
Abstract
A fast, easy, and cheap method for the simultaneous determination and quantification of aflatoxins (B1, B2, G1, G2), T-2 and HT-2 toxins, and fumonisins (B1, B2) in cereal-derived products was developed. This method involved a quick, easy, cheap, effective, rugged, and safe (QuEChERS) extraction coupled with liquid chromatography-tandem mass spectrometry. The method validation was performed by analyzing samples spiked at four levels, and the recoveries ranged from 83.6 to 102.9%, whereas the maximum values of repeatability and within-laboratory reproducibility were 14.3 and 15.7% following the performance criteria set by the European legislation. The method was then applied for the analysis of 21 cereal-derived products purchased on the Italian market, which were correctly packaged and labeled as intended for human consumption. The co-occurrence of more than one mycotoxin in the analyzed samples could represent a risk for consumers, and the described method could be a valid alternative for their simultaneous detection in the framework of official control.

ᅟ
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cereal-derived products constitute a great portion of the human diet, and in some countries like Italy, durum wheat is used nearly exclusively for pasta production. However, crops can be affected by mycotoxin-producing fungi and therefore consumer exposure to these compounds can be particularly high. Mycotoxins are secondary metabolites produced by different fungi along crop production both in the field and during harvest and storage. Moreover, these compounds can be very stable to food processing and therefore they can be found also in the final product.
Toxigenic fungi can be distinguished into aggressive or opportunistic plant pathogens belonging to Fusarium genera, and other fungi, i.e., Aspergillus spp. and Penicillium spp. that initially colonize plants but proliferate mostly during storage [1]. Fungi belonging to Fusarium genera mainly produce fumonisins and trichothecenes. Fumonisins can be found particularly in maize contaminated by Fusarium verticillioides and Fusarium proliferatum, prior to harvest or during the early stage of storage. Fumonisin B1 (FB1) is the most significant in terms of toxicity and occurrence, while fumonisin B2 (FB2) is a deoxy analog of FB1 [2]. The International Agency for Research on Cancer [3] classified fumonisins as possibly carcinogens to humans (group 2B). Trichothecenes are a group of secondary metabolites also produced by fungi included in the Fusarium genera. They have a sesquiterpenoid ring structure containing an epoxide at the C12–C13 position, which is responsible for their toxicological activity [4]. A new group of trichothecenes causing most concern for human health are T-2 and HT-2 toxins. These mycotoxins are commonly found in cereals, including wheat, barley, maize, oats, rye, and derived products, particularly in Europe and in hot, wet climatic regions. T-2 toxin inhibits protein, RNA, and DNA synthesis. Recent data also indicate that T-2 toxin induces apoptosis, and in some cell types, necrosis, as well as lipid peroxidation affecting cell membrane integrity [5]. Moreover, T-2 toxin induces hematotoxicity and myelotoxicity associated with impairment of hematopoiesis in bone marrow [6].
Aflatoxins are difuranocoumarins produced primarily by two species of Aspergillus which colonize plants in the field prior to harvest, but they can accumulate also in postharvest cereals [7]. Aspergillus flavus is ubiquitous and develops on the aerial parts of plants (leaves, flowers); moreover, it produces mainly B aflatoxins. Aspergillus parasiticus produces both B and G aflatoxins, is more adapted to a soil environment, and has a more limited distribution [8]. The order of acute and chronic toxicity is aflatoxin B1 (AFB1) > aflatoxin G1 (AFG1) > aflatoxin B2 (AFB2) > aflatoxin G2 (AFG2), reflecting the role played by epoxidation of the 8,9-double bond and the greater potency associated with the cyclopentenone ring of the B series, when compared with the six-membered lactone ring of the G series. Both AFB1 and AFM1 have been classified in group 1 (human carcinogen) and are also clearly genotoxic [9, 10]. The risk for primary liver cancer is considerably increased in geographical regions with a high prevalence of carriers of the hepatitis B serum antigen [11].
Maximum levels of these mycotoxins in foodstuffs have been established by different international agencies. In the European Union, these levels are laid down for aflatoxins and fumonisins in Commission Regulation (EU) No. 165/2010 [12] and Commission Regulation (EC) No. 1126/2007 [13], respectively. Regarding the T-2 and HT-2 toxins, the European Commission asked the European Food Safety Authority for a scientific opinion on the risk for human and animal health related to these mycotoxins in food and feed [5], but until now, no legal limits have been set. Following the Commission Recommendation 2013/165/EU [14], further investigations must be undertaken to collect information on the factors resulting in relative high levels of T-2 and HT-2 toxins in cereals and cereal-derived products. Indicative levels for T-2 and HT-2 toxins have been established based on the occurrence data available in the EFSA database [5]. Mycotoxins, either from the same or different fungal species, may occur simultaneously in plant products. Therefore, cereals and cereal-derived products can be contaminated with two or more mycotoxins at the same time. Many studies have been described aiming at single micotoxin detection [15, 16], but the most current research is directed to development of analytical methods for simultaneous mycotoxin determination using immunoaffinity cleanup [17,18,19] or without cleanup step by diluting sample extracts before injection in mass spectrometer [20,21,22]. Due to the variability of matrices susceptible to contamination, the possible co-occurrence [23], and the very wide range of mycotoxin concentrations, validated and versatile multi-mycotoxin and multi-matrix methods are strongly necessary. The Commission Regulation (EU) No. 519/2014 [24] established rules for the sampling and analysis for the official control of mycotoxins in food products. However, the analysis of complex foods, such as breakfast cereals, pre-cooked meals, and pastries, can be carried out for individual components of the complex food before processing or for the whole processed food. In the last case, interactions between the components and the toxins or possible formation of toxins during food processing can occur. Several analytical methods have been developed and validated to detect mycotoxins [25], but the heterogeneity of food matrices together with the need for a fast, simultaneous, and accurate determination of multiple mycotoxins generate great challenges for routine analysis [26]. The aim of the present study was to develop and validate a LC-MS/MS method for the rapid and simultaneous determination of B1, G1, B2, and G2 aflatoxins, T-2 and HT-2 toxins, and B1 and B2 fumonisins in cereal-derived products. The extraction and cleanup were performed by a quick, easy, cheap, effective, rugged, and safe (QuEChERS) method as firstly reported by Anastassiades et al. [27] for pesticide detection in fruits and vegetables. The method was then used to analyze different cereal-derived products collected from Italian markets to evaluate its possible application for mycotoxin identification and quantification in these food matrices.
Materials and methods
Reagents and materials
Methanol, acetonitrile, formic acid, all HPLC grade, as well as ammonium formiate were purchased from Sigma (St. Louis, MO, USA). The Original QuEChERS kits containing 4 g MgSO4 and 1 g NaCl were supplied by Agilent Technologies (Santa Clara, CA, USA). Polyvinylidene fluoride filters were obtained from Merck Millipore (Carrigtwohill, Co. Cork, Ireland). Ultrapure water was obtained by Elga Labwater (Wicombe, UK). HT-2 toxin was supplied by Sigma-Aldrich (St. Louis, MO, USA) as solution in acetonitrile at 100 μg/mL. All other mycotoxin reference standards were obtained from Biopure (Tullin, Austria) as solutions in acetonitrile at the following concentrations: AFB1 (2 μg/mL), AFB2 (0.5 μg/mL), AFG1 (2 μg/mL), and AFG2 (0.5 μg/mL) and T-2 toxin (100 μg/mL), while FB1 and FB2 were supplied in acetonitrile/water (50:50, v/v) at concentrations of 50 μg/mL. As internal standard (IS), 13C17-aflatoxin B1 (13C17-AFB1) was chosen, and it was supplied by Biopure at 0.5 μg/mL concentration. All stock solutions were stored at −18 °C, except for fumonisins and T-2 toxin solutions that were stored at 4 °C, until the expiration date stated by the manufacturer.
Preparation of working solutions
For the analysis, a working solution in acetonitrile was prepared by diluting each individual stock solution at the following concentrations: aflatoxins (25 ng/mL), T-2 and HT-2 toxins (62.5 ng/mL), and fumonisins (5000 ng/mL). Moreover, a working solution of the internal standard (13C17-AFB1) was prepared at the concentration of 25 ng/mL in acetonitrile.
Preparation of solvent calibration curves
Calibration standards were prepared from the working solutions by evaporating acetonitrile to dryness under a stream of nitrogen at 40 °C and dissolving the residue with a solution of methanol/water containing 5 mM ammonium formiate and 0.1% formic acid (10:90, v/v). Four calibration solutions were obtained at the following concentrations: 1.25–5–10–20 ng/mL for all aflatoxins (each solution contained 13C17-AFB1 at 5 ng/mL), 3.125–12.5–25–50 ng/mL for T-2 and HT-2 toxins, and 20–40–60–120 ng/mL for fumonisins.
Sample preparation
For the extraction of mycotoxins from cereal-derived products, the QuEChERS method optimized by Rubert et al. [28] for barley samples was used, with some modifications. Two grams of the homogenized sample of cereal-derived product were weighed into a 50-mL tube and spiked with internal standard 13C17-AFB1 at a concentration of 2 μg/kg. The fortified sample was kept in the dark for 15 min, to allow equilibration of IS with the matrix. The extraction was performed by adding 10 mL of water containing 0.1% formic acid and shaking the mixture for 3 min. Then 10 mL of acetonitrile were added and the sample was further shaken for 3 min. In the following step, 4 g MgSO4 and 1 g NaCl were added and the mixture was immediately shaken for 2 min to prevent agglomerates forming during MgSO4 hydration and then centrifuged at 3500×g for 10 min (Megafuge 1.0, Heraeus Instruments, Hanau, Germany). For the analysis of AFB1, AFB2, AFG1, and AFG2 and T-2 and HT-2 toxins, 2.5 mL of the extract were evaporated at 40 °C under a stream of nitrogen and the residue was completely dissolved by adding 200 μL of a solution of methanol/water containing 5 mM ammonium formiate and 0.1% formic acid (10:90, v/v). For the analysis of FB1 and FB2, 1 mL of extract was diluted with 1 mL of water containing 5 mM ammonium formiate and 0.1% formic acid. Before the instrumental analysis, the sample was filtered through a 0.45-μm polyvinylidene fluoride filter into a vial for autosampler equipped with insertion for small volume.
Matrix-matched calibration curves
The mycotoxins quantification was performed using matrix-matched calibration curves prepared by extracts of a blank sample of wheat flour spiked at four concentration levels: 0.5–2–4–8 μg/kg for AFB1, AFB2, AFG1, and AFG2, 1.25–5–10–20 μg/kg for T-2 and HT-2 toxins, and 200–400–600–800 μg/kg for FB1 and FB2.
Mass spectrometry conditions
The mass spectrometer was an API 3000 triple quadrupole equipped with an electrospray interface set in the positive ionization mode (Applied Biosystems, Toronto, ON, Canada). The LC-MS/MS system, data acquisition, and processing were managed by Analyst software 1.6 version supplied by Applied Biosystems. For mass spectrometer analyses, the following settings were used: temperature 400 °C; ion spray voltage +5500 V, curtain gas nitrogen 10 psi, nebulizer gas air 8 psi, and collision gas nitrogen 5 psi. The mass spectrometer was operated in multi-reaction monitoring (MRM) mode by monitoring two transitions (1 precursor ion, 2 product ions) for each compound with a dwell time of 40 ms. Tuning experiments were performed by direct infusion (flow rate of 10 μL/min) of 1 μg/mL standard solutions of individual mycotoxin in methanol/water containing 5 mM ammonium formiate and 0.1% formic acid (50:50, v/v). The infusion was carried out using a model 11Plus Infusion Pump (Harvard, South Natick, MA, USA). Declustering potential, collision energy and cell exit potential, and focusing and entrance potentials were optimized during infusion. All the optimized parameters are summarized in Table 1.
Chromatographic conditions
The chromatographic analysis was carried out by a Perkin Elmer HPLC system which constituted a model series 200 Micro Pump, a model Series 200 Autosampler, equipped with a degasser and a column oven (Perkin Elmer, Waltham, MA, USA). A reversed-phase separation of the analytes was performed at 30 °C on a Kinetex XB-C18 100 Å column, 100 mm × 2.1 mm internal diameter, 2.6 μm (Phenomenex, Torrance, CA, USA). The mobile phase was composed of methanol (eluent A) and water containing 5 mM ammonium formiate and 0.1% formic acid (eluent B). The injection volume was 20 μL. A gradient elution was performed setting the mobile phase composition as follows: after 2 min at 20% eluent A, linear increase to 95% in 14 min and kept constant for 4 min to rinse the column. Then the component A was linearly lowered to 20% in 2 min and the column re-equilibrated for 8 min.
Method validation
The developed method was validated as a quantitative confirmatory method. The evaluated parameters were instrumental linearity, specificity, precision, trueness, limit of detection (LOD), and limit of quantification (LOQ) following Regulation (EC) No. 882/2004 [29].
The instrumental linearity was assessed by four-point solvent calibration curves (containing IS at 5 ng/mL) chosen to cover the validation range. The specificity was evaluated by analyzing 20 blank samples of cereal-derived products to verify the absence of interfering compounds at the retention time of the checked analytes. The intra- and inter-day precision was evaluated by spiking three aliquots of a blank wheat flour sample at four concentration levels. This experiment was repeated in two different days for a total of 24 analyses.
The validation levels were chosen as follows: (i) for each aflatoxin, the spiking levels were 0.5–1–1.5–3 times the maximum level of 2 μg/kg fixed for AFB1 in cereals by Commission Regulation (EU) No. 165/2010; (ii) for T-2 and HT-2 toxins individually, the spiking levels were 0.5–1–1.5–3 times the LOQ (equal to 5 μg/kg) recommended by Commission Recommendation (EU) No. 165/2013; and (iii) for each fumonisin, the spiking levels were 0.25–0.5–0.75–1.5 times the maximum level of 800 μg/kg fixed for total fumonisins in maize-based breakfast cereals by Commission Regulation (EC) No. 1126/2007.
Trueness was expressed in terms of recovery (percentage of measured concentration versus spiked concentration) and precision (as relative standard deviation (%RSD)).
The LOD was fixed at half the first validation level while the LOQ was fixed at the first validation level.
Sample collection and analysis
The developed method was used to analyze 21 cereal-derived products for human consumption to monitor the mycotoxin levels in these kinds of food. These cereal-based foods were randomly purchased from local markets in central Italy. The samples were collected in duplicate and distinguished in six wheat flours, six dry pasta, five baked foods, two corn meals, and two breakfast cereals. All samples were homogenized using a laboratory mill and stored in a dark and dry place until the analysis. Each sample was analyzed in duplicate, and the results are expressed as mean values.
Results and discussion
Extraction procedure
In the present study, the QuEChERS method of Rubert et al. [28] was applied with some modifications. Different extraction procedures (i.e., matrix solid phase dispersion, QuEChERS, and solid–liquid extraction) were tested to demonstrate that the recoveries obtained by QuEChERS were comparable with those obtained applying the other two extraction techniques. However, QuEChERS resulted as the fastest and cheapest procedure because the pre-concentration and cleanup steps were not necessary, thus allowing to reduce the solvent consumption. Moreover, QuEChERS procedure presents several advantages over the most traditional methods of analysis in the following ways: high sample throughput of samples in a short time is possible, solvent usage and waste are very small, and reagents are few expensive.
LC-MS/MS
The LC-MS/MS method was developed following the performance criteria for mass spectrometric detection reported in the Commission Decision (EC) No. 657/2002 [30]. The analysis was performed in MRM mode, due to high sensitivity and specificity. This acquisition mode allowed to have the identification points required by the above-cited decision for the identification of the investigated mycotoxins.
During the tuning experiments, different mobile phases were tested. For the detection of aflatoxins, the use of water containing just only 0.1% formic acid enhanced the sodium ion adduct against the protonated molecular ion, whereas if adding 5 mM ammonium formiate in water containing 0.1% formic acid, the formation of sodium ion adduct was reduced promoting the exaltation of protonated molecular ion, as shown in Fig. 1 for AFB1. Therefore, the use of eluent B as mobile phase provided a higher instrumental response of the proton ion adduct and a better ionization reproducibility for all the examined aflatoxins.

Chromatograms obtained for AFB1 (as sodium ion adduct with m/z 334.9 and protonated molecular ion with m/z 313.2) using as mobile phase water containing 0.1% formic acid (left) and water containing 5 mM ammonium formiate and 0.1% formic acid (right)
Validation study
The instrumental linearity was estimated by using the least square regression line equation. Aflatoxin calibration curves in solvent were constructed using the area ratio of the analyte peak to the internal standard peak versus analyte concentration, while for the T-2 and HT-2 toxins and fumonisins, the internal standard was not taken into account. The correlation coefficient indicated a good fit for all the analytes with values included in the range of 0.996–1.000. The chromatogram of the first solvent calibration point is reported in Fig. 2. Regarding the specificity, for all the cereal-derived products, the absence of any interfering peaks around the analytes’ retention times was verified to demonstrate that there were no differences among the different analyzed matrices. Figure 3 showed the chromatogram of a representative blank wheat flour sample. The comparison between this chromatogram (Fig. 3) and that of a solvent standard (Fig. 2) showed the specificity of the method.

Chromatogram of the lowest solvent calibration point. For each analyte, the most intense product ion was shown

Chromatogram of a representative blank wheat flour sample. For each analyte, the most intense product ion was shown
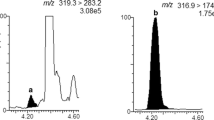
The applied method did not contemplate a cleanup step; therefore, in the analyzed samples, a high ion suppression effect was observed as shown in Fig. 4, where solvent and matrix-matched calibration curves at the same concentration were compared. The difference between the slope of solvent curve and the slope of matrix-matched calibration curve demonstrated the matrix ion suppression effect for each of the investigated mycotoxins. This effect could be due to the presence of endogenous substances in the sample that were retrieved in the final extract [31]. The chromatograms of a spiked wheat flour sample at the lowest validation level for the checked mycotoxins are shown in Figs. 5, 6, and 7. The signal of the lowest intense product ion appeared fully satisfactory with respect to signal-to-noise ratio.

Comparison of solvent and matrix-matched calibration curves

Chromatogram of a wheat flour sample spiked at the lowest validation level for IS, AFB1, and AFB2. For each analyte, all the studied ions were shown

Chromatogram of a wheat flour sample spiked at the lowest validation level for AFG1, AFG2, and T-2 toxin. For each analyte, all the studied ions were shown

Chromatogram of a wheat flour sample spiked at the lowest validation level for HT-2 toxin, FB1, and FB2. For each analyte, all the studied ions were shown
The precision was evaluated at four spiking levels (three replicates for level, two analytical series) as reported in Table 2. The matrix-matched calibration curve was applied to evaluate the actual recovery for each analyte. Only for aflatoxins the matrix-matched calibration curve was constructed using the area ratio of the analyte peak to the internal standard peak versus analyte concentration, while for the T-2 and HT-2 toxins and fumonisins the internal standard was not taken into account. As reported by Rubert et al. [28], the use of external matrix-matched calibration or internal standard calibration could minimize the variation among different samples. The best option should be the use of a suitable IS (isotopically labeled, deuterated, or analog) which could overcome ion suppression/enhancement. Nevertheless, it could be expensive to buy a specific IS for each one of the investigated mycotoxins, so in the present study, the matrix-matched calibration curves were applied to assess the extraction efficiency.
The recovery and precision were evaluated using the results of the matrix-matched calibration curves; the recovery was expressed in terms of percentage of measured concentration versus spiked concentration and precision as %RSD in terms of repeatability (intra-day, %RSDr) and within reproducibility (inter-day, %RSDR).
The validation data obtained for each mycotoxin are reported in Table 2. The mean recoveries for all the mycotoxins ranged between 83.6 and 102.9%. The maximum values of repeatability and within-laboratory reproducibility were 14.3 and 15.7%, fulfilling the requirements established by the Commission Regulation 519/2014/EU. The performance of the method allowed to achieve LOD and LOQ values (Table 3) lower than the official limits fixed by the European Commission for each mycotoxin in cereal-derived products. Since the experimental LOD values obtained for each mycotoxin were lower than half LOQ values, LOD were fixed at half LOQ concentrations. The performances of the described method were similar in terms of precision, repeatability, and reproducibility with other methods [17, 19] that used a stronger cleanup step. In particular, the developed method showed a good sensitivity and precision at low spiking levels for T2 and HT2 toxins if compared with methods that involved a simple dilution of sample extract [21]. Moreover, the method allowed a suitable determination of mycotoxins in cereals reducing both the quantity of solvent and the time and cost of analysis, especially in case of use of immunoaffinity columns [17,18,19].
Detection of mycotoxins in cereal-derived products from the Italian market
The developed and validated method was then applied to detect the investigated mycotoxins in cereal-derived products randomly purchased on the market. It allowed to detect mycotoxin concentrations below the maximum limits set by the European legislation. All the analyzed samples showed mycotoxin levels below the LOQ, except for a sample of dry pasta showing values of 3.0 μg/kg of HT-2 toxin, and a cornmeal sample, in which both AFB1 (3.3 ± 1.4 μg/kg) and B1 and B2 fumonisins (711 and 219 μg/kg, respectively) were detected. Fumonisin B1 was also found in the other examined cornmeal sample, at levels of 431 μg/kg. Regarding the sample positive for AFB1, the detected value could be considered compliant with the regulatory limit of 2 μg/kg because this result subtracted from the uncertainty was lower than the maximum limit, as established by the EURACHEM/CITAC Guide [32] and Commission Regulation (EU) No. 519/2014.
The results of this study showed that samples were positive for mycotoxins at low rates; AFB1 and FB2 were found just only in one (4.8%) cornmeal, and FB1 was detected in all the two (9.5%) investigated cornmeal samples. With regard to aflatoxins, our results were similar with those obtained in other studies, in which these mycotoxins were never detected [33] or only 0.28% of samples were positive [34]. The EFSA database [35] reported a total of 1341 cereals and their milling products and 842 processed cereal products analyzed for aflatoxin content, showing a maximum mean value at lower bound (LB) in unspecified grain milling products (2.21 μg/kg) while the maximum mean value at upper bound (UB) was found in oat milling products (2.60 μg/kg). For processed cereal products, the maximum mean value was found in fine bakery wares (0.45 μg/kg, LB) and raw pasta (1.87 μg/kg, UB). On the contrary, some other studies found high levels of AFB1 in maize samples intended for human consumption, up to 14.9 μg/kg [36] and 636 μg/kg [37]. In both cases, these cereals were harvested from tropical and subtropical zones with a climate characterized by strong sunshine and high rainfall, very favorable to fungi growth, especially of thermotolerant species such as Aspergillus spp. Aflatoxin contamination has been reported also in wheat. The percentages of positive samples for AFB1, AFB2, AFG1, and AFG2 were 42, 12, 37, and 12%, respectively, in 41 wheat samples grown and consumed in some regions of Turkey. The levels of AFB1 and AFB2 ranged from 0.0104 to 0.144 μg/kg and from 0.0129 to 0.0364 μg/kg, respectively, whereas the concentrations of AFG1 and AFG2 were found to be between 0.0210 and 0.446 μg/kg and between 0.0272 and 0.129 μg/kg, respectively [38]. Aflatoxin concentrations up to 7 μg/kg were found in 62% of wheat samples collected from experimental field located in northern Italy, probably due to bad climatic conditions at harvest time or inappropriate postharvest drying practices [39]. These data confirmed the widespread occurrence of aflatoxigenic strains of Aspergillus also in Italy and highlighted the importance of the postharvest care of grains.
In the present study, the cornmeal samples were found to be contaminated by both aflatoxins and fumonisins. The corn belongs to the commodities at high risk of mycotoxin contamination, whereas other cereals like wheat are more resistant or only moderately susceptible to fungal growth and toxin production in the field. Samples of corn flour from Morocco markets showed higher AFB1 levels than those detected in wheat flour, ranging from 0.23 to 11.2 μg/kg in corn flour and from 0.03 to 0.15 μg/kg in wheat flour [40]. In a previous study on cereals commercialized in Morocco, corn samples were found to be contaminated also by FB1 (50% of positive samples) with an average value of 1930 μg/kg, the highest value being 5960 μg/kg [41]. Another survey on the occurrence of Fusarium toxins in food collected in the European Union [42] showed that the most frequently contaminated cereals were maize, followed by wheat and oat, with an incidence of 20% (out of 3490 samples) and 14% (3032 samples) for T-2 and HT-2 toxins, respectively. Strains of Fusarium species commonly isolated from cereals in the Mediterranean area produced different mycotoxins, some at very high concentrations. A survey of fumonisin production by strains of F. verticillioides and F. proliferatum from Italy, Spain, and France showed that all strains produced FB1 and FB2, with the highest average production in decreasing order: corn, wheat, barley, and sorghum [43]. Due to the widespread production of fumonisins in Fusarium-infected plants in these countries, their occurrence in commercial cereals, especially corn-based products, could be expected. In contrast, it has been reported that fumonisin concentrations were often low in the central and northeastern Mediterranean areas [44].
During cereal harvest and storage, the fungi contamination together with a high humidity and temperature are key factors affecting mycotoxin formation. The most important tool for the prevention and limitation of appearance of mycotoxins in cereals is a good management in agricultural production and storage, such as proper irrigation, choice of genetically resistant crop strains and biopesticide control, as well as the sorting and disposal of visible moldy or damaged seeds, reducing the bioavailability of mycotoxins using clay and chemoprotection [45]. Drying seeds to the required moisture content immediately after harvest could be useful before cereal storage.
Conclusions
Cereals and cereal-derived products are among the most consumed food throughout the world, but they are susceptible to mycotoxin contamination. Certain regions present favorable conditions (e.g., climate, high humidity) for mycotoxin formation, representing a health risk particularly in people where the consumption of cereal-based food is extraordinary. For instance, small children are often more exposed than adults to mycotoxin intake due to their high consumption of cereal-derived products such as breakfast cereals, biscuits, etc. The results obtained in this study with regard to samples positive to more than one mycotoxin (e.g., AFB1 and both FB1 and FB2 in one cornmeal product) confirmed the need of analytical methods aiming at their simultaneous detection. The described method could be used in official as well as private laboratories in terms of time, cost, and instrumentation.
References
Reyneri A. The role of climatic condition on micotoxin production in cereal. Vet Res Commun. 2006;30:87–92.
European Food Safety Authority. Opinion of the scientific panel on contaminants in food chain on a request from the Commission related to fumonisins as undesirable substances in animal feed. The EFSA J. 2005;235:1–32.
International Agency for Research on Cancer. Summaries and evaluations: fumonisin B1. Lyon, France; 2002a. pp. 1–301.
Yang S, Wang Y, Beier RC, Zhang H, De Ruyck K, Sun F, et al. Simultaneous determination of type A and B trichothecenes and their main metabolites in food animal tissues by ultra-performance liquid chromatography coupled with triple quadrupole mass spectrometry. J Agr Food Chem. 2015;63(38):8592–600.
European Food Safety Authority. Scientific opinion on the risks for animal and public health related to the presence of T-2 and HT-2 toxin in food and feed. The EFSA J. 2011;9(12):2481. 1–187
Parent-Massin D. Haematotoxicity of trichothecenes. Toxicol Lett. 2004;153:75–81.
Liu Z, Gaoa J, Yuc J. Aflatoxins in stored maize and rice grains in Liaoning Province, China. J Stored Prod Res. 2006;42:468–79.
European Food Safety Authority. Opinion of the scientific panel on contaminants in the food chain on a request from the Commission related to the potential increase of consumer health risk by a possible increase of the existing maximum levels for aflatoxins in almonds, hazelnuts and pistachios and derived products. The EFSA J. 2007;446:1–127.
International Agency for Research on Cancer. IARC Monographs on the evaluation of carcinogenic risks to humans. Some naturally occurring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins. Lyon, France; 1993. pp. 397–488.
International Agency for Research on Cancer. IARC Monographs on the evaluation of carcinogenic risks to humans. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. Lyon, France; 2002b. pp. 1–556.
Henry SH, Bosch FX, Bowers JC, Portier CJ, Petersen BJ, Barraj L. Aflatoxins. In: Safety Evaluation of Certain Food Additives and Contaminants, prepared by the Forty-ninth Meeting of the Joint FAO/WHO Expert Committee. FAO/WHO Food Additives Series 1999;40:361–468.
Commission Regulation (EU) No. 165/2010 of 26 February 2010 amending Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuff as regards aflatoxins, Off J Eur Union, L 50, 8–12.
Commission Regulation (EC) No 1126/2007 of 28 September 2007 amending Regulation (EC) No 1881/2006 setting maximum levels for certain contaminants in foodstuff as regards Fusarium toxins in maize and maize products, Off J Eur Union. L 255, 14–17.
Commission Recommendation (EU) No 165/2013 of 27 March 2013 on the presence of T-2 and HT-2 toxin in cereals and cereal products. Off J Eur Union, L 91, 12–15.
Imperato R, Campone C, Piccinelli AL, Veneziano A, Rastrelli L. Survey of aflatoxins and ochratoxin a contamination in food products imported in Italy. Food Control. 2011;22:1905–10.
Ruadrew S, Craft J, Aidoo K. Occurrence of toxigenic Aspergillus spp. and aflatoxins in selected food commodities of Asian origin sourced in the west of Scotland. Food Chem Toxicol. 2013;55:653–8.
Vaclavikova M, MacMahon S, Zhang K, Begley TH. Application of single immunoaffinity clean-up for simultaneous determination of regulated mycotoxins in cereals and nuts. Talanta. 2013;117:345–51.
Zhang Z, Hu X, Zhang Q, Li P. Determination for multiple mycotoxins in agricultural products using HPLC-MS/MS via a multiple antibody immunoaffinity column. J Chrom B. 2016;1021:145–52.
Lattanzio VMT, Ciasca B, Powers S, Visconti A. Improved method for the simultaneous determination of aflatoxins, ochratoxin A and fusarium toxins in cereals and derived product by liquid chromatography—tandem mass spectrometry after multi-toxin immunoaffinity clean up. J Chrom A. 2014;1354:139–43.
Berthiller F, Sulyok M, Krska R, Schuhmacher R. Chromatographic methods for the simultaneous determination of mycotoxins and their conjugates in cereals. Int J Food Microbiol. 2007;119:33–7.
Soleimany F, Jinap S, Abas F. Determination of mycotoxins in cereals by liquid chromatography tandem mass spectrometry. Food Chem. 2012;130:1055–60.
Oueslati S, Romero-González R, Lasram S, Garrido Frenich A, Martínez Vidal JL. Multi-mycotoxin determination in cereals and derived products marketed in Tunisia using ultra-high performance liquid chromatography coupled to triple quadrupole mass spectrometry. Food Chem Toxicol. 2012;50:2376–81.
Alkadri D, Rubert J, Prodi A, Pisi A, Mañes J, Soler C. Natural co-occurrence of mycotoxins in wheat grains from Italy and Syria. Food Chem. 2014;157:111–8.
Commission Regulation (EU) No 519/2014 of 16 May 2014 amending Regulation (EC) No 401/2006 as regards methods of sampling of large lots, spices and food supplements, performance criteria for T-2, HT-2 toxin and citrinin and screening methods of analysis. Off J Eur Union, L 147, 29–43.
Bueno D, Istamboulie G, Muñoz R, Marty JL. Determination of mycotoxins in food: a review of bioanalytical to analytical methods. Appl Spectrosc Rev. 2015;50(9):728–74.
Köppen R, Koch M, Siegel D, Merkel S, Maul R, Nehls I. Determination of mycotoxins in foods: current state of analytical methods and limitations. Appl Microbiol Biotechnol. 2010;86(6):1595–612.
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and dispersive solid-phase extraction for the determination of pesticide residues in produce. JAOAC Int. 2003;86(2):412–31.
Rubert J, Dzuman Z, Vaclavikova M, Zacharisova M, Soler C, Hajslova J. Analysis of mycotoxins in barley using ultra high liquid chromatography high resolution mass spectrometry: comparison of efficiency and efficacy of different extraction procedures. Talanta. 2012;99:712–9.
Regulation (EC) No 882/2004 of the European Parliament and of the Council of 29 April 2004 on official controls performed to ensure the verification of compliance with feed and food law, animal health and animal welfare rules. Off J Eur Communities, L191, 1–64.
Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of result. Off J Eur Union, L221, 8–36.
Antignac JP, De Wasch K, Monteau F, De Brabander H, Andre F, Le Bizec B. The ion suppression phenomenon in liquid chromatography–mass spectrometry and its consequences in the field of residue analysis. Anal Chim Acta. 2005;529:129–36.
EURACHEM/CITAC Guide. Quantifying uncertainty in analytical measurement, laboratory of government chemist. 3rd ed. 2012. pp. 1–133.
Vidal A, Marin S, Ramos AJ, Cano-Sancho G, Sanchis V. Determination of aflatoxins, deoxynivalenol, ochratoxin a and zearalenone in wheat and oat based bran supplements sold in the Spanish market. Food Chem Toxicol. 2013;53:133–8.
Liu Y, Lu Y, Wang L, Chang F, Yang L. Survey of 11 mycotoxins in wheat flour in Hebei province, China. Food Addit Contam B. 2015;8:250–4.
European Food Safety Authority. Aflatoxins (sum of B1, B2, G1, G2) in cereals and cereal-derived food products. Supporting publications. 2013;EN:406, pp. 1–11.
Trung TS, Tabuc C, Bailly S, Querin A, Guerre P, Bailly JD. Fungal mycoflora and contamination of maize from Vietnam with aflatoxin B1 and fumonisin B1. World Mycotoxin J. 2008;1(1):87–94.
Warth B, Parich A, Atehnkeng J, Bandyopadhyay R, Schuhmacher R, Sulyok M, et al. Quantitation of mycotoxins in food and feed from Burkina Faso and Mozambique using a modern LC-MS/MS multitoxin method. J Agr Food Chem. 2012;60:9352–63.
Giray B, Girgin G, Engin AB, Aydin S, Sahin G. Aflatoxin levels in wheat samples consumed in some regions of Turkey. Food Control. 2007;18:23–9.
Perenzin M, Cattaneo M, Rizzi E, Pedrotti A, Tonesi R. Frumento tenero biologico: Risultati agronomici e qualitativi. L’Informatore agrario. 2001;33:31-3.
Zinedine A, Juan C, Soriano JM, Moltó JC, Idrissi L, Mañes J. Limited survey for the occurrence of aflatoxins in cereals and poultry feeds from Rabat, Morocco. Int J Food Microbiol. 2007;115:124–7.
Zinedine A, Brera C, Elakhdari S, Catano C, Debegnach F, Angelini S, et al. Natural occurrence of mycotoxins in cereals and spices commercialized in Morocco. Food Control. 2006;17:868–74.
Schothorst RC, van Egmond HP. Report from SCOOP task 3.2.10 collection of occurrence data of Fusarium toxins in food and assessment of dietary intake by the population of EU member states, subtask: trichothecenes. Toxicol Lett. 2004;153:133–43.
Visconti A, Doko MB. Survey of fumonisin production by Fusarium isolated from cereals in Europe. J AOAC. 1994;77:546–50.
Bottalico A. Fusarium diseases of cereals: species complex and related mycotoxins profiles, in Europe. J Plant Pathol. 1998;80:85–103.
Shuaib FMB, Ehiri J, Abdullahi A, Williams JH, Jolly PE. Reproductive health effects of aflatoxins: a review of the literature. Reprod Toxicol. 2010;29:262–70.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Annunziata, L., Stramenga, A., Visciano, P. et al. Simultaneous determination of aflatoxins, T-2 and HT-2 toxins, and fumonisins in cereal-derived products by QuEChERS extraction coupled with LC-MS/MS. Anal Bioanal Chem 409, 5143–5155 (2017). https://doi.org/10.1007/s00216-017-0462-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0462-z