
Abstract
RNA modification is a post-transcriptional process by which certain nucleotides are altered after their initial incorporation into an RNA chain. Transfer RNAs (tRNAs) is the most heavily modified class of RNA molecules. These modifications expand the chemical and functional diversity of tRNAs and enhance their structural stability. To date, more than 100 modifications have been identified, the majority of which are specific from one domain of life. However, few modifications are extensively present in the three domains of life. Among those, the m1A nucleotide, which consists in the methylation at position 1 of the adenine aromatic ring, is found in tRNAs and ribosomal RNAs. In tRNAs, the m1A modification occurs at position 9, 14, 22, 57 and 58. The enzyme TrmK catalyzes the m1A formation at position 22. Here we report the backbone 1H, 15N and 13C chemical shift assignments of TrmK from Bacillus subtilis obtained by heteronuclear multidimensional NMR spectroscopy as well as its secondary structure in solution as predicted by TALOS+. These assignments of TrmK pave the way for interaction studies with its tRNA substrates.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Biological context
The biosynthesis of transfer RNAs (tRNAs) is a complex process composed of several steps leading to the formation of mature tRNAs with correct structures and functionalities. Apart from the processing of their extremities, the most salient property of the maturation process consists in the post-transcriptional incorporation of a large number of chemical modifications by the so-called modification enzymes. Among the modification enzymes, the methyltransferases (MTases) are the most frequent and diverse (Hori 2014). They catalyze the transfer of a methyl group from a methyl donor, mostly the S-adenosyl-l-methionine (SAM), towards different positions of the nucleotides. The m1A modification, which consists in the incorporation of a methyl group at position 1 of adenines, occurs on nucleotides 9, 14, 22 and 58 of tRNAs. This modification brings a positive charge on the adenine aromatic ring. We focused our work on the methyltransferase TrmK from Bacillus subtilis that catalyzes the methylation of adenine 22 of tRNASer and tRNATyr (Roovers et al. 2008). B. subtilis TrmK belongs to the COG2384 (Cluster of orthologous groups). The members of this family are found in Gram-negative and Gram-positive bacteria. Their sequences are well-conserved in many bacterial pathogens (L. monocytogenes, V. cholerae, S. pneumoniae…). Since TrmK is essential for cell viability in S. pneumoniae and since no homologues are found in humans, it was proposed to be a good target for the discovery of novel antibiotics (Thanassi et al. 2002). We solved the X-ray structure of B. subtilis TrmK (Dégut et al. manuscript in preparation) and used NMR chemical shift mapping to get insight on the protein-RNA recognition mode. We report here the backbone chemical shift assignments of TrmK, assignments that were necessary to interpret the NMR chemical shift mapping with its tRNA substrate.
Methods and experiments
Protein expression and purification
Recombinant B. subtilis TrmK was expressed and purified with a protocol adapted from Roovers et al. (2008). Since wild-type TrmK is prone to aggregation through cysteine oxidation, we produced a protein variant with cysteine to serine mutations (i.e. C35S and C152S), which abolished protein aggregation and retained full enzymatic activity (Dégut et al., manuscript in preparation). Mutagenesis was performed by use of the Quickchange site-directed mutagenesis kit (Stratagen). The presence of the desired mutations in trmK was checked by sequencing. This variant was overexpressed in BL21(DE3) E. coli cells, in rich labeled media or minimum media (see below) supplemented with kanamycin at 30 μg·mL−1. The cells were grown at 37 °C to OD600 ~0.6, cooled down at 18 °C and induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM. Cells were harvested 24 h after induction by centrifugation and frozen at −80 °C until further use. The frozen cells were suspended in 1/50 of the culture volume of a 50 mM Tris-HEPES buffer pH 8.2 containing 500 mM NaCl, 5 % glycerol and 1 mM of phenylmethanesulfonylfluoride (PMSF). The suspension was sonicated and the lysate was centrifuged for 30 min at 45,000 g. The resulting supernatant was loaded to a 5 mL Nickel Sepharose column (HisTrap, GE Healthcare) previously equilibrated with a 50 mM Tris–HCl buffer pH 8.0 containing 500 mM NaCl and 5 % glycerol (equilibration buffer). The resin was then washed with 30 mL of buffer and the protein was eluted with a gradient of the equilibration buffer supplemented with 500 mM imidazole pH 8.0. The N-terminal His6-tag of TrmK was removed by thrombin cleavage (25 U thrombin/mg of protein) performed overnight at 4 °C. PMSF at 250 µM, and EDTA at 1 mM were then added to the protein sample. The sample was concentrated with Amicon 10,000 MWCO (Millipore) and injected on a size exclusion chromatography column (Superdex-75 26/60, GE Healthcare) equilibrated with a 50 mM sodium phosphate buffer pH 7.0 containing 500 mM NaCl and 2 % glycerol.
Doubly labeled (15N/13C) and triply labeled (2H/15N/13C) TrmK samples were obtained by growing the cells in Spectra-9CN and Spectra-9DCN media, respectively (Spectra Stable Isotopes, Inc.). Singly labeled (15N) TrmK samples were obtained by growing the cells in M9 minimum media supplemented with 15NH4Cl. To achieve high yield of protein in the fully deuterated medium, a double selection protocol of the strain was performed as previously described (Sivashanmugam et al. 2009). Besides, specifically unlabeled samples were produced in M9 minimum media supplemented with 15NH4Cl and with non-labeled histidine, arginine, or lysine at a final concentration of 1 mM, 1 h before induction (Rasia et al. 2012). NMR samples of TrmK at ~0.7 mM were prepared in a 50 mM sodium phosphate buffer pH 7.0, 500 mM NaCl, 2 % glycerol, 1 mM EDTA, 250 µM PMSF and 10 % D2O. The sample was put in a 3 mm-diameter NMR tube.
NMR experiments
All NMR spectra were recorded at 15 °C on Bruker 600, 800, or 950 MHz spectrometers equipped with cryogenic probes. Backbone assignment was performed using the following standard 3D NMR experiments (Salzmann et al. 1998): TROSY-HNCA, TROSY-HNCACB, TROSY-HN(CO)CACB and [1H–15N] NOESY-HSQC. A 3D (H)N(COCA)NH experiment was also measured (Bracken et al. 1997). Internal DSS standard was used for direct referencing of the 1H chemical shifts, and indirect referencing of 15N and 13C shifts (Wishart et al. 1995). Data processing was carried out with Topspin 3.2 for standard acquisition, and with MddNMR (Orekhov and Jaravine 2011) for non-uniform sampling acquisition. Analysis of spectrum and backbone assignment was performed with the Sparky suite (Goddard and Kneller). Table 1 summarizes the NMR experiments used for the assignment.
Resonance assignments and data deposition
Before tackling the assignment of TrmK NMR signals, we needed to cope with problems of precipitation and proteolysis of TrmK. Indeed, TrmK precipitated during concentrations steps, needed high concentrations of salt (500 mM NaCl) and was subjected to intensive proteolysis within a few days after purification. Numerous conditions were tested using dialysis buttons. They allowed us to test the stability of TrmK in various conditions of buffer and temperature while working at high concentrations of TrmK (15 mg·mL−1) and low volumes (50 µL). As a result, TrmK does not precipitate at high concentration (>15 mg·mL−1) in a phosphate buffer whereas we observed precipitation more rapidly in a Tris–HCl buffer. A high concentration of salt (400–500 mM NaCl) limited the precipitation of TrmK and its proteolysis. Anti-proteases (PMSF, Pepstatine A, Leupeptine, EDTA) were tested. PMSF and EDTA revealed to be efficient to stop proteolysis. PMSF was added in the lysis buffer and at different stages of purification and in the NMR tube. Lastly, glycerol was necessary to prevent precipitation when concentrating TrmK. 5 % of glycerol was an optimal concentration during purification steps and for storage of the protein. For the NMR studies, 2 % of glycerol was used as the best compromise between the stability of the protein and the linewidth of NMR signals.
The protein sample in this study consists in 238 residues after the cleavage of the N-terminal His-tag and contains 5 prolines. NMR assignments were based on 3D heteronuclear NMR experiments performed on 2H/15N/13C-labeled TrmK. Interestingly, the use of the (H)N(COCA)NH experiment (Bracken et al. 1997), which correlates each NH group to the nitrogen of the amide group of the following residue in the sequence of the protein, revealed to be very efficient for the achievement and the validation of the assignments. This experiment is not sensitive and is commonly used for unfolded protein. However, with a fully deuterated protein, this experiment turned out to be very helpful in the assignment procedure. Besides, we used several specifically unlabeled samples to confirm the assignment or to provide starting point for the assignment (Fig. 1). For instance, Fig. 1 shows the unlabeling of all the lysines of TrmK. In this sample, all the amide groups of TrmK are 15N-labeled except those of lysines. Consequently, the lysines are not observable in the red spectrum of Fig. 1. The superimposition with a reference TROSY spectrum (in black, Fig. 1) allows one to easily localize the peaks originating from lysine amide groups.

Superimposition of two 1H–15N TROSY spectra of TrmK in black for TrmK uniformly 15N-labeled and in red for TrmK 15N-labeled except on lysines. Lysines are thus not observable on the red spectrum confirming immediately the assignment of amide group for each lysine of the protein
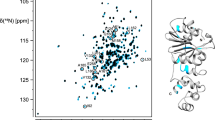
Backbone amide 1H–15N resonance assignment of TrmK was achieved for 222 of 233 non-proline residues, corresponding to 96 % of completeness (Fig. 2). Amide groups from residues M1, S28, H30, A31, L39, N40, H41, K42, T113, E116 and R117 could not be assigned the majority of which are found in loop regions. In addition, 98 % of Cα 98 % of Cβ and 92 % of CO were assigned. The backbone chemical shift assignments of TrmK were deposited in the BioMagResBank (www.bmrb.wisc.edu) under accession number 26744.

Two-dimensional 1H–15N TROSY spectrum of TrmK measured at 950 MHz and 15 °C on a protein sample uniformly deuterated and 15N/13C-labeled. Resonance assignments are indicated and reported in BMRB accession number 26744
An analysis of the chemical shifts of HN, N, CO, Cα and Cβ atoms was conducted with the TALOS+ webserver (Shen et al. 2009). Figure 3 compares the prediction of TrmK secondary structure obtained from TALOS+ with the secondary structure observed in the crystal structure of TrmK (Dégut et al. manuscript in preparation). The only light differences lie in the prediction from the NMR data of short α-helical turns (2 or three residues) around residue 68 and 165 that are not observed in the crystal structure. Therefore, both data are in very good agreement. The TrmK fold in solution and in the crystal is thus identical. Subsequently, the NMR footprint of binding of the tRNA on TrmK will be mapped on the X-ray structure of TrmK.

Prediction of TrmK secondary structure based on its backbone NMR chemical shifts using TALOS+ (Shen et al. 2009). The secondary structure prediction obtained with TALOS+ is shown as red bars for α-helices and yellow ones for β-strands, the height of the bars represent the probability of the secondary structure assigned by the software. Secondary structure of the TrmK X-ray structure is reported under the graph with yellow arrows for β-strands and red cylinders for α-helices
References
Bracken C, Palmer AG 3rd, Cavanagh J (1997) (H)N(COCA)NH and HN(COCA)NH experiments for 1H–15N backbone assignments in 13C/15N-labeled proteins. J Biomol NMR 9:94–100
Goddard TD, Kneller DG SPARKY 3. University of California, San Francisco
Hori H (2014) Methylated nucleosides in tRNA and tRNA methyltransferases. Front Genet 5:144. doi:10.3389/fgene.2014.00144
Orekhov VY, Jaravine VA (2011) Analysis of non-uniformly sampled spectra with multi-dimensional decomposition. Prog Nucl Magn Reson Spectrosc 59:271–292. doi:10.1016/j.pnmrs.2011.02.002
Rasia RM, Brutscher B, Plevin MJ (2012) Selective isotopic unlabeling of proteins using metabolic precursors: application to NMR assignment of intrinsically disordered proteins. ChemBioChem 13:732–739. doi:10.1002/cbic.201100678
Roovers M, Kaminska KH, Tkaczuk KL, Gigot D, Droogmans L, Bujnicki JM (2008) The YqfN protein of Bacillus subtilis is the tRNA: m1A22 methyltransferase (TrmK). Nucleic Acids Res 36:3252–3262. doi:10.1093/nar/gkn169
Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K (1998) TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci USA 95:13585–13590
Shen Y, Delaglio F, Cornilescu G, Bax A (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44:213–223. doi:10.1007/s10858-009-9333-z
Sivashanmugam A, Murray V, Cui C, Zhang Y, Wang J, Li Q (2009) Practical protocols for production of very high yields of recombinant proteins using Escherichia coli. Protein Sci 18:936–948. doi:10.1002/pro.102
Thanassi JA, Hartman-Neumann SL, Dougherty TJ, Dougherty BA, Pucci MJ (2002) Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res 30:3152–3162
Wishart DS et al (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR 6:135–140
Acknowledgments
Financial support from the TGIR-RMN-THC FR3050 CNRS for conducting the research is gratefully acknowledged. We thank Nelly Morellet for helpful discussions and technical advice.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Dégut, C., Barraud, P., Larue, V. et al. Backbone resonance assignments of the m1A22 tRNA methyltransferase TrmK from Bacillus subtilis . Biomol NMR Assign 10, 253–257 (2016). https://doi.org/10.1007/s12104-016-9680-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12104-016-9680-0