
Abstract
Juvenile dermatomyositis and juvenile scleroderma are rare multisystem autoimmune disorders. Although they share some pathognomonic hallmarks with adult onset myositis or scleroderma, there are significant differences in presentation, characteristics and associated features when the diseases present in childhood. In view of this, and the rarity of the conditions, it is important for care to be led by teams with expertise in pediatric rheumatology conditions. Prognosis has improved significantly in the West; likely due to early diagnosis and aggressive treatment with immunosuppressive medications. However, this trend is not replicated in the developing world. Early recognition of these diseases is crucial to achieve rapid and sustained remission and prevent disease or medication associated complications. This article aims to provide a practical overview for recognition, diagnosis and treatment of these conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Juvenile Dermatomyositis
Introduction
Juvenile dermatomyositis (JDM) is associated with significant mortality and morbidity in developing countries, with complications including calcinosis, lipodystrophy, contractures and muscle damage [1, 2]. Outlook can be improved by early recognition and aggressive treatment.
Epidemiology
JDM is the most frequent and best characterized juvenile Idiopathic Inflammatory Myopathy (IIM) with an incidence of 1.9–4 per million children per year and prevalence of 2.5/100,000 [3]. Peak onset is 7 y of age, with female predominance. Seasonal variations suggest environmental triggers (e.g., viruses / photosensitivity) which may instigate the disease in genetically predisposed individuals [4].
Clinical Features
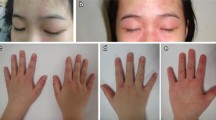
JDM is characterised by proximal muscle weakness and distinctive cutaneous findings (Fig. 1), but presenting features are variable and onset often insidious, hence classical features may not be seen at presentation [5–8]. Weakness is progressive and can become profound; starting with difficulty in climbing stairs or combing hair, progressing to an inability to roll over in bed. Children will often use compensatory manoeuvres. Neck flexor and abdominal muscle weakness should be looked for. Palatal / cricopharyngeal weakness results in nasal voice, swallowing difficulties or coughing during eating/ drinking, reflux into the nasopharynx or tracheal aspiration. Silent aspiration is recognised.

Typical cutaneous features associated with JDM: a Malar rash; b Erythema and dilatation of nailfold capillaries; c Heliotrope discoloration of the eyelids
Gowers’ sign is a useful screening test. Turning prone to rise from a supine position is a typical feature and is rarely seen in healthy children over 3 y of age [9]. Muscle strength should be assessed at diagnosis and serially during follow-up using the Childhood Myositis Assessment Scale (CMAS) and/ or Kendal Manual Muscle Testing (MMT8). Teaching videos are available on the International Myositis Assessment & Clinical Studies Group (IMACS) website [10].
Characteristic cutaneous features, reported in 72.9–97 % of cases at presentation, may be transient and precede muscle weakness [5, 7, 8]. The most frequent are heliotrope discoloration, often associated with periorbital edema, Gottron’s papules/ sign, and nailfold capillary changes (Table 1). An otoscope/ dermatoscope can help magnify nailbeds in clinic. Changes may be difficult to see in darker skin. Formal nailfold capillaroscopy can measure capillary density; a valuable marker of skin and muscle activity [11]. Other skin signs are common (Table 1) and should be looked for. Generalised subcutaneous edema or skin ulceration (reported in 5–30 % of patients) predicts a severe disease course with persistent weakness [5, 6, 8, 12].
Calcinosis, reported in 27.7 % in a recent Indian cohort [1], can be present at onset or later in the disease course (typically 1–3 y). It isassociated with delayed diagnosis, chronic disease or inadequate treatment. Calcium deposits may form plaques, nodules, sheets or a widespread exoskeleton, or liquefy to form pools of ‘milk of calcium’. These can extrude from the skin or become infected. They can cause pain, limit joint movement or cause tendons to shorten. Lipodystrophy (reported in 10–14 % patients), often difficult to reverse, can be associated with insulin-resistance or other metabolic abnormalities [1, 8, 12]. Skin disease is an important outcome measure in JDM, negatively impacting quality of life.
Fever, weight loss and fatigue are common constitutional features of JDM. Myalgia occurs frequently. Mouth ulcers, lymphadenopathy, Raynaud’s phenomenon, abdominal and chest pain are familiar symptoms, although non-specific.
Arthritis (typically polyarticular) or arthralgia occurs frequently [5, 7, 8, 12]. Children may not complain of pain but rather joint stiffness in the morning/ after rest. Contractures and muscle damage remain common in developing countries [1]; preventable by early aggressive treatment.
Gastrointestinal involvement includes dysphagia, abdominal pain, or ulceration. Gut vasculitis, although rare, can lead to perforation, and is an important cause of death [2]. Respiratory symptoms include dysphonia and dyspnea. A multicentre prospective study (n = 21) identified interstitial lung disease (ILD), aspiration pneumonia or respiratory muscle involvement in 76 % of patients [13]. Although less frequent in other cohorts [6, 7, 12], interstitial pneumonitis and aspiration pneumonia are important causes of death [2]. Cardiac involvement (pericarditis, myocarditis or arrhythmia) is rare in JDM but can be fatal [2]. Subclinical cardiovascular changes are recognised [14]. Cardiac symptoms may include chest pain, syncope or palpitations [12]. Neurological or ophthalmological involvement is rare. Major organ involvement, severe weakness or ulcerative skin disease puts a patient at high risk and warrants urgent transfer to a specialist centre (Table 2).
Investigations and Diagnosis
Diagnosis of definite JDM (using Bohan and Peter criteria) requires a characteristic rash (heliotrope, Gottron’s papules) plus three of the four muscle features: symmetrical muscle weakness, muscle biopsy evidence of myositis, elevation of serum levels of muscle-associated enzymes and electromyographic (EMG) triad of myopathy [15]. There has been a move away from using EMG or muscle biopsy in favour of MRI [4–7]. T2 weighted / STIR sequences detect edema in the myofascia and subcutaneous tissue. An MRI scoring system objectively defines acute inflammatory change [16]. Musculoskeletal ultrasound is a safe, inexpensive means of supporting diagnosis when MRI is unavailable or unsuitable, but requires operator expertise.
No one test is abnormal in all JDM cases. Investigations should be done to exclude systemic or neurological causes of myopathy, confirm diagnosis of IIM and define organ involvement. A muscle biopsy remains the gold standard and should be done where presentation is atypical; particularly when skin signs are absent, and to exclude muscular dystrophies/ mitochondrial cytopathies. Use of a standardized muscle biopsy tool helps to quantify severity of histological changes [17].
EMG or nerve conduction studies are no longer routinely carried out but remain important when diagnosis is uncertain to exclude neuropathy [5–7]. EMG does not reliably detect metabolic myopathies [18].
Measurement of muscle-derived enzymes should include creatinine phosphokinase (CPK), lactate dehydrogenase (LDH), alanine aminotransferase (ALT or SGPT), aspartate aminotransferase (AST or SGOT) and adolase (if available). Abnormalities are seen in 80–92 % of patients,but enzymes may be normal later in the disease course despite on-going disease activity or flare [6, 12]. More than 20 % of patients have normal CPK at presentation. CPK is moderately raised in IIM; if extremely high, consider other causes of myopathy.
Anti-nuclear antibody (ANA) is positive in approximately 70 % of patients but is non-specific and not diagnostic [12]. Other antibody tests may be useful in myositis overlap including anti-ENA, anti-dsDNA, Rheumatoid Factor (RF), and anti-thyroid antibodies. Myositis Specific Antibodies (MSA) such as anti-TIF 1-γ, anti-NXP2, anti-MDA5, anti-SRP, have the potential to aid diagnosis and prognosis by differentiating disease phenotypes but are not yet routinely available [4, 19].
Inflammatory markers (ESR, CRP) should be taken, but may be normal despite active disease. Likewise, raised serum von Willebrand factor may indicate disease exacerbations, but is not consistently elevated in active disease.
If clinically indicated, X-rays should be taken to determine extent of calcinosis and investigations done for ILD [chest radiograph, pulmonary function tests (PFTs), high resolution CT thorax (HRCT)], cardiac involvement (ECG, echocardiogram), or abdominal pathology (ultrasound). A speech and language assessment, videofluoroscopyand/or barium meal is indicated for dysphagia, dysphonia or symptoms of aspiration. Nailfold capillaroscopy is helpful when available.
Treatment
High dose corticosteroids (oral or intravenous) combined with methotrexate (MTX) has become the standard induction regimen for JDM [5–7]. This has been shown to result in shorter time to inactive disease compared to prednisolone alone in a randomised trial, with good safety profile [20]. If a newly diagnosed patient has inadequate response within the first 12 wk, intensification of treatment should be considered in consultation with an expert centre. Options may include addition of intravenous immunoglobulin (IVIG), or cyclosporin or switching MTX to mycophenolate mofetil (MMF) (Table 3) [4, 19, 21]. Consensus statements developed by Childhood arthritis and rheumatology research alliance (CARRA) propose combination therapies from time of diagnosis with steroid (oral plus / minus intravenous) with MTX, plus or minus IVIG [4].
Severe disease (such as skin ulceration, interstitial lung disease, gastrointestinal perforation) warrants treatment with intravenous cyclophosphamide [5–7, 19]. For refractory disease, B cell depletion therapy can be considered as adjunctive therapy if available but can take 26 wk to work [4]. Anti-TNF therapies are an alternative but infliximab and adalimumab may be more beneficial than etanercept [4, 19, 22].
Intensification of immunosuppressive therapy is recommended for developing or established calcinosis and may lead to regression of calcinosis over time. Anecdotal reports of treatments for calcinosis include diltiazem, infliximab, and bisphosphonates. Diclofenac can help inflammation around calcinotic deposits, and flucloxacillin can treat secondary infection that may prolong or worsen calcinosis. Topical tacrolimus (0.1 %) or topical corticosteroids may help localised skin disease [19]. However, expert opinion suggests that resistant skin disease reflects on-going systemic disease and should be treated by increasing systemic immunosuppression. JDM rashes can be triggered by ultraviolet light and adequate sun protection should be prescribed. Decreased bone mineral density is common in systemic rheumatic diseases and calcium/ vitamin D supplements, with or without bisphosphonates, may be required.
Steroid dose should be weaned as a patient shows clinical improvement. There is no high level evidence of when to stop therapy but consideration may be given to withdrawing treatment if a patient has been off steroids and in remission on MTX (or alternative disease modifying anti-rheumatic drug) for a minimum of 1 y.
Exercise for rehabilitation is extremely important to improve and maintain range of movement of the joints, muscle strength/ stamina and aerobic fitness [4, 19]. It should start at the time of diagnosis and form part of the treatment regime. A holistic approach is needed to deal with emotional and functional disease burden. Age appropriate patient reported outcome measures can be used to measure activity, participation and quality of life.
Outcomes
Over the last few decades, prognosis has significantly improved for JDM, but the disease continues to be associated with significant mortality and morbidity in developing countries. Delayed or inadequate steroid dose is one of the most important factors associated with poor prognosis and increased risk of calcinosis [19]. Persistence of skin rash is associated with longer time to remission and should be treated aggressively. Important causes of death in JDM are vasculopathic complications, major organ involvement and infections [2]. Early recognition and aggressive treatment is the key to improving outlook. All children with suspected IIM should be referred to a specialist centre, particularly those at high risk due to young age, major organ involvement, skin ulceration or disease complications.
Myositis Overlap and Mixed Connective Tissue Disease
Inflammatory myopathy may overlap with other autoimmune diseases in 3–11.2 % of cases [4, 8, 12]. The most common overlap is with scleroderma, but others include arthritis, lupus, or Sjögren’s. Patients are more likely to have Raynaud’s phenomenon, sclerodactyly, ILD, arthritis and gastro-intestinal symptoms than children with JDM [12]. ANA titres tend to be higher and anti-PM-Scl, anti-Ro, anti-La, anti-Sm or anti U1-RNP may be positive [12].
RNP positive mixed connective tissue disease (MCTD) is rare in children [23]. It has a female predominance. Features evolve over time; rashes of JDM / systemic lupus erythematosus (SLE), swollen hands, polyarthritis (frequently RF positive) and Raynaud’s phenomenon are common at onset, whilst scleroderma is more common later [23]. Vasculitis or vasculopathy can be severe. Hematological complications, such as resistant thrombocytopenia, are frequent. Sicca syndrome occurs in one-third of cases. Nephritis, seen in 25 % of patients is less severe than in SLE. Subclinical myositis is recognised. PFT abnormalities are common and can be asymptomatic. Cardiopulmonary disease and esophageal dysmotility are infrequent. Treatment should be directed towards the main symptoms. Long-term outlook is varied and unpredictable. Sclerodermatous features (sclerodactyly, vasculopathy and esophageal disease) can be resistant to treatment [23].
Juvenile Scleroderma
Juvenile scleroderma encompasses a spectrum of autoimmune diseases where chronic inflammation within the skin and subcutaneous tissues leads to fibrosis [24]. In children, localised scleroderma ismore common (93 %) than systemic scleroderma (7 %) [25]. Although the diseases share similar skin biopsy findings, fibrosis in localised scleroderma is limited to skin and subcutaneous tissues whereas organ fibrosis also occurs in systemic scleroderma.
Juvenile Localised Scleroderma
Juvenile localised scleroderma (JLS) subtypes include morphea (plaque, deep, pansclerotic or generalised) and linear scleroderma (affecting limbs and/or head). Atrophy of the skin and soft tissues is a complication. In growing children this can lead to joint contractures, limb length discrepancy and facial atrophy, which can have a marked psychological and functional effect (Fig. 2).

Child with JLS: a Linear scleroderma lesions affecting left arm leading to atrophy, limb length discrepancy and joint contracture (shown by asymmetry with non-affected right arm). b Same patient with patch of active morphea on left back with erythema and induration
Epidemiology
The reported incidence of JLS is 3.4 per million children per year, with two thirds of children having the linear subtype [25]. With female predominance, mean age of onset is 7.3 y [26]. There is often a significant delay in diagnosis of around one year and many cases are misdiagnosed initially [27, 28].
Clinical Features
Many children do not report symptoms but itching or abnormal sensation within the affected skin may suggest on-going disease activity. A thorough skin examination should be performed, preferably in natural light, to delineate extent of disease. New or extending lesions, induration, erythema or violaceous edge suggest active disease. Lesions can become tethered with dermal atrophy (skin may appear shiny, show visible vessels or cliff-drop sign) and subcutaneous atrophy (flattened or concave appearance) or may appear waxy and white. Hyper or hypopigmentation can occur.
Plaque morphea is characterised by round circumscribed areas of induration. Lesions may be superficial or deep with underlying subcutaneous fat atrophy. Generalised morphea has 4 or more large plaques involving two or more anatomical areas. Linear scleroderma presents with linear distribution of lesions that may extend from skin to subcutaneous tissues, muscle and bone. Linear lesions on the face can present as progressive facial hemiatrophy (also known as Parry Romberg Syndrome) or en coup de sabre lesions. Scalp lesions typically show loss of hair over affected skin and there may be loss of eyebrows/lashes. The temporomandibular joint and mouth should be examined for orthodontic abnormalities; with dental review if indicated.
In non-facial linear disease, the patient should be carefully assessed for gait disturbances, limb length discrepancy, differences in hand/foot size, joint contractures and associated arthritis.
Approximately one-fifth of patients with JLS have extra-cutaneous manifestations, which can be distant from the site of affected skin, most commonly articular, neurological and ocular [29]. The latter are more common in head and face lesions.
Investigations and Monitoring Disease Activity
Most children have normal inflammatory markers. ANA can be positive but is not diagnostically or prognostically useful [26]. Skin biopsy is unnecessary in most patients unless there is diagnostic uncertainty [30]. MRI and plain X-ray can help define depth of disease and associated bony changes. Serial photographs of skin lesions help monitor progression and response to treatment.
Because of the association with brain abnormalities, all children with head/facial lesions should have a baseline MRI brain [30]. Risk of uveitis in this group remains undefined but uveitis screening should be considered.
Many outcome measures have been studied in JLS but application in routine clinical practice is limited by availability e.g., thermography and laser Doppler [31, 32]. The Localised Scleroderma Cutaneous Assessment Tool (LoSCAT) is validated in JLS and is a useful, widely available tool [33]. It scores different anatomical areas based on clinician skin examination, with activity and damage indices.
Management
Children should be managed within a specialist team with expertise in JLS; joint clinics with pediatric rheumatologists and dermatologists are ideal.
There have been limited therapeutic trials within JLS with only one randomised placebo-controlled trial of systemic treatment. In this study, all children received oral prednisolone for 3 mo with either MTX or placebo. At 1 y, disease relapse occurred in 32.6 % of the MTX group compared to 70.8 % of the placebo group (p < 0.005). Use of other disease modifying anti-rheumatic drugs (DMARDs) in JLS is limited to a case series of MMF (n = 10) that appeared to be effective and well-tolerated. To date there are no published uncontrolled trials or case series of other DMARDs or biologics in JLS, although in practice these are used in refractory disease [34, 35]. Within the United Kingdom and United States of America, most pediatric rheumatologists use MTX and/or corticosteroids (oral or intravenous) as first line treatment [34, 36]. Topical therapies (e.g., corticosteroids, vitamin D analogues and tacrolimus) can be helpful adjuncts. Ultra-light therapies have also been used [37].
Physiotherapy and occupational therapy input is vital in children with functional impairment.
Outcomes
Studies of adults with childhood onset disease show persistent disease activity, although patients may experience long periods of disease inactivity [38, 39]. In one study, 56 % had permanent sequalae with 89 % having ongoing disease activity as adults [38], whereas in another study, 31 % of patients reported active disease after 10 y, with all but one patient having aesthetic sequelae and 38 % reporting functional limitations [39].
Juvenile Systemic Scleroderma
Juvenile systemic scleroderma or sclerosis (JSSc) is a rare and potentially life threatening disease. The hallmark features are inflammation, fibrosis and microvascular disease.
Epidemiology
The reported incidence of JSSc is 0.27 per million children per year [25]. Female to male ratio is approximately 4:1 [40, 41]. Mean age at onset in an international cohort was 8.1 y (range 0.4–15.6), although Indian children were older at 12 y with a longer time from onset to presentation (median 4 y) [40, 42].
Clinical Features
JSSc is characterised by skin induration that does not spare the fingers. Widespread and rapid skin thickening with early organ involvement is typical of diffuse cutaneous JSSc, whereas skin thickening is limited to distal extremities in limited cutaneous JSSc. Microvascular abnormalities manifest as Raynaud’s phenomenon, nailfold capillary changes, digital ulcers, pitting scars and ischemia. Compared to adults, children more commonly have overlap features with diseases such as JDM and SLE, with muscle involvement in one-third [41]. Table 4 shows the frequency of manifestations [40].
Investigations
Children may have positive auto-antibodies, including ANA (80 %), ENA (42 %), Scl-70 (34 %) or anti-centromere (7 %) [40]. Investigations at baseline should aim to identify organ involvement to define therapeutic decisions. Although cardiac involvement is uncommon, occurring in 5–10 %, it was the leading cause of death in JSSc [43]. Children should have a baseline ECG and echocardiogram, with consideration of 24 h cardiac monitoring for arrhythmias and cardiac MRI if symptomatic. Effective treatments exist for cardiopulmonary complications, so screening is vital even in asymptomatic children and should as a minimum include echocardiogram, ECG, PFT with diffusion capacity for carbon monoxide (DLCO) every 6 mo in early disease and annually in later disease.
Treatment
There are no therapeutic trials in JSSc; evidence is extrapolated from adult studies. Evidence based recommendations highlight organ-specific treatments (Table 4) [44]. In severe rapidly progressing disease, intravenous cyclophosphamide should be considered although biologics such as rituximab, tocilizumab and abatacept may also have a role.
Outcome
JSSc has a significant mortality, reported as 12 % in one study; all patients had diffuse disease, characterised by rapid progression and major organ involvement [43]. Adult survivors of JSSc appear to have similar organ involvement and survival compared to those with adult-onset SSc [45].
Abbreviations
- ALT:
-
Alanine aminotransferase
- ANA:
-
Anti-nuclear antibody
- AST:
-
Aspartate aminotransferase
- CARRA:
-
Childhood Arthritis and Rheumatology Research Alliance
- CMAS:
-
Childhood Myositis Assessment Scale
- CPK:
-
Creatinine phosphokinase
- CRP:
-
C-reactive protein
- DLCO:
-
Diffusion capacity for carbon monoxide
- DMARDS:
-
Disease modifying anti-rheumatic drugs
- dsDNA:
-
Double-stranded DNA
- ECG:
-
Electrocardiogram
- EMG:
-
Electromyography
- ENA:
-
Extractable nuclear antigens
- ESR:
-
Erythrocyte sedimentation rate
- HRCT:
-
High resolution computerised tomography
- IIM:
-
Idiopathic inflammatory myopathy
- ILD:
-
Interstitial lung disease
- IMACS:
-
International Myositis Assessment & Clinical Studies Group
- IVIG:
-
Intravenous immunoglobulin
- JDM:
-
Juvenile dermatomyositis
- JLS:
-
Juvenile localised scleroderma
- JSSc:
-
Juvenile systemic scleroderma
- LDH:
-
Lactate dehydrogenase
- LoSCAT:
-
Localised scleroderma cutaneous assessment tool
- MCTD:
-
Mixed connective tissue disease
- MMF:
-
Mycophenolate mofetil
- MMT:
-
Manual muscle testing
- MRI:
-
Magnetic resonance imaging
- MSA:
-
Myositis specific antibodies
- MTX:
-
Methotrexate
- PFTS:
-
Pulmonary function tests
- RCT:
-
Randomised controlled trial
- RF:
-
Rheumatoid factor
- SLE:
-
Systemic lupus erythematosus
- STIR:
-
Short T1 inversion recovery
- UK:
-
United Kingdom
References
Prasad S, Misra R, Agarwal V, Lawrence A, Aggarwal A. Juvenile dermatomyositis at a tertiary care hospital: is there any change in the last decade? Int J Rheum Dis. 2013;16:556–60.
Singh S, Suri D, Aulakh R, Gupta A, Rawat A, Kumar RM. Mortality in children with juvenile dermatomyositis: two decades of experience from a single tertiary care centre in North India. Clin Rheumatol. 2014;33:1675–9.
Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford). 2015;54:50–63.
Ernste FC, Reed AM. Recent advances in juvenile idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2014;26:671–8.
McCann LJ, Juggins AD, Maillard SM, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)—clinical characteristics of children recruited within the first 5 y. Rheumatology (Oxford). 2006;45:1255–60.
Robinson AB, Hoeltzel MF, Wahezi DM, et al. Clinical characteristics of children with juvenile dermatomyositis: the Childhood Arthritis and Rheumatology Research Alliance Registry. Arthritis Care Res (Hoboken). 2014;66:404–10.
Guseinova D, Consolaro A, Trail L, et al. Comparison of clinical features and drug therapies among European and Latin American patients with juvenile dermatomyositis. Clin Exp Rheumatol. 2011;29:117–24.
Ramanan AV, Feldman BM. Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin North Am. 2002;28:833–57.
Chang RF, Mubarak SJ. Pathomechanics of Gowers’ sign: a video analysis of a spectrum of Gowers’ maneuvers. Clin Orthop Relat Res. 2012;470:1987–91.
IMACS. International Myositis Assessment & Clinical Studies Group 2015. Available at: http://www.niehs.nih.gov/research/resources/imacs/diseaseactivity/index.cfm. Accessed on 21 April 2015.
Schmeling H, Stephens S, Goia C, et al. Nailfold capillary density is importantly associated over time with muscle and skin disease activity in juvenile dermatomyositis. Rheumatology (Oxford). 2011;50:885–93.
Shah M, Mamyrova G, Targoff IN, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Med (Baltimore). 2013;92:25–41.
Pouessel G, Deschildre A, Le Bourgeois M, et al. The lung is involved in juvenile dermatomyositis. Pediatr Pulmonol. 2013;48:1016–25.
Eimer MJ, Brickman WJ, Seshadri R, et al. Clinical status and cardiovascular risk profile of adults with a history of juvenile dermatomyositis. J Pediatr. 2011;159:795–801.
Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292:403–7.
Davis WR, Halls JE, Offiah AC, Pilkington C, Owens CM, Rosendahl K. Assessment of active inflammation in juvenile dermatomyositis: a novel magnetic resonance imaging-based scoring system. Rheumatology (Oxford). 2011;50:2237–44.
Varsani H, Charman SC, Li CK, et al. Validation of a score tool for measurement of histological severity in juvenile dermatomyositis and association with clinical severity of disease. Ann Rheum Dis. 2015;74:204–10.
Ghosh PS, Sorenson EJ. Diagnostic yield of electromyography in children with myopathic disorders. Pediatr Neurol. 2014;51:215–9.
Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371:2201–12.
Ruperto N, Pistorio A, Oliveira S, et al. A randomized trial in new onset juvenile dermatomyositis: prednisone versus prednisone plus cyclosporine versus prednisone plus methotrexate. [abstract]. Arthritis Rheum. 2012;64:2473.
Rouster-Stevens KA, Morgan GA, Wang D, Pachman LM. Mycophenolatemofetil: a possible therapeutic agent for children with juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2010;62:1446–51.
Riley P, McCann LJ, Maillard SM, Woo P, Murray KJ, Pilkington CA. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology (Oxford). 2008;47:877–80.
Tsai YY, Yang YH, Yu HH, Wang LC, Lee JH, Chiang BL. Fifteen-year experience of pediatric-onset mixed connective tissue disease. Clin Rheumatol. 2010;29:53–8.
Zulian F, Cuffaro G, Sperotto F. Scleroderma in children: an update. Curr Opin Rheumatol. 2013;25:643–50.
Herrick AL, Ennis H, Bhushan M, Silman AJ, Baildam EM. Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res (Hoboken). 2010;62:213–8.
Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford). 2006;45:614–20.
Hawley DP, Baildam EM, Amin TS, et al. Access to care for children and young people diagnosed with localized scleroderma or juvenile SSc in the UK. Rheumatology (Oxford). 2012;51:1235–9.
Weibel L, Laguda B, Atherton D, Harper JI. Misdiagnosis and delay in referral of children with localized scleroderma. Br J Dermatol. 2011;165:1308–13.
Zulian F, Vallongo C, Woo P, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52:2873–81.
Foeldvari I, Constantin T, Höger P, et al. Do we need a minimum standards in care for children with localized scleroderma- result of the consensus meeting in hamburg germany on the 11th of december 2011. Part I. Diagnosis and assessment of the disease. Arthritis Rheum. 2012;64:2004.
Martini G, Murray KJ, Howell KJ, et al. Juvenile-onset localized scleroderma activity detection by infrared thermography. Rheumatology (Oxford). 2002;41:1178–82.
Weibel L, Howell KJ, Visentin MT, et al. Laser doppler flowmetry for assessing localized scleroderma in children. Arthritis Rheum. 2007;56:3489–95.
Arkachaisri T, Vilaiyuk S, Torok KS, Medsger TA Jr. Development and initial validation of the localized scleroderma skin damage index and physician global assessment of disease damage: a proof-of-concept study. Rheumatology (Oxford). 2010;49:373–81.
Hawley DP, Pain CE, Baildam EM, Murphy R, Taylor AE, Foster HE. United Kingdom survey of current management of juvenile localized scleroderma. Rheumatology (Oxford). 2014;53:1849–54.
Li SC, Feldman BM, Higgins GC, Haines KA, Punaro MG, O’Neil KM. Treatment of pediatric localized scleroderma: results of a survey of North American pediatric rheumatologists. J Rheumatol. 2010;37:175–81.
Li SC, Torok KS, Pope E, et al. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res (Hoboken). 2012;64:1175–85.
Fett N, Werth VP. Update on morphea: part II. Outcome measures and treatment. J Am Acad Dermatol. 2011;64:231–42; quiz 43–4.
Saxton-Daniels S, Jacobe HT. An evaluation of long-term outcomes in adults with pediatric-onset morphea. Arch Dermatol. 2010;146:1044–5.
Piram M, McCuaig CC, Saint-Cyr C, et al. Short- and long-term outcome of linear morphoea in children. Br J Dermatol. 2013;169:1265–71.
Martini G, Foeldvari I, Russo R, et al. Systemic sclerosis in childhood: clinical and immunologic features of 153 patients in an international database. Arthritis Rheum. 2006;54:3971–8.
Scalapino K, Arkachaisri T, Lucas M, et al. Childhood onset systemic sclerosis: classification, clinical and serologic features, and survival in comparison with adult onset disease. J Rheumatol. 2006;33:1004–13.
Misra R, Singh G, Aggarwal P, Aggarwal A. Juvenile onset systemic sclerosis: a single center experience of 23 cases from Asia. Clin Rheumatol. 2007;26:1259–62.
Martini G, Vittadello F, Kasapcopur O, et al. Factors affecting survival in juvenile systemic sclerosis. Rheumatology (Oxford). 2009;48:119–22.
Kowal-Bielecka O, Landewe R, Avouac J, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR). Ann Rheum Dis. 2009;68:620–8.
Foeldvari I, Tyndall A, Zulian F, et al. Juvenile and young adult-onset systemic sclerosis share the same organ involvement in adulthood: data from the EUSTAR database. Rheumatology (Oxford). 2012;51:1832–7.
Contributions
Both authors contributed equally to the writing of this review and approved the final draft. LJM will act as guarantor for this paper.
Conflict of Interest
None.
Source of Funding
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
McCann, L.J., Pain, C.E. A Practical Approach to Juvenile Dermatomyositis and Juvenile Scleroderma. Indian J Pediatr 83, 163–171 (2016). https://doi.org/10.1007/s12098-015-1907-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-015-1907-z