
Abstract
Background
Keratoameloblastoma is a poorly characterized and rarely reported odontogenic neoplasm that can exhibit overlapping histopathologic features with conventional ameloblastoma and keratocystic odontogenic tumor (KCOT), with an ambiguous relationship to the so-called solid KCOT.
Methods
A peripheral maxillary tumor causing bone saucerization in a 54-year-old male is described and investigated with immunohistochemistry and Next-Generation Sequencing (NGS).
Results
Microscopically, the tumor comprised of a predominantly plexiform proliferation of odontogenic epithelium with central keratinization and evidence of surface origin. Peripheral cells exhibited nuclear palisading with variable reverse polarization, while stellate reticulum-like areas were observed internally. A few follicles and a few foci in the lining of cystic spaces revealed increased cellularity with cells exhibiting small but conspicuous nucleoli, focal nuclear hyperchromatism, and a few mitoses mostly seen in the peripheral outer cell layer. Nuclear staining for ki-67 was increased in those areas when compared with the other cystic, follicular, and plexiform areas. These features were interpreted as cytologic atypia suggesting also the possibility of a malignant process. Immunohistochemically, the tumor was positive for CK19 and negative for BRAF VE1, calretinin, and CD56. Ber-Ep4 was only focally positive. By sequencing, an ARID1A c.6527_6538delAG frameshift mutation (VAF: 5.8%), classified as likely oncogenic, and an FBXW7 c.1627 A > G missense mutation (VAF: 8.0%), classified as a variant of uncertain significance, were detected. Two mutations, probably germline (VAF ~ 50%), were recorded for RNF43 and FBXW7. No pathogenic variants were identified in PTCH1, BRAF, NRAS, HRAS, KRAS, FGFR2, or SMO genes.
Conclusion
The significance of an ARID1A variant in keratoameloblastoma is uncertain since this variant has not been reported in ameloblastoma or KCOT, to date. Alternatively, it may characterize malignant transformation in the present case since ARID1A mutations have been encountered in various cancers. Sequencing of additional cases is necessary to determine whether this may represent a recurrent genomic event.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Keratoameloblastoma (KA) is a rare odontogenic tumor that is thought to present with overlapping clinicopathologic features with the so-called solid keratocystic odontogenic tumor/odontogenic keratocyst (KCOT/OKC). The distinction between the two has been, at least to some, problematic or even impossible while others suggest that it is unnecessary to separate the two entities. There is also a thought that cases of KA represent ameloblastomatous transformation of KCOT/OKC. All the above postulates are discussed in a recent review paper by Liam Robinson et al. [1], where the clinicopathologic characteristics of apparent cases of KA are presented. These authors shared their personal experience of seven examples of KA and add them to the nineteen already published since the first report of the entity is under the name KA by Altini et al. [2]. One should note here that the first reported case is apparently by Ralph Spaulding in 1956 as correctly pointed by Professor Fumio Ide et al. [3].
Besides delineating the controversies in the KA and sKCOT/OKC, the significance of the work by Robinson et al. [1] lies on the fact that it provides evidence in support of KA as a variant of ameloblastoma (AMEL), apart from sKCOT/OKC and another rare variant of AMEL known as papilliferous keratoameloblastoma (PKA). Robinson et al. [1] presented as defining histomorphologic characteristics (a) the presence of variably solid and cystic epithelial follicular proliferations, (b) focal stellate reticulum-like central areas, (c) subnuclear vacuolization in the basal cell/ameloblastic layer, (d) overt and often pronounced lamellated-type central parakeratinization, and (e) occasionally, central areas of calcification [1]. Of interest was also the capricious immunohistochemical negativity of KA for calretinin [1], an immunohistochemical stain expected to be positive in ameloblastomas [1, 4]. There is, however, no reference on the evaluation for BRAFV600E [ Mendez et al.] or CD56 immunohistochemical reactivity. These antibodies have shown invariable positive staining for conventional and cystic ameloblastomas.
Progress in the characterization of molecular events of KCOT/OKC and AMEL has disclosed PTCH1 alterations nearly universally occurring in the former [5] and MAPK pathway alterations, in the absence of PTCH1 alterations, in almost 90% of cases in the latter [6]. BRAFV600E mutant ameloblastoma localizes to the mandible while maxillary ameloblastoma tends to be BRAF-wild type and enriched for SMO mutations [6, 7].
The mutually exclusive mutational profiles of KCOT/OKC and ameloblastoma raise the possibility that molecular characterization of KA may inform appropriate classification and treatment, given the more aggressive biologic behavior of ameloblastoma relative to KCOT. In fact, Zhang et al. [8] have tried to separate the two entities at the molecular level by investigating 9 cases diagnosed either as KA or sKCOT/OKC for PTCH1 and BRAF mutations. In their cohort there was lack of BRAF mutations and presence of PTCH1 missense mutation in 1 out of the 9 cases.
The vast majority of KA are, as expected, central tumors. A peripheral example KA has been published [9]. Herein, we report of an unusual peripheral tumor with ostensible features of KA and present some interesting, in our opinion, histopathologic, immunohistochemical, and targeted sequencing findings.
Case Report
Clinical Findings
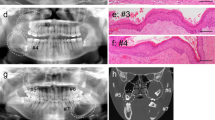
A 54-year-old male presented with a “tender” swelling in the area of the right maxillary premolars causing saucerization of the bone. A periapical radiograph revealed bone loss characterized by radiolucency with foci of subtle opacities (Fig. 1A). A mucosal flap was raised, and a conservative excision was performed.

A Radiolucent area in the right side of maxilla in the area of the premolars causing saucerization of the alveolar bone. Small radiopacities corresponded to calcified keratin. B Low-power microphotograph depicting interconnecting or plexiform cystic spaces of various sizes and scattered small follicles (H&E, original magnification × 40). C Plexiform pattern featuring a peripheral layer of frequently low columnar cells exhibiting reverse polarization. Internally, a stellate reticulum-like pattern was appreciated with abundant parakeratin exhibiting a lamellated pattern. Left and right, calcifying keratin (asterisk) is present. Also focally in the connective tissue, a few dystrophic calcifications (arrow) can be appreciated. (H&E, original magnification × 200). D Tadpole-like follicle with areas of reverse polarization of elongated basal cells with spongiosis, occasional subnuclear vacuolization, and central area of lamellated parakeratinization. Internally, the pattern may be interpreted as either condensed stellate-like reticulum or the spinous layer of keratocystic odontogenic tumor (odontogenic keratocyst), which explains the difficulty encountered. (H&E, original magnification × 400). E Small follicle revealing pseudoductal foci of early keratinization (arrow). (H&E, original magnification × 400). F Small nests featuring cells with small but conspicuous nucleoli (black arrow) and a few mitoses in the periphery (red arrow) (H&E, original magnification × 600)
Histopathology
At gross examination, the specimen comprised a soft tissue mass with calcified areas measuring 1.5 × 1.3 × 1.0 cm. Upon decalcification, the histologic findings were those of an epithelial mostly cystic tumor of apparent odontogenic origin characterized by interconnecting or plexiform cystic spaces and scattered small follicles both featuring areas of palisading in the peripheral basal cell layer with occasional reverse polarization, infrequent subnuclear vacuolization, and central areas of mostly lamellated keratinization (Fig. 1B–D). The cystic lining was a few layers in thickness akin to KCOT/OKC. A few follicles featured foci of early keratinization in form of keratin within pseudoductal structures (Fig. 1E). Also, in some follicles and in foci of the lining of cystic spaces, cells revealed small but conspicuous nucleoli (Fig. 1F), infrequent nuclear hyperchromatism, and a few mitoses mostly seen in the peripheral outer or basal cell layer. Basal cell hyperplasia was also occasionally noted. The stroma contained abundant keratin, deriving from burst cysts, which was calcified in many areas and accompanied by an intense plasmacytic inflammatory response.
Because the lesion was present throughout the submitted tissue and involved the surgical borders, a wider local excision was recommended which took place less than 3 months of the initial procedure. The surgical specimen measured 2.0 × 1.4 × 0.6 cm and included surface mucosa. The origin of the tumor from the surface epithelium was confirmed (Fig. 2A). Essentially similar histopathologic patterns of growth, i.e., plexiform, follicular, and areas were evident as were a few foci of calcifications. A heavy plasmacytic infiltrate was also noted. However, some plexiform areas and follicles featured foci of increased cellularity characterized by sheets of basophilic cells, with inconspicuous cytoplasm and round or oblong nuclei occasionally overlapping nuclei and small nucleoli. Also, foci of nuclear hyperchromatism and few scattered mitoses, the latter mostly in the periphery of the cellular areas, were observed (Fig. 2B, C).

A Neoplastic plexiform proliferation of cords with features of ameloblastoma and area of keratinization (H&E, original magnification × 40). Inset reveals palisading in the basal cell layer and stellate reticulum-like internal areas (H&E, original magnification × 200). B Plexiform pattern with areas of increased cellularity in the periphery. Also, small follicles are present. Pacinian-like keratin present in cystic areas (H&E, original magnification × 100). C High-power microphotograph of the periphery exhibiting nuclear hyperchromatism, cells with small conspicuous nucleoli, and mitotic activity primarily in the periphery. (H&E, original magnification × 600). D CK19 decorated mostly suprabasal cells, less often basal cells, and keratin. (CK19-ABC, original magnification × 100). E BeR-EP4 decorated the cytoplasmic membranes selectively and especially in extensions of the surface epithelium (Ber-EP4-ABC, original magnification × 200). F Variations in ki67 nuclear staining between areas of increased cellularity and cystic area (ki67-ABC, original magnification × 100) Inset reveals another area where ki67 positivity was increased in a cellular area but only present in the basal cell/peripheral cell layer in the cystic areas (ki67-ABC, original magnification × 200)
Immunohistochemical study of both surgical tissue materials was undertaken. Cytokeratin 19 (A53-B/A2.26, predilute, Cell Marque/Ventana) plasmalemmal staining was observed (Fig. 2D). Ber-EP4 (BerEP4, predilute, Cell Marque/Ventana) highlighted only focally the cytoplasm of the surface epithelium (Fig. 2E) exhibiting ameloblastomatous characteristics. There was no staining for calretinin (polyclonal, 1:100, Invitrogen), BRAF V600E (BRAFV600E; VE1, predilute, Ventana) and CD56 (MRQ-42, predilute, Cell Marque/Ventana). Immunohistochemical evaluation with Ki67 (30–9, predilute, Ventana) (Fig. 2F) revealed nuclear staining mostly in the basal and lower suprabasal cells of the surface epithelium in the areas of keratoameloblastomatous transformation, the basal cell layer lining of some keratin-filled cysts. In some nests, only rare positive cells were noted. However, increase in the number of nuclei stained was observed in the areas of increased cellularity where cytologic atypia was noted. The histopathologic features were interpreted as peripheral keratoameloblastoma with foci of at least cytologic atypia. The possibility of malignant transformation could not be excluded. Also, the surgical material of the second surgery, representing additional enucleation, was received in pieces and evaluation of tumor-free borders was impossible.
After the second surgery, repeated calls for patient reevaluation were unsuccessful and the patient remains lost to follow-up 25 months post second surgery.
Targeted Next-Generation Sequencing (NGS)
DNA extracted from FFPE tissue was subjected to multiplex PCR enrichment and next-generation sequencing at University Hospitals Cleveland Medical Center (Cleveland, OH) using the Oncomine Comprehensive Assay v3 (DNA) and Oncomine Focus Assay (RNA) on the ion Torrent S5 Prime platform (ThermoFisher Scientific, Waltham, MA), in which 150 cancer-associated genes were interrogated for SNVs, CNVs, indels, and 23 driver genes were analyzed for gene fusions. Neoplastic tissue accounted for about 10% of sample tissue due to extensive inflammatory infiltrate. Sequencing identified an ARID1A c.6527_6538delAG frameshift mutation (VAF: 5.8%) classified as likely oncogenic and an FBXW7 c.1627 A > G missense mutation (VAF: 8.0%) classified as a variant of uncertain significance, in addition to two other variants of uncertain significance which were detected at a VAF of roughly 50% (suspected germline) (Table 1). Paired normal tissue was not available for testing.
Discussion
KA was last included in the WHO classification in 1992, where it was considered a variant of ameloblastoma with extensive keratinization. However, it has since been omitted on account of its poorly defined diagnostic criteria and rarity [10]. Occasional examples, including cases with prominent papillary architecture, termed PKA, continue to be published, but an improved understanding of this unusual variant of ameloblastoma remains desirable. KA shows nearly equal male-to-female ratio and approximately 2:1 mandibular predilection, somewhat less dominant than conventional AMEL [1]. KA can occur at a wide age range, and has been treated variably by resection or enucleation, with recurrences noted following enucleation [11].
Only a peripheral example has appeared in the literature which was treated, apparently, with conservative enucleation since the mass was “well-encapsulated” [9]. Follow-up information was not provided. Histopathologically, the provided figures reveal similar pattern of growth with the present case. However, there are mostly low to medium power and of suboptical quality when it comes to the detailed depiction of the cytologic neoplastic characteristics. However, there is no reference of cytologic atypia as described herein.
Reports prior to the study by Robinson et al. [1] state that KA can present substantial histopathologic overlap with the rarely reported sKCOT/OKC with some authors postulating ameloblastomatous transformation in sKCOT/OKC [3, 8, 12]. Whitt et al. [13] proposed classifying KA into four different subtypes (a) papilliferous, (b) simple, (c) simple with OKC-like features, and (d) complex. Still, tumors reported as KA and sKCOT/OKC represent diagnostic challenges, despite the fact that this terminology implies apparent relationships to well-known entities. Histopathologically, nuclear palisading and hyperchromatism of peripheral/basal cells is seen in both conventional KCOT/OKC and conventional ameloblastoma, and parakeratosis is not unique to KCOT as acanthomatous change with keratinization can be seen in conventional ameloblastoma [1].
There is only one molecular study which tried to investigate gene aberrations in sKCOT/KA. Zhang et al. [8] reported 9 cases without subclassifying each case as KA or sKCOT, on account of this substantial histopathologic overlap. None of the 9 cases harbored a BRAFV600E mutation, in good agreement with the findings in the present case. Two cases with fresh tissue were subjected to PTCH1 sequencing, one of which harbored a missense mutation. These findings led the authors to conclude that solid KCOT and KAB should not necessarily be segregated into two separate entities but should be best classified altogether as solid KCOT; however, this opinion should be substantiated by stronger data. A study utilizing next-generation sequencing to identify the molecular characteristics of KA is necessary.
In the present tumor, sequencing failed to identify any pathogenic variants in BRAF, NRAS, HRAS, KRAS, FGFR2, or SMO, as seen in ameloblastoma, or any pathogenic variant in PTCH1, as seen in KCOT. Additionally, immunohistochemistry for BRAF VE1 calretinin and CD56 was negative, the latter being also positive in cases of ameloblastoma and occasionally in KCOT/OKC [14].
Interestingly, targeted NGS revealed an ARID1A alteration. ARID1A (AT-rich interactive domain-containing protein 1 A) encodes a large subunit of the SWI/SNF chromatin remodeling complexes. This gene family plays an important role in maintaining chromosome accessibility to tissue-specific enhancers and localizing SWI/SNF complexes to them [15]. SWI/SNF complexes are mutated in approximately 20% of human cancers, representing the most frequently mutated epigenetic regulator in cancer, and are among the most commonly mutated tumor suppressors in cancer [16, 17]. ARID1A mutations have been identified in endometrioid and clear cell ovarian carcinoma, gastric adenocarcinoma, urothelial carcinoma, and hepatocellular carcinoma, among several other tumor types [16].
The presence of an ARID1A alteration in the present case is of unclear clinical significance, as ARID1A alterations have not been reported in ameloblastoma or KCOT, to date, and SWI/SNF complexes are not known to modulate MAPK pathway signaling. However, ARID1A functions as an epigenetic regulator in dental root patterning and development, and can regulate sonic-hedgehog (SHH) signaling in this context [18,19,20]. More work is needed to determine if ARID1A alterations play any role in or modulate SHH signaling in odontogenic neoplasia. ARID1A mutations have been identified in 45% of basosquamous carcinomas, as a secondary event following PTCH1 alterations, and may confer a squamous phenotype in these tumors genetically related to basal cell carcinoma [21]. In the present case, there was no evidence of PTCH1 alterations. Thus, sequencing of examples of KA are necessary to determine if ARID1A inactivation is a recurrent genomic event or if it occurs secondarily to a more commonly occurring mutation, conferring plasticity in differentiation.
Of interest in the current case are the (a) increased cellularity in the periphery of a few follicles with the presence of nuclear hyperchromatism and few scattered mitoses, (b) the presence of keratin in the stroma, the apparent result of burst cystic areas with frequent calcification of the keratin as it interacted with the stroma, (c) intense plasmacytic infiltrate, (d) negative immunostaining for calretinin, BRAF V600E and CD56, and (e) increased ki67 nuclear reaction in areas of cytologic atypia. This may suggest either a possible malignant transformation (low-grade peripheral ameloblastic carcinoma with KA features?) or proliferation of cells to form new neoplastic follicles and nests. One should take into consideration the relatively short time between the first and the second surgery and a possible impact this may have had in the proliferative capacity of the neoplastic cells. As mentioned, the first specimen did not reveal the atypia noted in the second. The presence of occasionally calcified keratin in the stroma has been reported in examples of KA [1] and explains the radiopacities seen radiographically. The heavy plasmacytic infiltrate is apparently an inflammatory response to the ruptured cysts.
The pattern of growth in the present case was reminiscent to squamous cell carcinoma cuniculatum (CC) an entity that was discussed in the differential diagnosis of KA by Robinson et al. [1]. We are of the opinion that the histomorphologic features herein are overwhelmingly of an ameloblastomatous tumor and we do not know of any reports in the literature of oral CC exhibiting such distinct phenotype.
In conclusion, we presented a 54-year-old male with a gingival tumor fulfilling diagnostic criteria of KA with, however, foci of cytologic atypia or possible malignant transformation. The tumor was found to harbor an ARID1A mutation on targeted sequencing. No alterations in MAPK pathway signaling genes or in PTCH1 were identified. ARID1A mutations have not been reported in AMEL or KCOT/OKC to date, rendering interpretation of this pathogenic variant challenging. Sequencing of additional cases of KA will be necessary to determine if KA harbors a unique mutational profile relative to AMEL or KCOT/OKC or if additional, more commonly implicated genes are found to be consistently mutated. Additionally, sequencing of future cases of sKCOT will be necessary to establish if PTCH1 inactivation is a recurrent finding in that tumor, and to ultimately confirm if KA and sKCOT/OKC are separate entities.
Data Availability
The data generated during the study that include clinical notes, clinical photographs, pertinent radiographs, hematoxylin and eosin preparations, immunohistochemical stains, and Next-Generation Sequencing data are available on request from the corresponding author (IGK).
Code Availability
Non-applicable.
References
Robinson L, Smit C, Fonseca FP, Abrahão AC, Romañach MJ, Khurram SA et al (2022) Keratoameloblastoma: a report of seven new cases and review of literature. Head Neck Pathol. https://doi.org/10.1007/s12105-022-01470-5
Altini M, Lurie R, Shear M (1976) A case report of keratoameloblastoma. Int J Oral Surg 5(5):245–249
Ide F, Ito Y, Muramatsu T, Saito I, Abiko Y (2012) Histogenetic relations between keratoameloblastoma and solid variant of odontogenic keratocyst. Oral Surg Oral Med Oral Pathol Oral Radiol 114(6):812–813
Altini M, Coleman H, Doglioni C, Favia G, Maiorano E (2000) Calretinin expression in ameloblastomas. Histopathology 37:27–32
Stojanov IJ, Schaefer IM, Menon RS, Wasman J, Gokozan HN, Garcia EP et al (2020) Biallelic PTCH1 inactivation is a dominant genomic change in sporadic keratocystic odontogenic tumors. Am J Surg Pathol 44(4):553–560
Heikinheimo K, Huhtala H-T, Thiel A, Kurppa KJ, Heikinheimo H, Kovac M (2019) The mutational profile of unicystic ameloblastoma. J Dent Res 98(1):54–60
Mendez LD, Wolsefer NS, Asa SL et al (2022) The diagnostic utility of BRAF VE1 mutation-specific immunohistochemistry in ameloblastoma. Mod Pathol 35:1570–1577
Zhang R, Yang J, Zhang J, Hong Y, Xie X, Li T (2021) Should the solid variant of odontogenic keratocyst and keratoameloblastoma be classified as the same entity? A clinicopathological analysis of nine cases and a review of the literature. Pathology 53(4):478–486
Parikh N, Nandini C, Jain S, Mansata AV (2018) Peripheral keratoameloblastoma: a novel case report. J Oral Maxillofac Pathol 22(2):249–253
Kramer IRHPJ, Shear M (1992) World Health Organization International histological classification of tumors. Springer-Verlag, Heidelberg, p 13
Lee C, Park BJ, Yi WJ, Heo MS, Lee SS, Huh KH (2015) Keratoameloblastoma: a case report and a review of the literature on its radiologic features. Oral Surg Oral Med Oral Pathol Oral Radiol 120:e219-2255
Ide F, Mishima K, Saito I (2003) Solid-cystic tumor variant of odontogenic keratocyst: an aggressive but benign lesion simulating keratoameloblastoma. Virchows Arch 442(5):501–503
Whitt JC, Dunlap CL, Sheets JL, Thompson ML (2007) Keratoameloblastoma: a tumor sui generis or a chimera? Oral Surg Oral Med Oral Pathol Oral Radiol Endod 104:368–376
Cairns L, Naidu A, Robinson CM et al (2010) CD56 (NCAM) expression in ameloblastomas and other odontogenic lesions. Histopathology 57:544–548
Mathur R (2018) ARID1A loss in cancer: towards a mechanistic understanding. Pharmacol Ther 190:15–23
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J et al (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45(6):592–601
Shain AH, Pollack JR (2013) The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 8(1):e55119
Jing J, Feng J, Li J, Han X, He J (2019) Ho TV et al Antagonistic interaction between Ezh2 and Arid1a coordinates root patterning and development via Cdkn2a in mouse molars. eLife 8:e46426
Du J, Jing J, Yuan Y, Feng J, Han X, Chen S et al (2021) Arid1a-Plagl1-Hh signaling is indispensable for differentiation-associated cell cycle arrest of tooth root progenitors. Cell Rep 35(1):108964
Du J, Jing J, Chen S, Yuan Y, Feng J, Ho T-V et al (2021) Arid1a regulates cell cycle exit of transit-amplifying cells by inhibiting the Aurka-Cdk1 axis in mouse incisor. Development 148(8):dev198838
Chiang A, Tan CZ, Kuonen F, Hodgkinson LM, Chiang F, Cho RJ et al (2019) Genetic mutations underlying phenotypic plasticity in Basosquamous Carcinoma. J Invest Dermatol 139(11):2263–2271e2265
Funding
The study was funded by the University of Minnesota Oral Pathology Research Fund (internal funding).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare no conflict of interest.
Ethical Approval
Immunohistochemistry and new generation sequencing (NGS) were performed for investigative and diagnostic purposes and that part of the study was exempt from the Institutional Review Board. The NGS part of the study was carried out at the University Hospitals Cleveland Medical Center, Cleveland OH. The NGS part of the study was approved by the University of Minnesota Institutional Review Board (ID: 00012651). The University of Minnesota reviewed the study and determined that the proposed activity is not research involving human subjects as defined by DHHS and FDA regulations. To arrive to this determination, the University of Minnesota IRB used “WORKSHEET: Human Research (HRP-310).” Based on that, ongoing IRB review and approval for this activity is not required.
Consent to Participate
The histopathologic, immunohistochemical, and NGS parts of the study were exempt from the institutional review board (University of Minnesota IRB: ID: 00012651).
Consent for Publication
The patient’s information, excluding age and gender, cannot be identified. Please note that the patient was lost to follow-up. Repeated attempts to communicate with the patient were unsuccessful. The NGS part was performed for diagnostic/investigative purposes.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Stojanov, I.J., Ho, D., Huss, J. et al. An Unusual Gingival (Peripheral) Tumor with Features of Keratoameloblastoma with Cytologic Atypia or Possible Malignant Transformation Exhibiting ARID1A Mutation. Head and Neck Pathol 17, 808–814 (2023). https://doi.org/10.1007/s12105-023-01549-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-023-01549-7