
Abstract
Solitary fibrous tumors (SFTs) of the orbit are rare. In order to further characterize the clinical and pathologic features of solitary fibrous tumor arising at this anatomic site, 12 cases of orbital SFTs were analyzed in conjunction with a review of 263 cases reported from the English literature in order to develop a risk prediction model. SFTs of the orbit were equally distributed between males (n = 5) and females (n = 7) with a mean patient age of 46.8 years (median 44.5 years; range 18–76 years) at initial diagnosis. The patients typically presented with swelling or mass around the orbit, with proptosis (n = 10), ptosis (n = 5), and visual changes (n = 6). Tumors were orbital (n = 10) or upper eyelid (n = 2). Mean tumor size was 2.5 cm (median 2.6 cm). Microscopically, the tumors were characterized by cytologically bland spindle cells with patternless growth, hypocellular and hypercellular areas, variable amounts of collagen, and ectatic, branching blood vessels. By immunohistochemistry, all cases had a strong nuclear STAT6 expression. All patients were initially managed with excision or biopsy, three with presurgical embolization. The two patients with biopsy only had persistent disease (mean 37.2 months), but a third patient developed distant bone metastasis at 86.9 months. Overall mean follow-up was 73.1 months: 9 patients are alive or dead without disease (mean 77.9 months), two patients with persistent disease, and one patient with metastatic disease at last follow-up (102 months). Incorporating cases sufficiently reported in the literature, a risk prediction model based on age > 45 years, tumor size > 3 cm, tumor necrosis, mitoses of > 4/2 mm2, moderate to high cellularity, and moderate to severe pleomorphism allows for risk stratification for the development of local recurrence and distant metastasis. In conclusion, orbital SFTs are rare, but can be reliably diagnosed based on the presence of characteristic morphologic features and STAT6 immunohistochemistry. Orbital tumors tend to show a higher frequency of local recurrence than distant metastasis, which can be predicted by a risk stratification model unique to orbital tumors. With late disease common, long term clinical follow-up is recommended.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
First described in 1931 [1], solitary fibrous tumor (SFT) has been documented in many other organs after its original pleural description. It is much more common around body cavities, such as pleura, peritoneum, and meninges, and thus orbit tumors may be related to the meninges. The tumor has gone by many names over the years, including benign mesothelioma, pleural fibroma, and localized fibrous tumor. Hemangiopericytoma was thought to be of pericytic origin [2], but has been viewed more as a pattern diagnosis, with an open, patulous, staghorn vascular pattern as the most consistent feature. Giant cell angiofibroma and even fibrous histiocytoma are morphologically similar lesions considered in the diagnostic continuum. Over the past few decades, immunohistochemistry, cytogenetics, and molecular findings have shown that solitary fibrous tumor is the correct term for a spectrum of lesions, with hemangiopericytoma, giant cell angiofibroma, and even fibrous histiocytoma of the orbit now considered obsolete [3]. Extrapleural is usually applied to all other non-pleural sites for a fibroblastic mesenchymal neoplasm characterized by staghorn, thin-walled branching vessels, ovoid to elongated spindled cells in a background of wiry collagen and with a recurrent, characteristic NAB2-STAT6 gene fusion [4,5,6,7]. The fusion is represented by a strong nuclear STAT6 immunoreactivity [8, 9] although not evaluated systematically in a series of orbital tumors. The aim of this study was to present a clinicopathologic study of orbit SFT evaluated with STAT6 and to aggregate the findings of orbit SFTs reported in the literature to develop a more site-specific risk prediction model for orbit tumors.
Materials and Methods
Twelve cases of solitary fibrous tumors of the orbit, lacrimal gland, and eyelids were selected from a review of all solitary fibrous tumors (n = 17) identified in the pathology files between 2009 to 2019 from the head and neck region. Tumors identified within the central nervous system were excluded. These 12 cases were identified within a single healthcare delivery system treating more than 4 million patient members. As patients enter and exit the health delivery system frequently, a true incidence is difficult to determine. However, with 12 orbit cases diagnosed in 10 years, the incidence is approximately 0.2 patient/million population in any given year. Materials within the files were supplemented by a review of the patient demographics (sex, age, race) and symptoms at presentation (mass, swelling, proptosis/exophthalmos, visual changes, ptosis, headaches) including duration. Smoking and alcohol history were documented. Other concurrent clinical findings, including family history and possible paraneoplastic findings were identified. The medical history, imaging findings, surgical pathology, and operative reports were reviewed to obtain exact tumor location, lateralization and tumor size (greatest dimension in centimeters), procedures performed, and diagnostic evaluation. Follow-up data included specific treatment, the presence or absence of recurrent or persistent disease, and the current status of the disease and patient. This clinical investigation was conducted in accordance and compliance with all statutes, directives, and guidelines of an Internal Review Board authorization (#5968) performed under the direction of Southern California Permanente Medical Group relating to human subjects in research.
Hematoxylin and eosin-stained slides from all cases were reviewed. Mitotic figures were evaluated using a Olympus BX41 microscope per 2 mm2 using a field diameter of 0.5 mm with an area of 0.196 mm2, and with 10 consecutive fields counted, attempting to begin counting in hot-spot areas if any mitoses were identified, which equates to 1.96 mm2, which has been rounded to 2 mm2.
Immunophenotypic analysis was performed in all cases on a single block from each case by a standardized Envision™ method employing 4 µm-thick, formalin fixed, paraffin embedded sections. Evaluation of STAT6 (phosphor-Tyr641, LifeSpan BioSciences) was performed specifically, although CD34 (Dako-Agilent), bcl-2 (clone 124, Dako-Agilent), and CD99 (clone O13, Signet Laboratories) were also included in the initial evaluation of the tumors. Other immunohistochemistry studies reported were not standardized for each case, but were included during initial work-up of the case and thus varied considerably. Epitope retrieval was performed, as required by the manufacturer guidelines. Standard positive controls were used throughout, with serum used as the negative control.
A review of the English literature was based on a PubMed search from 1966 to 2019 with all cases of orbit solitary fibrous tumor reviewed [7, 10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107]. Cases were excluded if they did not include clinical information, imaging findings, pathology descriptions and/or images, and lacked clinical follow-up data. Cases were searched to include alternative diagnostic terminology (giant cell angiofibroma, hemangiopericytoma, fibrous histiocytoma, fibrous mesothelioma, and extrapleural solitary fibrous tumor, to name just a few). Specific attention was given to clinical series which included immunohistochemistry information. Duplicate cases were only included once [13, 21, 24, 37]. Cases secondarily involving the orbit from the brain or central nervous system (CNS) or the sinonasal tract (paranasal sinuses) were excluded, as were cases metastatic to the orbit from other sites [108, 109]. Cases of meningioma were excluded.
Risk Prediction Model
The specific information used to determine risk stratification was collected from literature reported orbit SFT cases and tabulated to assess the prediction from the three most widely used risk stratification prediction models for SFT, namely, by Pasquali et al. [110], Salas et al. [111], and refined Demicco et al. [112] and applying these same prediction models to the cases herein reported. Mitotic index is the only factor used in all models, and yet there is significant validity to tumor size, patient age, cellularity, pleomorphism, and tumor necrosis, all factors historically included in assessment of malignancy or recurrence. Thus, it was felt that inclusion of all the reported criteria may serve to more fully capture the unique nature of orbit tumors, recognizing that radiation exposure is exceptionally rare (only a single reported case [43]), and thus was not included. However, considering the younger patient age, smaller tumor size, and tumor site of orbit only, different cutoff points were proposed, recognizing that statistical modeling for only 12 cases in this report would not be valid for either univariate or multivariable analysis. While features reported in the literature are incorporated, these findings could not be independently validated or confirmed and so an attempt to include these cases as an external validation model would be unwise. The risk stratification model was developed to include all events, whether local recurrence and/or distant metastasis, using the following definition of recurrence: development of tumor at the site of the original tumor at least 12 months after initial treatment was completed and documented to be disease free. This definition is specifically employed to exclude persistent disease due to incomplete initial management (ie., biopsy only; embolization only; excision, but not wide excision). Patient age was scored 0 if ≤ 45 years and 1 if > 45 years of age at initial presentation. Tumor size was scored 0 if ≤ 3.0 cm and 2 if > 3 cm. Mitotic activity was scored as 0 if ≤ 4 mitoses/2 mm2 and 3 if > 4 mitoses/2 mm2, based on methodology described in Materials and Methods. Cellularity was interpreted to be moderate to high if there was no space between cells, with overlapping and crowding (nuclei in contact with each other). Moderate to high cellularity was scored as 1 if present. Pleomorphism was defined as variation in size and shape of the cells or nuclei and increased hyperchromasia of the nuclei. No or limited pleomorphism was scored as 0, while moderate to high pleomorphism was scored as 1. No tumor necrosis was scored as 0, while any tumor necrosis was scored as 1. The criteria incorporated in the published risk models and the proposed criteria are presented in Table 1.
Results
Clinical
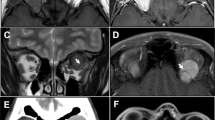
The clinicopathologic information is summarized in Table 2. The patients included 7 females and 5 males who ranged in age from 18 to 76 years, with a mean age at presentation of 46.8 years (median 44.5 years). Five of the patients were 55 years or older at presentation. There was no statistically significant difference in mean age at presentation between females (49.0 years) and males (43.6 years; p = 0.629). All patients were white. All of the patients presented with a swelling or mass, present for a duration of 3 to 60 months (mean 21.3; median 12.0 months). Men experienced symptoms for longer than women (27 months vs. 17.3 months), but this was not a statistically significant difference (p = 0.474). The majority of patients experienced exophthalmos (proptosis; Fig. 1) with visual changes (n = 6) and headaches (n = 2). Visual changes included double vision (n = 3), blurred vision (n = 3), and/or flashing lights. Ptosis was experienced by 5 patients. One patient each had glaucoma and cataract. One patient each had vertigo, upward gaze restriction, redness, hyperopia, astigmatism, and CREST syndrome with Raynaud. No patients reported any previous radiation. No family members experienced a similar tumor. One patient had a history of prostate carcinoma. Two patients were ever smokers and two patients were ever drinkers. Ten tumors involved the orbit, one the lacrimal gland exclusively, and two involved the upper eyelid. Of the orbital tumors, 5 were intraconal and 5 were extraconal. Eight tumors involved the left side and 4 the right side. Imaging studies (computed tomography and/or magnetic resonance imaging) were performed in 11 patients. All imaging studies documented a well circumscribed, ovoid mass, showing a soft tissue density (Fig. 1) and enhancement with contrast, resulting in displacement of the globe in 10 patients, but without evidence of bone destruction. Angiography was performed to guide presurgical embolization in 3 patients (Fig. 1).

a) Clinical photo showing left globe displacement and a superior eyelid swelling. b) Coronal T1 SE FS MRI of an extraconal medial right orbit bright signal mass (arrow). C) Computed tomography of a large retrobulbar mass (arrow) resulting in significant proptosis. d) Embolic material (arrow) can be seen in this medial left orbit mass
Pathologic Features
Macroscopic
The tumors ranged in size from 0.6 up to 3.7 cm, with a mean of 2.5 cm (median 2.6). There was no average difference in size between females and males (2.5 vs 2.4 cm, respectively). Tumors of the eyelid were statistically significantly smaller (mean 1.2 cm) than orbital tumors (mean 2.7 cm; p = 0.009). On gross examination, the tumors were white, tan, and received as multiple fragments of tissue, focally associated with cystic change.
Microscopic
The tumors entrapped the adjacent soft tissues and minor salivary-gland type tissue, but were usually well circumscribed (Fig. 2). The tumor cells entrapped nerves, such that perineural invasion was simulated. Expansion into the adjacent fat was noted in a few cases (Fig. 2), but true lipomatous differentiation within the tumor proliferation was not seen. The tumors showed a spectrum of hypo- to hypercellularity (Fig. 2), set within an easily identified, wiry, keloid-like collagen. The collagen could be thick and keloid-like in areas, but thin, refractile to wiry collagen was much more common. Collagen amount varied both within and between cases (Fig. 3). The architecture was haphazard or patternless, yielding a streaming quality in some cases, to a vaguely fascicular appearance in others (Fig. 3). The classical and characteristic hemangiopericytoma-like patulous, open, staghorn vessels could be seen, but was more easily identified in resection samples than incisional biopsies (Fig. 3). These vessels did not show peritheliomatous hyalinization. Myxoid change of the stroma could be seen, but was not a prominent finding (Fig. 3). Neoplastic giant cells were not identified. Mitoses were inconspicuous in most of the cases, although increased in one case. Tumor necrosis was absent, but degeneration around embolic material was noted in the three cases previously treated with embolization (Fig. 4). Nuclear pleomorphism was only identified in a single case, which also showed increased cellularity and increased mitoses of 5/2 mm2. In the remaining cases, the neoplastic cells were bland and lacking pleomorphism. The lesional cells were spindled to elongated, with round to oval nuclei with cells that have spindled pale to eosinophilic cytoplasm (Figs. 1–4). The cells lacked wavy nuclei or blunt-ended nuclei. Perinuclear vacuoles were absent. Dedifferentiation or anaplasia was not seen. Lymphovascular invasion was not seen.

a) Very well circumscribed tumor with a predominantly heavy, keloid-like collagen deposition. b) This solitary fibrous tumor expanded into the adjacent fat, but seemed to incorporate it, rather than being a lipomatous variant. c) Cellularity could vary from one area to another within the same tumor. d) Hypocellular regions with a more edematous appearance to the stroma

Various patterns in SFT. a) Hemangiopericytoma-like vessels with a non-descript spindled cell proliferation. b) Short, tight, cellular fascicles could be seen. c) Classical appearance of bland spindled cells set within a collagenized stroma with open vessels. d) Myxoid change within the stroma could be seen in some tumors

a) Strong, diffuse nuclear STAT6 reaction in a classical SFT. b) Strong and diffuse cytoplasmic CD34 immunoreactivity. c) Embolic material within a classical SFT pattern. d) STAT6 (left side) immunoreactivity is lost in the rest of the tumor adjacent to areas of embolization
Immunohistochemical Results
All tumors (n = 12) demonstrated a strong and diffuse nuclear reaction with STAT6 (Fig. 4). During original evaluation, CD34 (Fig. 4), bcl-2, CD99, and vimentin were positive to a variable degree in the neoplastic cells. CD68 and CD10 were also noted in isolated cells. However, pancytokeratin (AE1/AE3), S100 protein, SOX10, SMA, MSA, desmin, nuclear ß-catenin, CD31, epithelial membrane antigen, HMB45, CD56, glial fibrillary acidic protein, FXIIIA, FLI1, TLE1, neural filament (NF), and somatostatin receptor 2 were negative.
Treatment and Follow-up
All patients were managed by surgery, with three treated by pre-surgical embolization. Biopsy was performed in two patients, both of whom have persistent disease at 25.4 and 49.1 months after the original diagnosis. In one patient, local recurrence and distant metastasis to the femur were noted 86.9 months after the original resection. After surgery for diagnostic and therapeutic reasons, radiation (300 Gy to sphenoid and 200 Gy to femur/hip) and concurrent chemotherapy (combination of temozolomide [Temodar] and bevacizumab [Avastin]) were used for the metastatic disease. This patient is alive with disease at 102 months after the original diagnosis. All other patients (n = 9) are alive without disease or have died of unrelated causes an average of 77.9 months after initial diagnosis (range 13.8 to 264 months; median 44.6 months). The patient who died, died of metastatic prostate cancer to bone without any residuum of the orbit solitary fibrous tumor.
Discussion
Orbit SFTs are uncommon tumors displaying a generally benign clinical behavior, but with a small subset of cases showing local recurrence and/or distant metastatic disease. These tumors have been shown to have a characteristic histologic appearance of a fibroblastic population set within a variably collagenized stroma and associated with branching, patulous, slit-like, or staghorn type vessels. The NGFI-A binding protein 2 (NAB2) fuses with signal transducer and activator of transcription 6 (STAT6) as a result of a paracentric inversion of chromosome 12q.
In other anatomic sites, at least 12 fusion gene variants have been reported, with correlation to site and clinical behavior as well as specific histomorphology appearance of the tumors [113,114,115]. The most common fusion (NAB2ex4-STAT6ex2/3) was identified in pleuropulmonary tumors with dense fibrosis, benign behavior, and older patients, while the second most common fusion (NAB2ex6-STAT6ex16/17 or 16/18) was found in younger patients with deep soft tissue tumors and a more aggressive phenotype and clinical behavior [113, 114]. Biologically aggressive tumors have also been shown to have secondary alterations that include TERT promoter mutations and deletions or mutations of TP53 [116, 117]. While these specific molecular findings were not evaluated in this series, all of the tumors reported were from non-traditional sites (i.e., non-pleuropulmonary), and were, in general, in younger patients (median, 44.5 years), who all had small tumors (< 5 cm).
Risk Prediction Modeling
There are several risk stratification proposals for SFT (Table 1), based on clinical and pathology findings, although each system employs different criteria, with mitotic index the only criterion used in all. Before attempting risk stratification, the English literature reporting SFT as identified in the materials and methods was tabulated and summarized (Table 3). For risk stratification, patient age at presentation, site of the tumor, size of the tumor, tumor necrosis, cellularity, nuclear pleomorphism, and previous radiation are variably employed to develop a risk assessment for recurrence, metastasis, or overall survival [9, 110,111,112, 118]. These models each rely on different weighting for each parameter, obtained using a competing risks framework and prognostic modeling for multivariate models based on age, creating different prognostic groups, which were then internally validated through bootstrapping datasets, and externally validated by an independent cohort validation. All models establish risk for metastatic disease specifically, although risk for local recurrence is also predicted in one model [111]. These models were applied to all patients in this series (Table 2). Only one patient (8%) in this clinical series developed metastatic disease: she was 55 years old with a 2.4 cm orbital tumor, showing increased cellularity, 5 mitoses/2 mm2, pleomorphism, but no tumor necrosis at initial evaluation and without previous radiation therapy. By the Demicco, et al., model, there would be a low prediction for metastasis (2 points for ≥ 4 mitoses/10 HPFs; 1 point for age ≥ 55 years; total points = 3) [112]; by the Salas, et al. model, there would be a low risk for metastasis (1 point of > 4 mitoses; total points = 1) [111]; and by the Pasquali, et al. model, there would be a high risk for metastatic disease (3 points for > 4 mitoses/10 HPFs; 2 points for high cellularity; 2 points for nuclear pleomorphism; total points = 7). The latter criteria have been applied specifically to non-pleural primaries, without taking age, site, size or necrosis into consideration. Thus, modeling risk for orbital tumors may still need further evaluation [118] perhaps including factors already employed, but using a slightly different weighting to account for differences in age at presentation, overall tumor size, and the high rate of local recurrence, but low rate of metastatic disease.
In a critical review to determine factors employed in risk stratification, only 47 cases report all of the criteria used in the risk models, with only one patient documented to have metastatic disease to the lung; 12 cases with local recurrence and three interpreted to be “histologically” malignant based on increased mitoses, tumor necrosis, increased cellularity, and marked pleomorphism (Table 4) [10, 11, 29, 31, 35, 40, 43, 45, 47, 50, 52,53,54,55, 57, 61, 64,65,66,67, 74, 78, 79, 85, 87, 99, 102, 104]. Thus, 2.1% of orbital cases develop metastatic disease, and 26% develop local recurrence, an inverted finding to that reported for SFT in general, where 26% develop metastatic disease and 10% have local recurrence [99, 112, 118]. As demonstrated, the risk stratification for recurrence is not reliable, with many cases that develop recurrence missed in the current models, with the Salas, et al. model least likely to predict disease for orbital tumors. Attempts to create a risk stratification model of metastasis when prevalence is so low is bound to be fraught with difficulty and fail. However, perhaps a risk stratification for recurrence would be more helpful. Still, does one bias to over- or under-prediction? Generally, medicine is biased to a lower positive predictive value, while accepting false positives that overestimate the potential for developing disease. The Demicco refined risk stratification uses a ≤ 20% false positive rate for the low risk category as being acceptable [112]. With this bias in mind, an orbital risk stratification model was developed to predict local recurrence and/or metastatic disease (Table 1), recognizing some patients would be predicted to have recurrence, even though not yet detected (Table 4), and some patients with recurrence may not be risk stratified as high risk.
A younger age cut-off (> 45 years) was included since overall orbit tumors tend to develop in younger patients, so the 55- and 60-year thresholds used in other systems are insufficiently discriminating. With a median size of 2.9 cm from the selected literature cases, a size cutoff of > 3 cm more accurately reflects the relatively smaller size of orbital tumors in general and how this particular feature must be relatively smaller to be meaningful. The reported risk stratification models use 4 mitoses as a cutoff, and so > 4 mitoses/2 mm2 was included. From sarcoma data, tumor necrosis is recognized to guide grading, and while not frequently present in orbit SFTs, when necrosis is documented it is given a 1-point value. Given the much smaller tumor size, a 10% tumor necrosis cutoff was not used, and instead absent or present was applied. Increased (moderate to high) tumor cellularity and moderate to severe pleomorphism are each ascribed 1 point, recognizing these features are generally included in soft tissue tumor evaluation. These parameters yield a slightly more robust outcome modeling, although still not completely predictive. It is important to note that some cases in the literature had recurrences but were not documented to be completely excised, and so represents persistence and not true recurrence. However, if using local recurrence as a cutoff, then a total score of ≥ 3 correctly predicts recurrence in 13 of 16 patients with recurrence (81%), while not predicting recurrence in 3 patients (19% false negative rate) [11, 79, 99]. Further, this model would predict an intermediate risk of recurrence in 3 of the remaining 31 patients (9.7% false positive rate [61, 99]). Still, these latter 3 patients have been followed for an average of 72.7 months, while the average time to recurrence is 109.5 months, suggesting that a longer time horizon is needed to completely validate such a modeling. This latter finding strongly supports the recommendation that long-term follow-up of all orbit SFTs is required, as local recurrence is seen after a significant time interval from the initial presentation, reported as late as 33 years after initial presentation [10, 85,86,87, 99].
The unique nature of the site is further reinforced, when a series of previously reported sinonasal tract SFT [122] are evaluated, 3 of 6 patients would be classified as intermediate risk of recurrence, and at last follow-up (mean 80.3 months), none had developed recurrence. Thus, the criteria should only be applied to orbit tumors and not other head and neck sites.
Review of Orbital SFTs
About 20% of SFTs develop primarily in the head and neck, with sinonasal tract tumors more common than orbit [122], but with about half of head and neck tumors arising from the meninges. In general, orbital SFTs affect both sexes equally and present in a younger age group (median 42 years) than the 5th to 7th decades for other anatomic sites [7, 9, 21, 29, 39, 48, 88, 94, 99, 110, 119, 120], although similar to meningeal tumors [115, 121]. Tumors are usually present for about a year before diagnosis, related to the initial non-specific presentation. In the orbit, an expanding swelling or mass involving the orbit or eyelid is the most common finding, with exophthalmos/proptosis seen in many patients. Changes in vision, blurred vision, or double vision are seen less often, with pain, epiphora (excessive tearing), ptosis, and headache much less commonly reported. None of our cases nor any orbital tumors aggregated from the literature demonstrated paraneoplastic syndrome, where refractory hypoglycemia (Doege-Potter syndrome) is reported, usually due to large tumors which secrete insulin-like growth factor 2 (IGF2) [123, 124]. This finding is supported by the NAB2-STAT6 fusion resulting in a feedforward loop of constitutive EGR1-mediated transactivation of proliferation factors, which include IGF2 and FGRF1 [6].
There is no specific orbital subsite affected, although there are a few tumors of the eyelid and lacrimal sac specifically. While the tumors have a very wide size range (0.4 up to 15 cm), the median size is 2.6 cm (Table 3). Tumors of the head and neck are usually smaller than their soft tissue and cavity-lined counterparts, no doubt due to the anatomic confines of the region and symptoms that result in earlier clinical detection [99]. Review of the reported size highlights that 95.6% of all cases are ≤ 5 cm. As such, tumors in this anatomic location are smaller than other sites. Imaging studies are most helpful in delimiting size, disease extent, and exact location, which can inform management by presurgical embolization or operative approach. The lesions usually appear as homogenous, isodense to muscle, well-circumscribed, lobulated soft tissue masses, occasionally demonstrating internal cystic areas. Pressure remodeling of bone and displacement of the globe and associated intrinsic muscles of eye movement can be seen, but generally bone destruction or orbit infiltration is not present [125]. Post-contrast enhancement may be seen. Depending on the extent of collagen, the tumors may have a low T2-weighted MRI finding. Tumors are well known to strongly enhance after gadolinium injection [119]. So many tumors of the orbit are removed piecemeal that an accurate description is more challenging. However, tumors tend to be firm, white to tan with occasional cystic or degenerated areas. In patients managed with presurgical embolization, hemorrhage and degeneration may be present.
Importantly in such close proximity to the sinonasal tract, sinonasal glomangiopericytoma is a clinically, histologically, and molecularly distinctive sinonasal tract tumor, showing a sheet-like non-specific distribution of round to ovoid to spindled neoplastic cells set within a vascular stroma with peritheliomatous hyalinization, and a rich background of extravasated erythrocytes and mast cells and a characteristic CTNNB1 mutation and nuclear ß-catenin immunohistochemistry expression [126, 127]. These tumors are not reactive with STAT6 [8]. In the literature, SFT are frequently mis-diagnosed as cavernous hemangioma, schwannoma (benign peripheral nerve sheath tumor), pleomorphic adenoma, myofibroma, spindle cell lipoma, angiofibroma, perineurioma, and even biphenotypic sinonasal sarcoma. As none of the tumors in the differential diagnosis are STAT6 immunoreactive, it is worthwhile in small biopsies of spindled cell tumors from the orbit to include STAT6 in the panel of immunohistochemistry stains performed (ie., pancytokeratin, SOX10, S100 protein, SMA, ß-catenin).
Pre-surgical embolization is effective in reducing intraoperative bleeding [86, 89, 91, 104, 128], with embolic material noted in the subsequent excision samples. Importantly, interpretation of immunohistochemistry studies in and immediately adjacent to areas of embolization may result in a loss or reduction of the reactivity, as seen with STAT6 in the cases managed with embolization. However, the immunohistochemistry findings are still preserved away from these areas.
Conclusions
Orbital SFTs are rare neoplasms, more common in younger patients who present with small tumors that have been present for some time. The histologic features alone cannot be used to predict local recurrence or metastasis, both of which can be seen to develop after long disease-free intervals. An orbital risk stratification for SFT is suggested, modifying current extrapleural schemes to account for a significantly higher local recurrence risk rather than metastatic disease while also taking into consideration the generally younger age at presentation and the smaller tumor size. Further refinement of this risk stratification is encouraged to more accurately and adequately treat these STAT6 positive neoplasms.
References
Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385–412.
Stout AP, Murray MR. Hemangiopericytoma: a vascular tumor featuring Zimmermann's pericytes. Ann Surg. 1942;116(1):26.
Gengler C, Guillou L. Solitary fibrous tumour and haemangiopericytoma: evolution of a concept. Histopathology. 2006;48(1):63–74.
Chmielecki J, Crago AM, Rosenberg M, et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet. 2013;45(2):131–2.
Mohajeri A, Tayebwa J, Collin A, et al. Comprehensive genetic analysis identifies a pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic imbalances, and a characteristic gene expression profile in solitary fibrous tumor. Genes Chromosomes Cancer. 2013;52(10):873–86.
Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet. 2013;45(2):180–5.
Kao YC, Lin PC, Yen SL, et al. Clinicopathological and genetic heterogeneity of the head and neck solitary fibrous tumours: a comparative histological, immunohistochemical and molecular study of 36 cases. Histopathology. 2016;68(4):492–501.
Demicco EG, Harms PW, Patel RM, et al. Extensive survey of STAT6 expression in a large series of mesenchymal tumors. Am J Clin Pathol. 2015;143(5):672–82.
Demicco EG, Park MS, Araujo DM, et al. Solitary fibrous tumor: a clinicopathological study of 110 cases and proposed risk assessment model. Mod Pathol. 2012;25(9):1298–306.
Rice CD, Kersten RC, Mrak RE. An orbital hemangiopericytoma recurrent after 33 years. Arch Ophthalmol. 1989;107(4):552–6.
Dorfman DM, To K, Dickersin GR, Rosenberg AE, Pilch BZ. Solitary fibrous tumor of the orbit. Am J Surg Pathol. 1994;18(3):281–7.
Westra WH, Gerald WL, Rosai J. Solitary fibrous tumor. Consistent CD34 immunoreactivity and occurrence in the orbit. Am J Surg Pathol. 1994;18(10):992–8.
Fukunaga M, Ushigome S, Nomura K, Ishikawa E. Solitary fibrous tumor of the nasal cavity and orbit. Pathol Int. 1995;45(12):952–7.
Lucas DR, Campbell RJ, Fletcher CDM, et al. Solitary fibrous tumor of the orbit. Int J Surg Pathol. 1995;2:193–8.
DeBacker CM, Bodker F, Putterman AM, Beckmann E. Solitary fibrous tumor of the orbit. Am J Ophthalmol. 1996;121(4):447–9.
McElvanney AM, Noble JL, O'Donovan DG, Bonshek RE, Banerjee SS. Solitary fibrous tumour: an atypical presentation within the orbit. Eye (Lond). 1996;10(Pt 3):396–9.
Ramdial PK, Nadvi S. An unusual cause of proptosis: orbital solitary fibrous tumor: case report. Neurosurgery. 1996;38(5):1040–3.
Ruska KM, Westra WH. Pathologic quiz case 1. Solitary fibrous tumor (SFT) of the orbit. Arch Otolaryngol Head Neck Surg. 1996;122(10):1130, 2.
Sciot R, Goffin J, Fossion E, Wilms G, Dom R. Solitary fibrous tumour of the orbit. Histopathology. 1996;28(2):188–91.
Char D, Weidner N, Ahn J, Harbour JW. Solitary fibrous tumor of the orbit. Orbit. 1997;16:113–8.
Fukunaga M, Naganuma H, Nikaido T, Harada T, Ushigome S. Extrapleural solitary fibrous tumor: a report of seven cases. Mod Pathol. 1997;10(5):443–50.
Karcioglu ZA, Nasr AM, Haik BG. Orbital hemangiopericytoma: clinical and morphologic features. Am J Ophthalmol. 1997;124(5):661–72.
Cho NH, Kie JH, Yang WI, Jung WH. Solitary fibrous tumour with an unusual adenofibromatous feature in the lacrimal gland. Histopathology. 1998;33(3):289–90.
Ing EB, Kennerdell JS, Olson PR, Ogino S, Rothfus WE. Solitary fibrous tumor of the orbit. Ophthalmic Plast Reconstr Surg. 1998;14(1):57–61.
Lanuza A, Lazaro R, Salvador M, et al. Solitary fibrous tumour of the orbit. Report of a new case. Int Ophthalmol. 1998;22(5):265–8.
Miyagi N, Sugita Y, Terasaki M, et al. Solitary fibrous tumor of the orbit: a case report. Jpn J Neurosurg. 1998;7:243–7.
de Saint Aubain Somerhausen N, Rubin BP, Fletcher CD. Myxoid solitary fibrous tumor: a study of seven cases with emphasis on differential diagnosis. Mod Pathol. 1999;12(5):463–71.
Festa S, Lee HJ, Langer P, Klein KM. Solitary fibrous tumor of the orbit: CT and pathologic correlation. Neuroradiology. 1999;41(1):52–4.
Hasegawa T, Matsuno Y, Shimoda T, et al. Extrathoracic solitary fibrous tumors: their histological variability and potentially aggressive behavior. Hum Pathol. 1999;30(12):1464–73.
Hayashi N, Borodic G, Karesh JW, et al. Giant cell angiofibroma of the orbit and eyelid. Ophthalmology. 1999;106(6):1223–9.
Kim HY, Lee SY, Kang SJ, Kim HJ. Solitary fibrous tumor of the orbit: a poorly-recognized orbital lesion. Acta Ophthalmol Scand. 1999;77(6):704–8.
Alexandrakis G, Johnson TE. Recurrent orbital solitary fibrous tumor in a 14-year-old girl. Am J Ophthalmol. 2000;130(3):373–6.
Guillou L, Gebhard S, Coindre JM. Orbital and extraorbital giant cell angiofibroma: a giant cell-rich variant of solitary fibrous tumor? Clinicopathologic and immunohistochemical analysis of a series in favor of a unifying concept. Am J Surg Pathol. 2000;24(7):971–9.
Havlik DM, Farnath DA, Bocklage T. Solitary fibrous tumor of the orbit with a t(9;22)(q31;p13). Arch Pathol Lab Med. 2000;124(5):756–8.
Carrera M, Prat J, Quintana M. Malignant solitary fibrous tumour of the orbit: report of a case with 8 years follow-up. Eye (Lond). 2001;15(Pt 1):102–4.
Fenton S, Moriarty P, Kennedy S. Solitary fibrous tumour of the orbit. Eye (Lond). 2001;15(Pt 1):124–6.
Gigantelli JW, Kincaid MC, Soparkar CN, et al. Orbital solitary fibrous tumor: radiographic and histopathologic correlations. Ophthalmic Plast Reconstr Surg. 2001;17(3):207–14.
Giuffre I, Faiola A, Bonanno E, Liccardo G. Solitary fibrous tumor of the orbit. Case report and review of the literature. Surg Neurol. 2001;56(4):242–6.
Goldsmith JD, van de Rijn M, Syed N. Orbital hemangiopericytoma and solitary fibrous tumor: a morphologic continuum. Int J Surg Pathol. 2001;9(4):295–302.
Hayashi S, Kurihara H, Hirato J, Sasaki T. Solitary fibrous tumor of the orbit with extraorbital extension: case report. Neurosurgery. 2001;49(5):1241–5.
Lucci LM, Anderson RL, Harrie RP, et al. Solitary fibrous tumor of the orbit in a child. Ophthalmic Plast Reconstr Surg. 2001;17(5):369–73.
Takamura H, Kanno M, Yamashita H, Maeda K. A case of orbital solitary fibrous tumor. Jpn J Ophthalmol. 2001;45(4):412–9.
Holbach LM, Colombo F, Schlotzer-Schrehardt U, Kirchner T. Solitary fibrous tumor of the orbit presenting 20 years after Hodgkin's disease. Orbit. 2002;21(1):49–544.
O'Donovan DA, Bilbao JM, Fazl M, Antonyshyn OM. Solitary fibrous tumor of the orbit. J Craniofac Surg. 2002;13(5):641–4.
Polito E, Tosi GM, Toti P, Schurfeld K, Caporossi A. Orbital solitary fibrous tumor with aggressive behavior. Three cases and review of the literature. Graefes Arch Clin Exp Ophthalmol. 2002;240(7):570–4.
Bernardini FP, de Conciliis C, Schneider S, Kersten RC, Kulwin DR. Solitary fibrous tumor of the orbit: is it rare? Report of a case series and review of the literature. Ophthalmology. 2003;110(7):1442–8.
Johnson TE, Onofrey CB, Ehlies FJ. Echography as a useful adjunct in the diagnosis of orbital solitary fibrous tumor. Ophthalmic Plast Reconstr Surg. 2003;19(1):68–74.
Krishnakumar S, Subramanian N, Mohan ER, et al. Solitary fibrous tumor of the orbit: a clinicopathologic study of six cases with review of the literature. Surv Ophthalmol. 2003;48(5):544–54.
Luo SH, Kao SC, Pan CS. Solitary fibrous tumor of the orbit. J Formos Med Assoc. 2003;102(10):726–8.
Schellini SA, Hoyama E, Marques ME, Abreu ES, Yamashita S. Orbital solitary fibrous tumor: report of two cases and literature review. Jpn J Ophthalmol. 2003;47(4):415–8.
Hsu SS, Lai PH, Wang JS, Yip CM. Solitary fibrous tumor of the orbit. J Chin Med Assoc. 2004;67(9):483–6.
Galie M, Tieghi R, Cavazzini L, Clauser L. Solitary fibrous tumor of the orbit: a case report. Int J Oral Maxillofac Surg. 2005;34(3):331–3.
Ness GO, Lybaek H, Arnes J, Rodahl E. Chromosomal imbalances in a recurrent solitary fibrous tumor of the orbit. Cancer Genet Cytogenet. 2005;162(1):38–44.
Romer M, Bode B, Schuknecht B, Schmid S, Holzmann D. Solitary fibrous tumor of the orbit–two cases and a review of the literature. Eur Arch Otorhinolaryngol. 2005;262(2):81–8.
Cerda-Nicolas M, Lopez-Gines C, Gil-Benso R, et al. Solitary fibrous tumor of the orbit: morphological, cytogenetic and molecular features. Neuropathology. 2006;26(6):557–63.
Farah-Klibi F, Ferchichi L, Zairi I, et al. Lipomatous hemangiopericytoma (adipocytic variant of solitary fibrous tumor) of the orbit. A case report with review of the literature. Pathologica. 2006;98(6):645–8.
Mascarenhas L, Lopes M, Duarte AM, et al. Histologically malignant solitary fibrous tumor of the orbit. Neurochirurgie. 2006;52(5):415–8.
Meyer D, Riley F. Solitary fibrous tumor of the orbit: a clinicopathologic entity that warrants both a heightened awareness and an atraumatic surgical removal technique. Orbit. 2006;25(1):45–50.
Warraich I, Dunn DM, Oliver JW. Solitary fibrous tumor of the orbit with epithelioid features. Arch Pathol Lab Med. 2006;130(7):1039–41.
Kim HJ, Kim HJ, Kim YD, et al. Solitary fibrous tumor of the orbit: CT and MR imaging findings. AJNR Am J Neuroradiol. 2008;29(5):857–62.
Leoncini G, Maio V, Puccioni M, et al. Orbital solitary fibrous tumor: a case report and review of the literature. Pathol Oncol Res. 2008;14(2):213–7.
Miller NR, Agrawal N, Sciubba JJ, Lane AP. Image-guided transnasal endoscopic resection of an orbital solitary fibrous tumor. Ophthalmic Plast Reconstr Surg. 2008;24(1):65–7.
Mukherjee B, Biswas J. Solitary fibrous tumor of the orbit. Indian J Pathol Microbiol. 2008;51(3):453–5.
Tam ES, Chen EC, Nijhawan N, et al. Solitary fibrous tumor of the orbit: a case series. Orbit. 2008;27(6):426–31.
Das JK, Sharma AS, Deka A, Das D. Solitary fibrous tumor of the orbit presenting in pregnancy. Indian J Ophthalmol. 2009;57(3):238–40.
Demirci H, Shields CL, Eagle RC Jr, Shields JA. Giant cell angiofibroma, a variant of solitary fibrous tumor, of the orbit in a 16-year-old girl. Ophthalmic Plast Reconstr Surg. 2009;25(5):402–4.
Girnita L, Sahlin S, Orrego A, Seregard S. Malignant solitary fibrous tumour of the orbit. Acta Ophthalmol. 2009;87(4):464–7.
Pitchamuthu H, Gonzalez P, Kyle P, Roberts F. Fat-forming variant of solitary fibrous tumour of the orbit: the entity previously known as lipomatous haemangiopericytoma. Eye (Lond). 2009;23(6):1479–81.
Savino G, Aliberti S, Colucci D, Perrotta V, Balestrazzi E. Atypical presentation of a case of solitary fibrous tumor of the orbit. Orbit. 2009;28(2–3):176–8.
Adeleye AO, Ogun OA, Ogun GO. Orbital solitary fibrous tumor. Another rare case from Africa. Int Ophthalmol. 2010;30(3):315–8.
Feuerman JM, Flint A, Elner VM. Cystic solitary fibrous tumor of the orbit. Arch Ophthalmol. 2010;128(3):385–7.
Bandyopadhyay R, Ghosh AK, Roy R, et al. Solitary fibrous tumour of the orbit: an unusual presentation. J Indian Med Assoc. 2011;109(9):676–7.
Furusato E, Valenzuela IA, Fanburg-Smith JC, et al. Orbital solitary fibrous tumor: encompassing terminology for hemangiopericytoma, giant cell angiofibroma, and fibrous histiocytoma of the orbit: reappraisal of 41 cases. Hum Pathol. 2011;42(1):120–8.
Young TK, Hardy TG. Solitary fibrous tumor of the orbit with intracranial involvement. Ophthalmic Plast Reconstr Surg. 2011;27(3):e74–e7676.
Chen H, Xiao CW, Wang T, et al. Orbital solitary fibrous tumor: a clinicopathologic study of ten cases with long-term follow-up. Acta Neurochir (Wien). 2012;154(2):249–55.
Kitamura Y, Akiyama T, Hirose S, Yoshida K. Optic nerve sheath solitary fibrous tumor. Acta Neurochir (Wien). 2012;154(4):633–5.
Ribeiro SF, Chahud F, Cruz AA. Orbital hemangiopericytoma/solitary fibrous tumor in childhood. Ophthalmic Plast Reconstr Surg. 2012;28(3):e58–60.
Blandamura S, Alaggio R, Bettini G, et al. Four cases of solitary fibrous tumour of the eye and orbit: one with sarcomatous transformation after radiotherapy and one in a 5-year-old child's eyelid. J Clin Pathol. 2014;67(3):263–7.
Griepentrog GJ, Harris GJ, Zambrano EV. Multiply recurrent solitary fibrous tumor of the orbit without malignant degeneration: a 45-year clinicopathologic case study. JAMA Ophthalmol. 2013;131(2):265–7.
Meyer TN, Matos BH, Oliveira LR, Mendonca AT. Report of a case of solitary fibrous tumour of the orbit. Oral Maxillofac Surg. 2013;17(3):225–7.
Parrozzani R, Fusetti S, Montesco C, Favero V, Midena E. Biphasic solitary fibrous tumor of the orbit with distant metastases. Int Ophthalmol. 2013;33(6):701–5.
Polomsky M, Sines DT, Dutton JJ. Solitary fibrous tumor of the orbit with multiple cavities. Ophthalmic Plast Reconstr Surg. 2013;29(5):e117–e11919.
Rose AM, Kabiru J, Rose GE. A rare case of orbital haemangiopericytoma arising in childhood. Orbit. 2013;32(6):384–6.
Warner EJ, Burkat CN, Gentry LR. Orbital fibrous histiocytoma mimicking cavernous hemangioma on dynamic contrast-enhanced MRA imaging. Ophthalmic Plast Reconstr Surg. 2013;29(1):e3–5.
Graue GF, Schubert HD, Kazim M. Correlation between clinical features, imaging and pathologic findings in recurrent solitary fibrous tumor of the orbit. Orbit. 2013;32(6):375–80.
Kishimoto I, Shinohara S, Fujiwara K, et al. A case of intraorbital solitary fibrous tumor resected successfully with preoperative arterial embolization. Nihon Jibiinkoka Gakkai Kaiho. 2014;117(12):1477–82.
Le CP, Jones S, Valenzuela AA. Orbital solitary fibrous tumor: a case series with review of the literature. Orbit. 2014;33(2):145–51.
Liu Y, Li K, Shi H, Tao X. Solitary fibrous tumours in the extracranial head and neck region: correlation of CT and MR features with pathologic findings. Radiol Med. 2014;119(12):910–9.
Wallace KM, Alaraj A, Aakalu VK, Aletich V, Setabutr P. Endovascular preoperative embolization of orbital hemangiopericytoma with n-butyl cyanoacrylate glue. Ophthalmic Plast Reconstr Surg. 2014;30(4):e97–100.
Genc A, Toktas Z, Azman C, Bozkurt SU, Kilic T. Solitary fibrous tumor of the orbit: a case report and review of the literature. Turk Neurosurg. 2015;25(6):984–7.
Hashemi N, Ling JD, Soparkar C, et al. Transarterial onyx embolization of an orbital solitary fibrous tumor. Ocul Oncol Pathol. 2015;1(2):98–102.
Rahman T, Ahmed K, Sarmah J, Das A. Solitary fibrous tumor of orbit: a rare entity. Indian J Cancer. 2015;52(3):396–7.
Tenekeci G, Sari A, Vayisoglu Y, Serin O. Giant solitary fibrous tumor of orbit. J Craniofac Surg. 2015;26(5):e390–e392392.
Kunzel J, Hainz M, Ziebart T, et al. Head and neck solitary fibrous tumors: a rare and challenging entity. Eur Arch Otorhinolaryngol. 2016;273(6):1589–98.
Tata A, Cohen-Inbar O, Sheehan JP. Treatment of orbital solitary fibrous tumour with gamma knife radiosurgery and systematic review of literature. BMJ Case Rep. 2016;2016.
Feijo ED, Nery ACS, Caiado FR, Limongi RM. Solitary fibrous tumor of the lacrimal gland mimicking pleomorphic adenoma. Arq Bras Oftalmol. 2017;80(3):189–91.
Jung SK, Paik JS, Park GS, Yang SW. CD34 + tumours of the orbit including solitary fibrous tumours: a six-case series. BMC Ophthalmol. 2017;17(1):59.
Moriyama M, Kodama S, Hirano T, Suzuki M. Endoscopic-modified medial maxillectomy and its limitation for a solitary fibrous tumor of the lacrimal sac and nasolacrimal duct. Auris Nasus Larynx. 2017;44(3):370–4.
Smith SC, Gooding WE, Elkins M, et al. Solitary fibrous tumors of the head and neck: a multi-institutional clinicopathologic study. Am J Surg Pathol. 2017;41(12):1642–56.
Vu AF, Chundury RV, Blandford AD, Perry JD. Recurrent orbital solitary fibrous tumor in a 12-year-old. Ocul Oncol Pathol. 2017;3(2):83–6.
Alam S, Backiavathy V, Mukherjee B, Subramanian K. A rare case of giant multicystic solitary fibrous tumor of the orbit. Orbit. 2018;37(1):69–72.
Tanaka K, Yano H, Hayashi H, Hirano A. Total resection combined with osteotomy is more effective for orbital solitary fibrous tumor excision: a report of three cases. Int Ophthalmol. 2018;38(1):345–51.
Zheng L, McCluskey P, Ghabrial R. Largest reported orbital solitary fibrous tumour. Clin Exp Ophthalmol. 2018;46(3):301–3.
Demura M, Hayashi Y, Sasagawa Y, et al. Intraorbital solitary fibrous tumor requiring preoperative embolization of feeding artery. Asian J Neurosurg. 2019;14(2):593–7.
Hyde RA, Liu Y, Aakalu VK, Setabutr P. Solitary fibrous tumor of the orbit with growth during pregnancy: a case report. Orbit. 2019;38(3):256–8.
Sagiv O, Bell D, Guo Y, et al. Pathological features and clinical course in patients with recurrent or malignant orbital solitary fibrous tumor/hemangiopericytoma. Ophthalmic Plast Reconstr Surg. 2019;35(2):148–54.
Sayit AT, Elmali M, Gul A, Sullu Y. Solitary fibrous tumor of the orbit: Computed tomography and histopathological findings. J Cancer Res Ther. 2019;15(3):719–21.
Patel MM, Jakobiec FA, Zakka FR, et al. Intraorbital metastasis from solitary fibrous tumor. Ophthalmic Plast Reconstr Surg. 2013;29(3):e76–e7979.
Glazer-Hockstein C, Syed NA, Warhol M, Gausas RE. Malignant solitary fibrous tumor metastatic to the orbit. Ophthalmic Plast Reconstr Surg. 2004;20(6):471–3.
Pasquali S, Gronchi A, Strauss D, et al. Resectable extra-pleural and extra-meningeal solitary fibrous tumours: a multi-centre prognostic study. Eur J Surg Oncol. 2016;42(7):1064–70.
Salas S, Resseguier N, Blay JY, et al. Prediction of local and metastatic recurrence in solitary fibrous tumor: construction of a risk calculator in a multicenter cohort from the French Sarcoma Group (FSG) database. Ann Oncol. 2017;28(8):1979–87.
Demicco EG, Wagner MJ, Maki RG, et al. Risk assessment in solitary fibrous tumors: validation and refinement of a risk stratification model. Mod Pathol. 2017;30(10):1433–42.
Akaike K, Kurisaki-Arakawa A, Hara K, et al. Distinct clinicopathological features of NAB2-STAT6 fusion gene variants in solitary fibrous tumor with emphasis on the acquisition of highly malignant potential. Hum Pathol. 2015;46(3):347–56.
Barthelmess S, Geddert H, Boltze C, et al. Solitary fibrous tumors/hemangiopericytomas with different variants of the NAB2-STAT6 gene fusion are characterized by specific histomorphology and distinct clinicopathological features. Am J Pathol. 2014;184(4):1209–18.
Fritchie K, Jensch K, Moskalev EA, et al. The impact of histopathology and NAB2-STAT6 fusion subtype in classification and grading of meningeal solitary fibrous tumor/hemangiopericytoma. Acta Neuropathol. 2019;137(2):307–19.
Dagrada GP, Spagnuolo RD, Mauro V, et al. Solitary fibrous tumors: loss of chimeric protein expression and genomic instability mark dedifferentiation. Mod Pathol. 2015;28(8):1074–83.
Demicco EG, Wani K, Ingram D, et al. TERT promoter mutations in solitary fibrous tumour. Histopathology. 2018;73(5):843–51.
Demicco EG, Griffin AM, Gladdy RA, et al. Comparison of published risk models for prediction of outcome in patients with extrameningeal solitary fibrous tumour. Histopathology. 2019;75(5):723–37.
Chick JF, Chauhan NR, Madan R. Solitary fibrous tumors of the thorax: nomenclature, epidemiology, radiologic and pathologic findings, differential diagnoses, and management. AJR Am J Roentgenol. 2013;200(3):W238–W248248.
Font RL, Hidayat AA. Fibrous histiocytoma of the orbit. A clinicopathologic study of 150 cases. Hum Pathol. 1982;13(3):199–209.
Schweizer L, Koelsche C, Sahm F, et al. Meningeal hemangiopericytoma and solitary fibrous tumors carry the NAB2-STAT6 fusion and can be diagnosed by nuclear expression of STAT6 protein. Acta Neuropathol. 2013;125(5):651–8.
Thompson LDR, Lau SK. Sinonasal tract solitary fibrous tumor: a clinicopathologic study of six cases with a comprehensive review of the literature. Head Neck Pathol. 2018;12(4):471–80.
Meng W, Zhu HH, Li H, et al. Solitary fibrous tumors of the pleura with Doege-Potter syndrome: a case report and three-decade review of the literature. BMC Res Notes. 2014;7:515.
Fung EC, Crook MA. Doege-Potter syndrome and 'big-IGF2': a rare cause of hypoglycaemia. Ann Clin Biochem. 2011;48(Pt 2):95–6.
Ganly I, Patel SG, Stambuk HE, et al. Solitary fibrous tumors of the head and neck: a clinicopathologic and radiologic review. Arch Otolaryngol Head Neck Surg. 2006;132(5):517–25.
Thompson LD, Miettinen M, Wenig BM. Sinonasal-type hemangiopericytoma: a clinicopathologic and immunophenotypic analysis of 104 cases showing perivascular myoid differentiation. Am J Surg Pathol. 2003;27(6):737–49.
Lasota J, Felisiak-Golabek A, Aly FZ, et al. Nuclear expression and gain-of-function beta-catenin mutation in glomangiopericytoma (sinonasal-type hemangiopericytoma): insight into pathogenesis and a diagnostic marker. Mod Pathol. 2015;28(5):715–20.
Yammine K, Nasser HA, Hadi U, et al. Salvage preoperative embolization of an infratemporal solitary fibrous tumor: A case report with review of the literature. Medicine (Baltimore). 2018;97(13):e0251.
Acknowledgement
Data presented as Abstract #2, Poster #97 at the 109th Annual Meeting of the United States and Canadian Academy of Pathology, Los Angeles, CA: March 3, 2020.
Funding
No external funding was obtained for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest as it relates to this research project
Ethical Approval
All procedures performed in this retrospective data analysis involving human participants were in accordance with the ethical standards of the institutional review board (IRB #5968), which did not require informed consent. The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of Southern California Permanente Medical Group.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Thompson, L.D.R., Liou, S.S. & Feldman, K.A. Orbit Solitary Fibrous Tumor: A Proposed Risk Prediction Model Based on a Case Series and Comprehensive Literature Review. Head and Neck Pathol 15, 138–152 (2021). https://doi.org/10.1007/s12105-020-01184-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-020-01184-6