
Abstract
This paper reports the production of a novel magnetic nanocomposite based on multi-walled carbon nanotubes (MWCNTs) decorated with magnetic core–shell Fe3O4@SiO2 nanoparticles which were used to fabricate a modified carbon paste electrode (Fe3O4@SiO2/MWCNT-CPE). The Fe3O4@SiO2/MWCNT-CPE was investigated for the simultaneous determination of sunset yellow (SY) and tartrazine (TT) in 0.1 mol/L phosphate buffer solution (pH 6.0) using square wave voltammetry (SWV). The synthesized nanocomposite was characterized by transmission electron microscopy, scanning electron microscopy, energy dispersive spectroscopy and Fourier transform infrared spectroscopy. The proposed electrode exhibits linear ranges of 0.5–100 μmol/L SY and TT with detection limits of 0.05 and 0.04 μmol/L for SY and TT, respectively. The novel proposed voltammetric method was successfully applied in the simultaneous determination of SY and TT in food products, with results similar to those obtained using a HPLC method at 95 % confidence level.

A magnetic nanocomposite based on MWCNTs decorated with core–shell Fe3O4@SiO2 was prepared and showed good ability to distinguish the response of SY and TT
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Color is a vital constituent of foods and probably the first characteristic perceived by the human senses. Almost all foods—from raw agricultural commodities to finished products—have an associated color. Further, many tests have shown that color can organoleptically dominate the flavor of a food. Today, synthetic dyes have widely replaced natural colors. These dyes are used to supplement and enhance the natural colors destroyed during processing or storage, and substantially increase the appeal and acceptability of foodstuffs where no natural colors exist, for example, soft drinks or ice cream. But, synthetic dyes are also used to mask decay, to redye food, to mask aging effects, or to disguise poor products. Colors permitted for food use can be divided in three categories: (1) synthetic dyes, (2) natural colors (for example, caramel, or beetroot) and naturally identical colors (canthaxanthine), and (3) inorganic pigments. Synthetic colors in food products are predominantly azo and triarylmethane dyes (Schuster and Gratzfeld-Hüsgen 1995). These colorants are present in many common food products, such as sugar candies, sugar-coated pills, jelly beans, powdered drinks, sweets, nutrient-enhanced sports beverages, ice creams, and gelatins (Nevado et al. 1997). On the other hand, some of these substances pose potential risks to human health, especially if they are excessively consumed. For instance, some synthetic azo-dyes can be harmful to human health and when in contact with some drugs can cause allergic and asthmatic reactions, as well as induce the development of cancer and other diseases. Consequently, the use of synthetic colorants in food products is strictly controlled by different national legislations (European Parliament and Council Directive 94/36/EC 1994; Medeiros et al. 2012a, b).
For analysis, food colorants usually have to be extracted quantitatively from solid food matrices. Depending on the kind of food and chemical structures of colorants used, simple extraction with water (Berzas Nevado et al. 1998a, b), ion-pair extraction (Lau et al. 1995; Lehmann et al. 1986), or adsorption on solid materials, such as wool threads (Thaler and Sommer 1953), alumina (Maslowska and Janiak 1997), octadecylsilyl silica (Lehmann et al. 1970), or polyamide (Ni and Gong 1997; Sabir et al. 1980), have been described. In the latter cases, desorption of colorants using mixtures of solvents makes possible their analysis.
Quantitative analysis of colorants resulting from these procedures can be performed along various routes. Spectrophotometry allows simultaneous quantitative analyses of mixtures of colorants having similar absorption spectra (Capitán-Vallvey et al. 1998; Bozdogan et al. 2000). The use of high-performance liquid chromatography (HPLC) was also reported for quantification of colorants. Ion-exchange HPLC (Chen et al. 1998), ion-pair reversed-phase chromatography (Kiseleva et al. 2003; Gennaro et al. 1997; DeVilliers et al. 2003), or reversed-phase chromatography (Chaytor and Heal 1986; Prado and Godoy 2002; Berzas Nevado et al. 1998a, b; Kirschbaum et al. 2003) have been described. Recently, capillary zone electrophoresis (Berzas Nevado et al. 1999; Masár et al. 1996) and micellar electrokinetic capillary chromatography (Desiderio et al. 1998; Chou et al. 2002) have been shown to be suitable for analysis of colorants.
Some electrochemical approaches for determining these food colorants have been previously reported (Nevado et al. 1997; Medeiros et al. 2012a, b; Song et al. 2010; Song 2010; Silva et al. 2007; Ghoreishi et al. 2011; López-de-Alba et al. 2002; Medeiros et al. 2012a, b; Ni and Bai 1997; Kapor et al. 2001). Song et al. (2010) studied the electrochemical reduction of TT at a MWCNT-modified pyrolytic graphite electrode; the validity of the proposed method was assessed by determining TT in soft drinks, in which a detection limit (LOD) of 0.5 mg/L (0.9 μmol/L) was obtained. Song (2010) also used a MWCNT-modified glassy carbon electrode to investigate the electrochemical reduction of SY by cyclic voltammetry (CV). The proposed method was satisfactorily applied in the determination of SY in soft drinks in two concentration ranges, with a lower LOD value of 0.5 mg/L. Quite recently, Ghoreishi et al. (2011) used a carbon paste electrode chemically modified with MWCNTs for the determination of brilliant blue (BB) using differential pulse voltammetry (DPV) or SWV, in the concentration range 0.05–22.0 μmol/L.
Moreover, few electroanalytical methods have been reported for the simultaneous determination of food colorants. Medeiros et al. (2012a, b) used DPV and a cathodically pretreated boron-doped diamond electrode to simultaneously determine two pairs of synthetic food colorants commonly found mixed in food products: TT and SY or BB and SY. The proposed method was applied in the simultaneous determination of these synthetic colorants in food products, with results similar to those obtained using a HPLC method at 95 % confidence level. Kapor et al. (2001) used a hanging mercury drop electrode for the simultaneous voltammetric determination of TT and indigo carmine in soft drinks and candies, using their cathodic redissolution voltammetric waves. Ni and Bai (1997) reported the use of a ratio derivative voltammetric method for resolving overlapping voltammograms without a pre-separation; this method was successfully applied to resolve binary mixtures of amaranth and SY.
Nanomaterials have drawn tremendous attention along with their unique physical and chemical properties. Several methods have been tried to obtain excellent nanocomposite containing kinds of nanomaterials, which can combine the advantages of them (Yan et al. 2008). Up to now, carbon nanotubes (CNTs) are one of the best candidates for the preparation of nanocomposite materials.
At present, significant interest has been generated in research that mixes CNTs with different materials such as metals, metal oxides, complex metal oxides, and polymers (Vasilios et al. 2005; Cao et al. 2006; Sepahvand et al. 2008) by physical and chemical approaches to prepare multifunctional nanocomposites. Nanoparticulate hybrid materials made from inorganic solids and CNTs are highly promising for applications in nanoscale devices and nanoelectronics (Campbell and Compton 2010; Yanez-Sedeno et al. 2010).
Magnetic nanoparticles (MNPs) have many superior characteristics such as small size, easy synthesis, high surface area, high coercivity, facile separation by magnetic forces, as well as low toxicity and low cost, and therefore, have widely applications in a variety of fields such as catalysis, bioseparation, bioengineering, magnetic separation, and sensors (Gardimalla et al. 2005; Shahrokhian et al. 2012). Magnetite (Fe3O4) nanoparticles are one of the magnetic nanoparticles in common use due to their good biocompatibility, strong super paramagnetic property, low toxicity, easy preparation, and high adsorption ability. Fe3O4 nanoparticles exhibit high surface area and low mass transfer resistance. However, magnetic nanoparticles are facile agglomerates, easily oxidized in air, and unstable in aqueous solution during the synthesis and applications (Lu et al. 2007). For many applications, it is thus crucial to develop protection strategies to chemically stabilize the naked magnetic nanoparticles. Silica (SiO2) has been known as one of the most ideal coating layers for Fe3O4 nanoparticles due to its reliable chemical stability, rich surface chemistry, high thermal stability, simplicity of preparation, and low-temperature encapsulation compatibility and reactivity with various coupling agents (Hu et al. 2011). Currently, there is extensive interest in the fabrication of core–shell particles with unique and tailored properties for various applications in material science (Hu et al. 2013). These strategies comprise grafting of or coating with organic species, coating with an inorganic layer, such as silica or carbon (Chang et al. 2012). Therefore, investigations on the synthesis of magnetic core–shell particles have gained much attention due to the harvest of combined advantages of synergistic structural/electronic effects of core–shell structure and the inherent properties of the magnetic element.
Decoration of carbon-based materials with magnetic nanoparticles, such as coating or loading CNTs with Fe3O4 (Cao et al. 2006; Jia et al. 2007), can combine the advantages of the CNTs and magnetite nanoparticles. On the other hand, decoration of CNTs with magnetic iron oxide and preparing magnetic nanocomposites are expected to exhibit different physical properties than those of neat nanotubes (Qian et al. 2010; Peng et al. 2009).
In the present study, in order to couple the multiwall carbon nanotubes (MWCNTs) with the core–shell magnetic nanoparticles properties of Fe3O4@SiO2, a new, simple, and cheap nanocomposite of MWCNTs decorated with Fe3O4@SiO2 nanoparticles (Fe3O4@SiO2/MWCNTs) was prepared and used as a carbon paste electrode modifier. To the best of our knowledge, no study has reported the simultaneous determination of TT and SY by using the core–shell Fe3O4@SiO2/MWCNTs nanocomposite carbon paste electrode (Fe3O4@SiO2/MWCNTs-CPE). So, in continuation of our studies, the suitability of Fe3O4@SiO2/MWCNTs-CPE as a working electrode in electrochemical methods was used for simultaneous determination of SY and TT in food products without expensive and time-consuming pretreatments.
Experimental
Reagents and Solutions
Multi-walled carbon nanotubes (purity >95 %) was purchased from US Research Nanomaterials, Inc. Sunset yellow, tartrazine (Scheme 1), and graphite powder (average particle size 30 μm) were from Aldrich (St. Louis, MO, USA). Standard 1.0 mmol/L SY and TT solutions were prepared with a 0.1 mol/L phosphate buffer solution (PBS, pH 6.0). Ferric chloride (FeCl3.6H2O), ferrous sulphate (FeSO4.7H2O), tetraethoxysilane (TEOS), and ammonium hydroxide (NH4OH) were from Fluka (Buchs, Switzerland). All other reagents and solvents were purchased from Aldrich or Merck and used without further purification. Aqueous solutions of colorants were prepared fresh at the time of experiments. Double distilled water was used in the experiments.

Chemical structure of the tartrazine and sunset yellow
Apparatus
Electrochemical measurements were performed with Autolab potentiostat PGSTAT 30 (Eco Chemie BV, Utrecht, the Netherlands) driven by the General Purpose Electrochemical Systems data processing software (software version 4.9). A conventional three-electrode system was used, consisted of a working electrode (unmodified or modified), a saturated Ag/AgCl reference electrode and a platinum wire counter electrode, respectively. All the pH values were measured with a Metrohm pH meter (model 827, Swiss made).
Transmission electron microscopic (TEM) images were obtained by using (TEM; Philips -model CM10). The morphology of Fe3O4@SiO2/MWCNTs nanocomposite was investigated by TEM and scanning electron microscopy (SEM) along with energy-dispersive X-ray spectroscopy (EDX; Electroanalyzer Sama 500). Fourier transform infrared (FT–IR) spectra were recorded in KBr pellets in the frequency range of 400–4,000 cm−1 using Tensor 27, Bruker.
Chromatographic determinations of SY and TT were carried out using a Agilent 1100 series HPLC (Germany) consisting of a gradient pump, a YMC Pack ODS-AM (5 μm, 250 mm × 4.6 mm ID) end-capped column coupled with an UV–Vis detector and computer. The eluent consisted of an aqueous 0.13 mol/L ammonium acetate solution, brought to pH 7.5 by the drop wise addition of a 1.5 mol/L sodium hydroxide solution (mobile phase A), and a mixture of (80:20 v/v) methanol:acetonitrile (mobile phase B) at a flow rate of 1.0 mL/min, while the injection volume was 20 μL. The detections were performed at 484 and 430 nm for SY and TT, respectively. All separations were carried out at 25 ± 1 °C (Minioti et al. 2007).
Synthesis of Fe3O4@SiO2/MWCNTs Nanocomposite
The preparation process of Fe3O4@SiO2/MWCNTs nanocomposite was according to our previous report (Arvand and Hassannezhad 2014). Briefly, the carboxylated MWCNTs (MWCNTs–COOH) were prepared by refluxing the MWCNTs in a mixture of concentrated sulfuric acid and nitric acid (3:1 v/v ratio) at 70 °C for 6 h. After cooling to room temperature the mixture was filtered and washed with double distilled water until pH of filtrate became ∼7 and finally dried at 60 °C overnight.
About 0.1 g of MWCNTs–COOH was dissolved in 70 mL of water by ultrasonic irradiation (Sono Swiss SW3-H, 38 kHz, Switzerland) for 20 min. The mixture was further stirred vigorously for 30 min at 60 °C. Then 177 mg of FeCl3.6H2O was added under stirring. After the mixture was stirred vigorously for 30 min under N2 atmosphere, 95 mg of FeSO4.7H2O was added and keep stirring under N2 atmosphere for 30 min. At last, 30 mL of 6 % NH4OH aqueous solution was added into the mixture drop by drop at 60 °C during 1 h and reacted for another 2 h. N2 atmosphere was used during the reaction to prevent critical oxidation. The reaction mixture was then centrifuged, washed with double distilled water, and dried. The obtained black precipitate was Fe3O4/MWCNTs nanoparticles and was ready for use.
Core–shell Fe3O4@SiO2/MWCNTs nanocomposites were prepared by growing silica layers onto the surface of the Fe3O4/MWCNTs as described by Lu et al. (2008). Twenty-five milliliters of ethanol, 1 mL water, 1 mL ammonium hydroxide, and 150 μL of TEOS were added in a 250-mL three-neck flask in a 40 °C water bath. Fe3O4/MWCNTs were added to the above solution under mechanical stirring. Aliquots of the mixture were takes out after 12 h by centrifugation and washed with water and vacuum-dried at 60 °C overnight.
Preparation of Working Electrodes
Prior to preparation of modified electrodes, glassy carbon electrode (as electrical contact) was carefully polished with alumina until a mirror finish was obtained. The electrode was rinsed thoroughly with distilled water. The prepared electrode was dried and used immediately for modification. Graphite powder (0.06 g) and 0.006 g Fe3O4@SiO2/MWCNTs nanocomposite were blended by hand in a mortar with a pestle. Two drops of paraffin oil were added to the above mixture with a syringe and mixed well to obtain a modified uniform wetted paste. A portion of this modified carbon paste was packed into the end of a Teflon tube with a hole (2 mm in diameter and 3 mm deep). Electrical contact was made by forcing a glassy carbon electrode down the Teflon tube and into the back of the paste to prepare the Fe3O4@SiO2/MWCNTs-CPE. The new surface was obtained by pushing an excess of paste out of the tube and polishing it on a piece of weighing paper just before use. Unmodified carbon paste electrode was prepared in the same way, in the absence of Fe3O4@SiO2/MWCNTs nanocomposite.
Treatment of Commercial Food Samples
All samples (powdered juice drinks, gelatins, and nutrient enhanced sports drink beverages), obtained from a local supermarket, were pretreated as described in other similar works (Medeiros et al. 2012a, b; Minioti et al. 2007; Vidotti et al. 2006). The solid samples were homogenized. One gram portions of powdered juice drinks or gelatin were accurately weighed and dissolved in 10.0 mL of a 0.1 mol/L H2SO4 solution. To dissolve the gelatin, the sulfuric acid solution was gently heated. Each sample solution was placed in an ultrasonic bath for 10 min for complete extraction of the colorants. This solution was then filtered through a folded paper filter. Subsequently, 0.5 mL aliquots of this solution were transferred to the electrochemical cell containing 9.5 mL of a 0.1 mol/L PBS (pH 6.0). In the case of the samples of sports drink beverages, 0.5 mL aliquots were directly transferred to the electrochemical cell containing 9.5 mL of a 0.1 mol/L PBS. Voltammograms were obtained after each aliquot addition.
Results and Discussion
FT–IR Characterization
Figure 1 shows the FT–IR spectra of (a) MWCNTs–COOH, (b) Fe3O4/MWCNTs, and (c) Fe3O4@SiO2/MWCNTs. In Fig. 1(a), the peaks at 1,636 and 1,381 cm−1 correspond to C=O and C–O stretching, respectively. The two weak peaks at 2,928 and 2,850 cm−1 correspond to the –CH stretching mode and a broad band peak at 3,439 cm−1 are attributed to the COOH groups onto the external surface of MWCNTs (Singh et al. 2008). In addition, in Fig. 1(b), the peak at 605 cm−1 is the stretching vibration due to the interactions of Fe–O–Fe in Fe3O4. Compared with the two spectra (b and c), the existence of the characteristic Si–O–Si peak at 1,080 cm−1 in Fig. 1(c) is direct evidence to verify the formation of the silica shell (Baby and Ramaprabhu 2010).

FT–IR spectra of a MWCNTs-COOH, b Fe3O4/MWCNTs, and c Fe3O4@SiO2/MWCNTs
Morphology of Fe3O4@SiO2/MWCNTs Nanocomposite
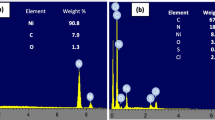
The structural morphology of the Fe3O4@SiO2/MWCNTs nanocomposite was examined by TEM and SEM. Figure 2(a) shows the typical TEM image of the core–shell Fe3O4@SiO2/MWCNTs nanocomposite, which demonstrates that the core–shell Fe3O4@SiO2 nanoparticles with black color shows Fe3O4 and ash color is the SiO2 shell, was uniformly attached to the surface of MWCNTs. In addition, Fig. 2a shows that the surface morphology of MWCNTs is different when MWCNTs are decorated with Fe3O4@SiO2 nanoparticles.

TEM image of Fe3O4@SiO2/MWCNTs nanocomposite (a), SEM images of Fe3O4@SiO2/MWCNTs nanocomposite (b), Fe3O4@SiO2/MWCNTs-carbon paste (d), and EDS spectrum (c) taken from Fe3O4@SiO2/MWCNTs nanocomposite
Figure 2b displays a typical morphology of Fe3O4@SiO2/MWCNTs nanocomposite characterized by SEM. This figure shows that Fe3O4@SiO2 nanoparticles well distributed on the surface of MWCNTs and it did not change the morphology of MWCNTs. However, it can be clearly seen that SiO2 coated Fe3O4 magnetic nanoparticles were dispersed in MWCNTs, thus enhancing the effective surface area. The EDS shown in Fig. 2(c) confirms the presence of iron and silicon in the Fe3O4@SiO2/MWCNTs nanocomposite. Also, the surface morphology of modified carbon paste with Fe3O4@SiO2/MWCNTs nanocomposite was investigated by SEM and depicted in Fig. 2(d).
Cyclic Voltammetry Characterization of Modified Electrodes
Electrochemical sensing characteristics of the CPE, MWCNTs-CPE, and Fe3O4@SiO2/MWCNTs-CPE were studied with a solution of 5 mmol/L K3[Fe(CN)6] probe at scan rate of 0.1 V/s. As seen in Fig. 3, a pair of reversible redox peaks with separation of peak potential (ΔE p) of 0.503, 0.803, and 0.109 V were observed at (a) CPE, (b) MWCNTs-CPE, and (c) Fe3O4@SiO2/MWCNTs-CPE, respectively. The anodic peak current (I pa) of Fe3O4@SiO2/MWCNTs-CPE was found to be 2.7 and about 4.6 times higher than MWCNTs-CPE and CPE, respectively. This behavior can be attributed to the fact that Fe3O4@SiO2/MWCNTs-CPE exhibits faster electron-transfer kinetics and larger electroactive surface area compared to MWCNTs-CPE and CPE.

CVs of 5 mmol/L K3Fe(CN)6 at CPE (dotted line), MWCNTs-CPE (dashed line), and Fe3O4@SiO2/MWCNTs-CPE (solid line). Scan rate, 0.1 V/s
The effective surface area of the CPE, MWCNTs-CPE, and Fe3O4@SiO2/MWCNTs-CPE were estimated by the K3[Fe(CN)6] redox system. For a reversible process, the Randles–Sevcik formula is as follows:
where I p refers to the anodic peak current, n is the total number of electron transferred (n = 1), A is the effective surface area of the electrode, D is the diffusion coefficient for K3[Fe(CN)6] = 7.6 × 10−6 cm2/s, C * is the concentration of K3[Fe(CN)6], and ν is the scan rate. The electrochemically effective surface areas (A eff) of CPE, MWCNTs-CPE, and Fe3O4@SiO2/MWCNTs-CPE were calculated 0.01, 0.02, and 0.06 cm2, respectively.
Cyclic Voltammetric Studies of SY and TT on Fe3O4@SiO2/MWCNTs-CPE
Figure 4(a) illustrates the cyclic voltammograms of SY and TT at a concentration of 85 μmol/L on Fe3O4@SiO2/MWCNTs-CPE in PBS (pH 6.0), within the potential window of −0.05–−0.45 V and at scan rate of 0.1 V/s. According to cyclic voltammograms in Fig. 4(a), SY and TT showed well-behaved reduction peaks at the Fe3O4@SiO2/MWCNTs-CPE. The reduction of SY and TT on the modified electrode is an electrochemically irreversible process. Furthermore, SY and TT reduction at presented modified electrode results in a well-defined and peak-shaped signal at about −0.2 and −0.31 V, respectively.

a CVs of the Fe3O4@SiO2/MWCNTs-CPE in presence of SY (blue line), TT (green line), and SY and TT mixture (red line) in 0.1 mol/L PBS. b CVs of SY and TT mixture at CPE (dotted line), MWCNTs-CPE (dashed line), and Fe3O4@SiO2/MWCNTs-CPE (solid line). Concentration of each analyte is 85 μmol/L; E acc = 0.2 V; t acc = 180 s; scan rate, 0.1 V/s
Cyclic voltammetric response for the mixture of SY and TT at the CPE, MWCNTs-CPE, and Fe3O4@SiO2/MWCNTs-CPE in the presence of 85 μmol/L SY and TT (PBS, pH 6.0) are shown in Fig. 4(b). As can be seen, SY and TT showed a reduction peak at the CPE electrode (dotted line) with reduction potential of −0.23 and −0.35 V, respectively. In contrast, at MWCNTs-CPE (dashed line), the peak currents increased and the cathodic peak potential of SY and TT were about −0.21 and −0.31 V, respectively. The reduction of two molecules became facile on the MWCNTs-CPE. According to Fig. 4(b), the peak currents of SY and TT at Fe3O4@SiO2/MWCNTs-CPE (solid line) are remarkably enhanced compared to the CPE and MWCNTs-CPE. These results indicated that Fe3O4@SiO2/MWCNTs nanocomposite had great improvement to the increase of the electro-chemical response, which was due to excellent characteristics of nanocomposite such as good electrical conductivity, high surface area, and more electroactive interaction sites that can provide increased mass transport and easier accessibility to the active sites. Thus, the simultaneous determination of SY and TT is feasible at Fe3O4@SiO2/MWCNTs-CPE.
Effect of Solution pH
In most cases, the supporting electrolyte pH is one of the important parameters in the electrochemical reaction. Hence, standard solutions of SY and TT at a concentration of 85 μmol/L were used to find the optimum pH of the supporting electrolyte best suited for determination of SY and TT by Fe3O4@SiO2/MWCNTs-CPE. The effect of solution pH on the electrochemical response of the Fe3O4@SiO2/MWCNTs-CPE towards the simultaneous determination of SY and TT was investigated in the pH range of 4.5–8.0 applying phosphate buffer solution (Fig. 5(a)). It was observed that the peak current (I pc) reached its maximum value at pH 5.0 for SY and 6.0 for TT reduction (Fig. 5(b)). Thus, pH = 6.0 was employed for further studies due to the proximity to physiological pH (6–7).

a CVs of 85 μmol/L SY and TT at the Fe3O4@SiO2/MWCNTs-CPE in PBS with different pH values (4.5, 5.0, 6.0, 7.0, and 8.0). b Dependence of cathodic peak currents and c cathodic peak potentials as a function of solution pH
Based on the related Refs. (Song et al. 2010; Medeiros et al. 2012a, b), the electrochemical reduction of SY and TT involves a four-proton and four-electron process. In this study, it is observed that the reduction peak potential of SY and TT shift positively with the proton concentration in the range of 4.5–8.0, and the pH dependence of E pc can be expressed by the linear regression equations as: E pc = 0.062pH – 0.583, R 2 = 0.997 and E pc = 0.057pH – 0.662, R 2 = 0.998 for SY and TT, respectively (Fig. 5(c)). The calculated slope of 0.062 (SY) and 0.057 V (TT) are close to the theoretical value of 0.0586 V according to the Nernst equation, suggesting that the electrochemical reduction of SY and TT at the proposed electrode is equal proton–electron process (Scheme 2).

Reduction mechanism for the SY and TT colorants at the Fe3O4@SiO2/MWCNTs-CPE surface
Effect of Scan Rate
To obtain the kinetic parameters, the cyclic voltammograms of Fe3O4@SiO2/MWCNTs-CPE in the PBS solution at pH 6.0 in the presence of 85 μmol/L SY and TT were recorded at various scan rates (Fig. 6(a)). As shown in Fig. 6b, the reduction peak currents of SY and TT increase linearly with scan rates in the range from 0.01 to 0.28 V/s, suggesting that electrode reactions of SY and TT on the Fe3O4@SiO2/MWCNTs-CPE are controlled by adsorption. The linear regression equations are expressed as, I pc (μA) = 13.21ν (V/s) + 0.491, R 2 = 0.992 and I pc (μA) = 14.93ν (V/s) + 0.051, R 2 = 0.991 for SY and TT, respectively. In addition, there was a linear relation between logI pc and logν, corresponding to the following equations: logI pc (μA) = 0.712logν (V/s) + 0.997, R 2 = 0.990 and logI pc (μA) = 0.958logν (V/s) + 1.146, R 2 = 0.988 for SY and TT, respectively (Fig. 6(c)). The slopes of 0.712 (SY) and 0.958 (TT) are close to the theoretically expected value of 1.0 for an adsorption controlled process.

a CVs of the reduction 85 μmol/L SY and TT at the Fe3O4@SiO2/MWCNTs-CPE in 0.1 mol/L PBS (pH 6.0) with different scan rates, 0.01, 0.02, 0.04, 0.06, 0.08, 0.1, 0.12, 0.14, 0.16, 0.18, 0.2, 0.22, 0.24, 0.26, and 0.28 V/s. b Relationship between cathodic peak current and potential scan rate. c Variation of the logIpc vs. logν. d Variation of the Epc vs. logν
For an irreversible and adsorption-controlled reduction process, the electron transfer rate constant (k s) can be estimated using the Laviron’s equations (Laviron 1979). As shown in Fig. 6d, plotting the E pc vs. logν produces a straight line with the slope of 2.3RT/(1 − α)nF, and the linear regression equations are expressed as: E pc (V) = −0.039logν – 0.240, R 2 = 0.984 and E pc (V) = −0.064logν – 0.388, R 2 = 0.993 for SY and TT, respectively. From the value of the slope, the electron transfer coefficient (α) is estimated to be 0.39 (SY) and 0.54 (TT). According to the Laviron equation:
the k s is calculated to be 0.12 and 0.10 s−1 for SY and TT, respectively. These results demonstrate that the Fe3O4@SiO2/MWCNTs nanocomposite significantly facilitates the electron transfer kinetics and promotes the electrochemical reduction of SY and TT.
Effect of Accumulation Potential and Accumulation Time
Accumulation step is usually a simple and effective way to enhance the determining sensitivity. Since the electrode process of SY and TT is adsorption controlled on the Fe3O4@SiO2/MWCNTs-CPE, it is very important to investigate the effects of accumulation potential (E acc) and accumulation time (t acc). Keeping the accumulation time as 180 s at a concentration of 100 μmol/L SY and TT, the accumulation potential was investigated in the range of 0.5–−0.1 V. The results prove that varying accumulation potential shows the influence on the increase of SY and TT reduction current (Fig. 7(a)). The peak current reached its maximum value at an accumulation potential of 0.2. Herein, 0.2 V was chosen as the optimum accumulation potential. Keeping E acc = 0.2 V constant at the mixture of 100 μmol/L SY and TT, the reduction peak current of SY and TT gradually increased with increasing accumulation time from 0.0 to 180 s, and reached the maximum current response at 180 s (Fig. 7(b)). Also, there is no significant increase in the current response when increasing the accumulation time. This phenomenon is probably due to the saturated adsorption of SY and TT on the Fe3O4@SiO2/MWCNTs-CPE surface. Therefore, an accumulation time of 180 s was used for each voltammetric measurement of SY and TT.

a The dependence of cathodic peak current of SY and TT at a concentration of 100 μmol/L on accumulation potential in the range of 0.5–−0.1 V at Fe3O4@SiO2/MWCNTs-CPE, keeping the accumulation time as 180 s. b The effect of accumulation time on the cathodic peak current of SY and TT, keeping E acc = 0.2 V
Calibration Curve
The determination of SY and TT at Fe3O4@SiO2/MWCNTs-CPE was performed under the optimal SWV conditions. For this purpose, in a mixture of SY and TT, the concentration of one specie changes while the concentration of the other remains constant. The results are shown in Fig. 8. As shown in Fig. 8(a), the peak currents of SY increased with increasing the concentration of SY in the presence of TT. The same appears to be the case when TT coexists with a constant concentration of SY in the various concentrations (Fig. 8(b)). Simultaneous determination of various concentrations of SY and TT was carried out on the Fe3O4@SiO2/MWCNTs-CPE under the optimized conditions (Fig. 8(c)). The results and the analytical parameters including linear regression equation (LRE), correlation coefficient (r), linear working range (LWR), and limit of detection (LOD) for individual and simultaneous determination of SY and TT are reported in Table 1. Thus, it could be concluded that, by employing the proposed method, the simultaneous determination of two molecules was as efficient as their individual determinations.

a SWVs obtained at the Fe3O4@SiO2/MWCNTs-CPE for SY at different concentrations in the presence of 5.0 μmol/L TT. SY concentrations (from 0.5 to 100), 0.5. 0.8, 1.5, 2.5, 3.5, 4.5, 5.5, 6.5, 8, 10, 12, 14, 20, 30, 35, 45, 55, 65, 85, and 100 μmol/L. Inset calibration curve of SY. b SWVs obtained at the Fe3O4@SiO2/MWCNTs-CPE for TT at different concentrations in the presence of 5.0 μmol/L SY. TT concentrations (from 0.5 to 100), 0.5, 0.95, 1.4, 2.7, 4.5, 5.4, 6.3, 7.2, 8.3, 10, 14, 19, 28, 37, 46, 56, 71, 82, 91, and 100 μmol/L. Inset calibration curve of TT. Other conditions were as in Fig. 4. c SWVs obtained for the reduction of SY and TT at concentrations of (from 0.5 to 100), 0.5, 0.95, 2.5, 4, 6, 8, 10, 20, 30, 40, 50, 60, 75, 95, and 100 μmol/L. Other conditions were as in Fig. 4
Stability and Repeatability of the Modified Electrode
The stability and repeatability of the Fe3O4@SiO2/MWCNTs-CPE were also explored. The SWV experiments were carried out using the modified electrode. The cathodic peak current of SY and TT did not change for two weeks, showing an elevated stability of the Fe3O4@SiO2/MWCNTs-CPE. Also, the relative standard deviation (RSD) of the modified electrode in response to 5.0 μmol/L SY and TT for five measurements was 2.36 and 2.15 %, respectively, indicating the good repeatability of proposed modified electrode.
Interference Study
The influence interferences on voltammetric determination of SY and TT at the Fe3O4@SiO2/MWCNTs-CPE were investigated by SWV under the above optimized conditions. The limit of interference was taken as the maximum concentration of the foreign substances that caused an approximately ±10 % relative error in the determination of SY and TT. The results of these measurements are summarized in Table 2, which indicate that the concentrations of K+, Na+, Fe2+, Zn2+, Cu2+, Br−, I−, Cl−, and NO3 − and some coexistent substances have not significantly influenced the height of the peak currents.
Application of Fe3O4@SiO2/MWCNTs-CPE for Determination of SY and TT in Real Samples
From the preceding observation, it is apparent that Fe3O4@SiO2/MWCNTs-CPE possesses a high sensitivity and a good detection limit for the determination of SY and TT in an ideal laboratory sample. In order to test its practical application, this modified electrode was used to determine SY and TT in food products. The results are presented in Table 3. These results confirm the good precision and accuracy of the proposed method. By analyzing these results obtained for the commercial food products, one can conclude that the values obtained by our novel proposed method agree quite well with those obtained by the reference HPLC method. In fact, applying the paired t-test to the results obtained by both methods, the resulting t values are smaller than the critical one (2.78, α = 0.05), indicating that there is no difference between the obtained results at a confidence level of 95 %.
A comparison between the analytical performance of the present modified electrode and some previous literature electrodes for the determination of SY and TT are given in Table 4. It can be seen that the Fe3O4@SiO2/MWCNTs-CPE offers reasonable linear range for simultaneous detection of SY and TT and the LOD is lower than some of previous reports. These results certify that this electrochemical sensor for SY and TT determination is an appropriate and acceptable platform.
Conclusions
In this study, we reported the synthesis of a new magnetic nanocomposite of multiwall carbon nanotube, decorated with core–shell Fe3O4@SiO2 nanoparticles could be used as a modifier in carbon paste electrode and its application to simultaneous determination of SY and TT in real samples. The electrochemical behavior of these compounds has been studied by cyclic voltammetry and square wave voltammetry. Very wide linear dynamic ranges, very low detection limits, high sensitivity, very good repeatability, and high stability, together with simple procedures for preparation and determination can be presented as the advantages of the prepared electrode. Hence, the proposed electrode was used as a selective, simple, and precise voltammetric sensor for determination of SY and TT in food products.
References
Arvand M, Hassannezhad M (2014) Mater Sci Eng C 36:160–167
Baby TT, Ramaprabhu S (2010) Talanta 80:2016–2022
Berzas Nevado JJ, Guiberteau Cabanillas C, Contento Salcedo AM (1998a) Anal Lett 31:2513–2535
Berzas Nevado JJ, Guiberteau Cabanillas C, Contento Salcedo AM (1999) Anal Chim Acta 378:63–71
Berzas Nevado JJ, Rodríguez Flores J, Guiberteau Cabanillas C, Villaseñor Llerena MJ, Contento Salcedo A (1998b) Talanta 46:933–942
Bozdogan A, Ozgur MU, Koyuncu I (2000) Anal Lett 33:2975–2982
Campbell FW, Compton RG (2010) Anal Bioanal Chem 396:241–259
Cao H, Zhu M, Li Y (2006) J Solid State Chem 179:1208–1218
Capitán-Vallvey LF, Fernández MD, De Orbe I, Avidad R (1998) Talanta 47:861–868
Chang C, Wang X, Bai Y, Liu H (2012) Trends Anal Chem 39:195–206
Chaytor JP, Heal RL (1986) J Chromatogr 368:450–455
Chen QC, Mou SF, Hou XP, Riviello JM, Ni ZM (1998) J Chromatogr A 827:73–81
Chou SS, Lin YH, Cheng CC, Hwang DF (2002) J Food Sci 67:1314–1318
Desiderio C, Marra C, Fanali S (1998) Electrophoresis 19:1478–1483
DeVilliers A, Alberts F, Lynen F, Crouch A, Sandra P (2003) Chromatographia 58:393–397
European Parliament and Council Directive 94/36/EC (1994) OJEC L 237:13–29
Gan T, Sun J, Cao S, Gao F, Zhang Y, Yang Y (2012) Electrochim Acta 74:151–157
Gan T, Sun J, Meng W, Song L, Zhang Y (2013) Food Chem 141:3731–3737
Gardimalla HMR, Mandal D, Stevens PD, Yen M, Gao Y (2005) Chem Commun 35:4432–4434
Gennaro MC, Gioannini E, Angelino S, Aigotti R, Giacosa D (1997) J Chromatogr A 767:87–92
Ghoreishi SM, Behpour M, Golestaneh M (2011) Anal Methods 3:2842–2847
Ghoreishi SM, Behpour M, Golestaneh M (2012) Food Chem 132:637–641
Hu F, Chen S, Yuan R (2013) Sensors Actuators B 176:713–722
Hu Y, Li J, Zhang Z, Zhang H, Luo L, Yao S (2011) Anal Chim Acta 698:61–68
Jia BP, Gao L, Sun J (2007) Carbon 45:1476–1481
Kapor MA, Yamanaka H, Carneiro PA, Zanoni MVB (2001) Ecletica Quim 26:1–14
Kirschbaum J, Krause C, Pfalzgraf S, Brückner H (2003) Chromatographia Suppl 57:S115–S119
Kiseleva MG, Pimenova VV, Eller KI (2003) J Anal Chem 58:685–690
Lau OW, Poon MMK, Mok SC, Wong FMY, Luk SF (1995) Int J Food Sci Technol 30:793–798
Laviron E (1979) J Electroanal Chem 101:19–28
Lehmann G, Binkle B, Scheller A (1986) Fresenius J Anal Chem 323:355–358
Lehmann G, Collet P, Hahn HG, Ashworth MFR (1970) J AOAC 53:1182–1189
López-de-Alba PL, López-Martínez L, De-León-Rodríguez LM (2002) Electroanalysis 14:197–205
Lu Z, Dai J, Song X, Wang G, Yang W (2008) Colloids Surf A 317:450–456
Lu AH, Salabas EL, Schüth F (2007) Angew Chem Int Ed 46:1222–1244
Masár M, Kaniansky D, Madajová V (1996) J Chromatogr A 724:327–336
Maslowska J, Janiak J (1997) Dtsch Lebensm Rundsch 93:224–227
Medeiros RA, Lourencao BC, Rocha-Filho RC, Fatibello-Filho O (2012a) Talanta 99:883–889
Medeiros RA, Lourencao BC, Rocha-Filho RC, Fatibello-Filho O (2012b) Talanta 97:291–297
Minioti KS, Sakellariou CF, Thomaidis NS (2007) Anal Chim Acta 583:103–110
Nevado JJB, Flores JR, Llerena MJV (1997) Fresenius J Anal Chem 357:989–994
Ni YN, Bai JL (1997) Talanta 44:105–109
Ni Y, Gong X (1997) Anal Chim Acta 354:163–171
Peng X, Chen J, Misewich J, Wong S (2009) Chem Soc Rev 38:1076–1098
Prado MA, Godoy HT (2002) J Liq Chromatogr Relat Technol 25:2455–2472
Qian H, Greenhalgh ES, Shaffer M, Bismarck A (2010) J Mater Chem 20:4751–4762
Sabir DM, Edelhauser M, Bergner KG (1980) Dtsch Lebensm Rundsch 76:314–317
Schuster R, Gratzfeld-Hüsgen A (1995) Analysis of synthetic dyes in food samples by capillary zone electrophoresis. Agilent Technologies, Publication Number 5964-0280E
Sepahvand R, Adeli M, Astinchap B, Kabiri R (2008) J Nanoparticle Res 10:1309–1318
Shahrokhian S, Rastgar S, Amini MK, Adeli M (2012) Bioelectrochemistry 86:78–86
Silva MLS, Garcia MBQ, Lima JLFC, Barrado E (2007) Talanta 72:282–288
Singh BP, Singh D, Mathur RB, Dhami TL (2008) Nanoscale Res Lett 3:444–453
Song YZ (2010) Can J Chem 88:676–681
Song X, Shi Z, Tan X, Zhang S, Liu G, Wu K (2014) Sensors Actuators B 197:104–108
Song YZ, Xu JM, Lv JS, Zhong H, Ye Y, Xie JM (2010) Indian J Chem A 49:1030–1034
Thaler H, Sommer G (1953) Z Lebensm Unters Forsch A 97:441–446
Vasilios G, Vasilios T, Dimitrios G, Dimitrios P (2005) Chem Mater 17:1613–1617
Vidotti EC, Costa WF, Oliveira CC (2006) Talanta 68:516–521
Yan J, Zhou H, Yu P, Su L, Mao L (2008) Electrochem Commun 10:761–765
Yanez-Sedeno P, Riu J, Pingarron JM, Rius FX (2010) Trends Anal Chem 29:939–953
Yang X, Qin H, Gao M, Zhang H (2011) J Sci Food Agric 91:2821–2825
Ye X, Du Y, Lu D, Wang C (2013) Anal Chim Acta 779:22–34
Zhang W, Liu T, Zheng X, Huang W, Wan C (2009) Colloids Surf B 74:28–31
Acknowledgments
The authors are thankful to the post-graduate office of Guilan University for the support of this work.
Compliance with Ethical Standards
ᅟ
Funding
This study was funded by Guilan University.
Conflict of Interest
M. Arvand, Y. Parhizi, and S.H. Mirfathi declare that they have no conflict of interest.
Ethical Approval
All procedures performed in the studies were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This article does not contain any studies with humans and animals performed by any of the authors.
Informed Consent
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Arvand, M., Parhizi, Y. & Mirfathi, S.H. Simultaneous Voltammetric Determination of Synthetic Colorants in Foods Using a Magnetic Core–Shell Fe3O4@SiO2/MWCNTs Nanocomposite Modified Carbon Paste Electrode. Food Anal. Methods 9, 863–875 (2016). https://doi.org/10.1007/s12161-015-0253-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-015-0253-0