey on sterigmatocystin in cereals, cereals-based products, beer and nuts. World Mycotoxin J 9:633–642
Murphy PA, Hendrich S, Landgren C, Bryant CM (2006) Food mycotoxins: an update. J Food Sci 71:51–65
Ozden S, Akdeniz AS, Alpertunga B (2012) Occurrence of ochratoxin A in cereal-derived food products commonly consumed in Turkey. Food Control 25:69–74
Peraica M, Radic B, Lucic A, Pavlovic M (1999) Toxic effects of mycotoxins in humans. Bull World Health Organ 77:754–766
Plattner RD (1999) HPLC/MS analysis of fusarium mycotoxins, fumonisins and deoxynivalenol. Nat Toxins 7:365–370
Prandini A, Tansini G, Sigolo S, Filippi L, Laporta M, Piva G (2009) On the occurrence of aflatoxin M1 in milk and dairy products. Food Chem Toxicol 47:984
Ren Y, Zhang Y, Shao S, Cai Z, Feng L, Pan H, Wang Z (2007) Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J Chromatogr A 1143:48–64
Righetti L, Paglia G, Galaverna G, Dall’Asta C (2016) Recent advances and future challenges in modified mycotoxin analysis: why HRMS has become a key instrument in food contaminant research. Toxins 8:361
Rocha MEB, Freire FCO, Maia FEF, Guedes MIF, Rondina D (2014) Mycotoxins and their effects on human and animal health. Food Control 36:159–165
Romagnoli B, Ferrari M, Bergamini C (2010) Simultaneous determination of deoxynivalenol, zearalenone, T-2 and HT-2 toxins in breakfast cereals and baby food by high-performance liquid chromatography and tandem mass spectrometry. J Mass Spectrom 45:1075–1080
Rubert J, Dzuman Z, Vaclavikova M, Zachariasova M, Soler C, Hajslova J (2012a) Analysis of mycotoxins in barley using ultra high liquid chromatography high resolution mass spectrometry: comparison of efficiency and efficacy of different extraction procedures. Talanta 99:712–719
Rubert J, Soler C, Mañes J (2012b) Application of an HPLC–MS/MS method for mycotoxin analysis in commercial baby foods. Food Chem 33:176–183
Ryan TA, Joiner BL (1976) Normal probability plots and tests for normality. Pennsylvania State University, The State College
Saeger S (2011) Determining mycotoxins and mycotoxigenic fungi in food and feed. Woodhead publishing series in food science, technology and nutrition: number 203. Woodhead Publishing Limited, Cambridge
Sartori AV, Mattos JS, Moraes MHP, Nóbrega AW (2015a) Determination of aflatoxins M1, M2, B1, B2, G1, G2 and ochratoxin A in UHT and powdered milk by modified QuEChERS method and ultra-high performance liquid chromatography-tandem mass spectrometry. Food Anal Methods 8:2321–2330
Sartori AV, Mattos JS, Souza YP, Santos RP, Moraes MHP, Nóbrega AW (2015b) Determination of aflatoxins M1, M2, B1, B2, G1 and G2 in peanut by modified QuEChERS method and ultra-high performance liquid chromatography-tandem mass spectrometry. Revista Visa em Debate 3:115–121
Shotwell OL, Goulden ML, Hesseltine CW (1976) Aflatoxin M1. Occurrence in stored and freshly harvested corn. J Agric Food Chem 24:683–684
Souza SVC, Junqueira RG (2005) A procedure to assess linearity by ordinary least squares method. Anal Chim Acta 552:25–35
Sulyok M, Krska R, Schuhmacher RA (2007) Liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semi-quantitative screening of moldy food samples. Anal Bioanal Chem 389:1505–1523
Tam J, Mankotia M, Mably M, Pantazopoulos P, Neil RJ, Calway P, Scott PM (2006) Survey of breakfast and infant cereals for aflatoxins B1, B2, G1 and G2. Food Addit Contam 23:693–699
Tamura M, Uyama A, Mochizuki N (2011) Development of a multi-mycotoxin analysis in beer-based drinks by a modified QuEChERS method and ultra-high-performance liquid chromatography coupled with tandem mass spectrometry. Anal Sci 27:629–635
Tamura M, Matsumoto K, Watanabe J, Lida J, Nagatomi Y, Mochizuki N (2012) High throughput quantitative analysis of multi-mycotoxin in beer-based drinks using UHPLC-MS/MS. Shimadzu corporation, First edition
Tamura M, Matsumoto K, Watanabe J, Iida J, Nagatomi Y, Mochizuki N (2014) Minimization of carryover for high-throughput liquid chromatography with tandem mass spectrometry analysis of 14 mycotoxins in corn grits. J Sep Sci 37:1552–1560
Turner NW, Subrahmanyam S, Piletsky SA (2009) Analytical methods for determination of mycotoxins: a review. Anal Chim Acta 632:168–180
Turner NW, Bramhmbhatt H, Szabo-Vezse M, Poma A, Coker R, Piletsky SA (2015) Analytical methods for determination of mycotoxins: an update (2009-2014). Anal Chim Acta 901:12–33
Vesonder R, Haliburton J, Stubblefield R, Gilmore W, Peterson S (1991) Aspergillus flavus and aflatoxins B1, B2, and M1 in corn associated with equine death. Arch Environ Contam Toxicol 20:151–153
Zachariasova M, Cajka T, Godula M, Malachova A, Veprikova Z, Hajslova J (2010) Analysis of multiple mycotoxins in beer employing (ultra)-high-resolution mass spectrometry. Rapid Commun Mass Spectrom 24:3357–3367
Zain ME (2011) Impact of mycotoxins on humans and animals. J Saudi Chem Soc 15:129–144
Zhang K, Wong JW, Krynitsky AJ, Trucksess MW (2014) Determining mycotoxins in baby foods and animal feeds using stable isotope dilution and liquid chromatography tandem mass spectrometry. J Agric Food Chem 62(36):8935–8943
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
André Victor Sartori declares that he has no conflict of interest. Maria Heloísa Paulino de Moraes declares that she has no conflict of interest. Rosana Pereira dos Santos declares that she has no conflict of interest. Yuri Pereira Souza declares that he has no conflict of interest. Armi Wanderley da Nóbrega declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(DOC 222 kb)
Rights and permissions
About this article

Cite this article
Sartori, A.V., de Moraes, M.H.P., dos Santos, R.P. et al. Determination of Mycotoxins in Cereal-Based Porridge Destined for Infant Consumption by Ultra-High Performance Liquid Chromatography-Tandem Mass Spectrometry. Food Anal. Methods 10, 4049–4061 (2017). https://doi.org/10.1007/s12161-017-0965-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-017-0965-4