
Abstract
Background
Transmembrane protein 92 (TMEM92) has been implicated in the facilitation of tumor progression. Nevertheless, comprehensive analyses concerning the prognostic significance of TMEM92, as well as its role in immunological responses across diverse cancer types, remain to be elucidated.
Methods
In this study, data was sourced from a range of publicly accessible online platforms and databases, including TCGA, GTEx, UCSC Xena, CCLE, cBioPortal, HPA, TIMER2.0, GEPIA, CancerSEA, GDSC, exoRBase, and ImmuCellAI. We systematically analyzed the expression patterns of TMEM92 at both mRNA and protein levels across diverse human organs, tissues, extracellular vesicles (EVs), and cell lines associated with multiple cancer types. Subsequently, analyses were conducted to determine the relationship between TMEM92 and various parameters such as prognosis, DNA methylation, copy number variation (CNV), the tumor microenvironment (TME), immune cell infiltration, genes with immunological relevance, tumor mutational burden (TMB), microsatellite instability (MSI), mismatch repair (MMR), and half-maximal inhibitory concentration (IC50) values.
Results
In the present study, we observed a pronounced overexpression of TMEM92 across a majority of cancer types, which was concomitantly associated with a less favorable prognosis. A notable association emerged between TMEM92 expression and both DNA methylation and CNV. Furthermore, a pronounced relationship was discerned between TMEM92 expression, the TME, and the degree of immune cell infiltration. Intriguingly, while TMEM92 expression displayed a positive correlation with macrophage presence, it inversely correlated with the infiltration level of CD8 + T cells. Concurrently, significant associations were identified between TMEM92 and the major histocompatibility complex, TMB, MSI, and MMR. Results derived from Gene Set Enrichment Analysis and Gene Set Variation Analysis further substantiated the nexus of TMEM92 with both immune and metabolic pathways within the oncogenic context.
Conclusions
These findings expanded the understanding of the roles of TMEM92 in tumorigenesis and progression and suggest that TMEM92 may have an immunoregulatory role in several malignancies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The worldwide incidence of cancer is both significant and on the rise [1]. The once-prevailing perception of cancer, primarily centered around tumor cells, has evolved to recognize the intricate interplay and reliance on the tumor microenvironment (TME). The TME profoundly influences the pathophysiological characteristics and therapeutic outcomes in numerous malignancies [2].
TMEMs are a type of protein that traverse the entirety of the lipid bilayer, firmly anchoring themselves within it [3]. Current research has identified numerous TMEMs that play pivotal roles in a spectrum of cellular activities. These include involvement in neurological disorders [4], epidermal keratinization [5], and glycosylation processes [6]. Notably, several TMEMs, including TMEM200A [7], TMEM205 [8], and TMEM9B [9], have been pinpointed as key regulators within the tumor immune microenvironment (TIME), further underscoring their significance in the dynamics of cancer progression.
The transmembrane protein 92 (TMEM92) is situated on the human chromosome within the 17q21.33 region and is classified as a member of the TMEM protein family. Recent research has suggested that TMEM92 functions potentially as a transmembrane adaptor, overseeing the regulation of membrane-associated proteins, notably cell surface receptors [10]. Specifically, TMEM92 has been identified to mediate the degradation of key proteins like β-catenin and E-cadherin by serving as an adaptor for E3 ubiquitin ligase [11]. Furthermore, in the context of pancreatic adenocarcinoma (PAAD), TMEM92 has emerged as a pioneering prognostic marker, providing valuable insights into clinical outcomes and the efficacy of immunotherapeutic interventions [12]. Past studies have posited that TMEM92 may operate as an oncogene, bolstering breast tumor cell proliferation, invasiveness, and motility through the modulation of epithelial-mesenchymal transition (EMT)-associated proteins [13]. Despite the mounting evidence positioning TMEM92 as a promising biomarker across diverse malignancies, a comprehensive understanding of the mechanisms underlying its role in tumorigenesis and progression, as well as its interplay with the TME, remains to be elucidated.
In the present study, we embarked on a meticulous exploration of multiple public databases to execute a thorough pan-cancer analysis centered on TMEM92. These encompassed evaluations of its expression profile, prognostic attributes, genetic modifications, DNA methylation patterns, and associated functional enrichments. Furthermore, we scrutinized the interrelationships between TMEM92 expression and immune-centric genes, the extent of immune cell infiltration, as well as the sensitivity to pharmacological agents.
Materials and methods
Data collection and TMEM92 expression analysis
The expression data for RNA-sequencing, alongside clinical information from the Cancer Genome Atlas (TCGA), Cancer Cell Line Encyclopedia (CCLE), and Genotype-Tissue Expression (GTEx), were procured from the UCSC XENA database (https://xenabrowser.net/datapages/). Utilizing this acquired data, an evaluation of TMEM92 expression was undertaken across 31 standard tissues, 33 tumor tissues, and an array of cancer cell lines. To ascertain differential expression between malignant and standard tissues in 33 cancer types, a comprehensive integration of data from both the GTEx and TCGA repositories was carried out. P < 0.05 was deemed statistically significant for discerning expression discrepancies between the aforementioned tissue types. All statistical analyses were executed using the R software platform, with subsequent visualization facilitated by the “ggplot2” package. Moreover, the expression profiles of TMEM92 within extracellular vesicles (EVs) were deduced from RNA-sequencing datasets encompassing diverse human bodily fluids, as provided by ExoRBase 2.0 (http://www.exorbase.org/). In addition, using the “Pathological Stage Plot” module available on GEPIA (http://gepia.cancer-pku.cn/), violin plots delineating TMEM92 expression across distinct pathological stages were obtained. Immunohistochemical visual representations showcasing TMEM92 protein expression in clinical cancer samples, as well as its subcellular localization within neoplastic cells, were sourced from the HPA database (https://www.proteinatlas.org/).
TMEM92 prognostic analysis in pan-cancer
Survival outcomes, specifically overall survival (OS), were extracted from the TCGA dataset. Based on the median expression value of TMEM92, patients were stratified into two distinct groups: high-TMEM92 and low-TMEM92. Survival analyses were subsequently conducted utilizing the Kaplan–Meier estimator coupled with the log-rank test. The generation of Kaplan–Meier survival curves was facilitated by the deployment of the “survival” and “survminer” packages within the R statistical environment.
Mutation analysis and methylation correlation
Data was procured from the cBioPortal platform (https://www.cbioportal.org/). Specifically, we selected the “TCGA Pan-Cancer Atlas Studies” under the “Quick select” category and input “TMEM92” to investigate its genetic alteration characteristics. Subsequent analysis of alteration frequency across all TCGA tumors was undertaken using the “Cancer Types Summary” module. Additionally, a comprehensive representation of the protein structure, inclusive of mutated sites within the 3D structure of TMEM92, could be discerned via the “Mutations” module. Methylation patterns and copy number variation (CNV) values related to TMEM92 were also extracted from the cBioPortal repository. Correlational analysis examining the relationship between TMEM92 methylation and prognostic implications in tumors was depicted through Kaplan–Meier survival plots.
TME analysis
Utilizing the computational algorithm formulated by Zeng et al., we probed the association between TMEM92 and TME [14]. The relationship between the expression levels of TMEM92 and the scores of gene signatures pertinent to TME was elucidated using the designated R package.
Utilizing the “ESTIMATE” package, we computed the following metrics within the TCGA cohort: ESTIMATEScore, ImmuneScore, StromalScore, and TumorPurity. The “ESTIMATE” algorithm, grounded on gene expression signatures, is designed to extrapolate the proportions of stromal and immune cells present within tumor samples [15]. Within the TME, an augmented presence of immune or stromal components typically leads to elevated ImmuneScore or StromalScore values, respectively.
Immune infiltration analysis
We leveraged three distinct datasets to explore the association between TMEM92 expression and the abundance of various immune cell infiltrates across diverse cancers. Initially, the panImmune feature matrix detailing immune characteristics, as delineated by Thorsson et al., published in Immunity, served as our primary data source for immune cell infiltrates [16]. Subsequently, data was retrieved from the ImmuCellAI database (http://bioinfo.life.hust.edu.cn/ImmuCellAI#!/), which similarly employed the CIBERSORT algorithm. In the final phase, we utilized TIMER2.0 (http://timer.cistrome.org/) to illustrate the interrelations between TMEM92 expression and various immune cell infiltrates. This was executed using an array of immune deconvolution algorithms, specifically: CIBERSORT, CIBERSORT_ABS, EPIC, MCPCOUNTER, QUANTISEQ, TIMER, and XCELL. Through the integration of these three datasets, a consistent narrative emerged, delineating the correlation between TMEM92 and the infiltration levels of the pertinent immune cells.
In our research, we further investigated the concomitant association between TMEM92 expression and various immune-related genes. This included genes associated with chemokines, chemokine receptors, the major histocompatibility complex (MHC), as well as immunosuppressive and immunostimulatory genes across a spectrum of cancers. For this analysis, we employed the “limma” package within the R statistical environment. To facilitate a visual representation of our findings, the “reshape2” and “RcolorBrewer” packages were used.
TMB, MSI, and MMR
In this study, the somatic mutation data related to tumor mutation burden (TMB) were sourced from the Genomic Data Commons (GDC) data portal, specifically from the UCSC Xena repository (https://portal.gdc.cancer.gov/). For each tumor sample, TMB assessment was conducted using the “mafTools” package. Microsatellite instability (MSI) data were derived based on methodologies outlined in a prior published study [17]. We used TCGA expression profile datasets to evaluate the expression levels of MMR genes—namely, MLH1, MSH2, MSH6, PMS2, and EPCAM—across various cancer types. Subsequently, to ascertain the association between TMEM92 expression and TMB, MSI, or MMR genes, a Spearman’s correlation analysis was conducted.
GSEA and GSVA analysis
To elucidate the potential signaling pathways influenced by TMEM92 in tumorigenesis, a Gene Set Enrichment Analysis (GSEA) was performed using the “clusterProfiler” package. From this analysis, the most significant 20 terms—drawn from both the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways—with an adjusted P < 0.05, were highlighted. Additionally, for a comprehensive analysis, the Gene Set Variation Analysis (GSVA) gene set was retrieved from the “hallmark gene sets” module available in the Molecular Signatures Database (MsigDB) accessible at (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). In furtherance of this, GSVA scores were computed to determine the correlations between TMEM92 expression levels and 50 well-established biological processes or states across all tumor samples. A synthesis of these correlation outcomes was provided for 33 tumor types.
Functional states analysis
In our study, the relationship between TMEM92 and 14 distinct cancer functional states was examined by leveraging single-cell sequencing data. This analysis was facilitated by the utilization of the “correlation plot” module accessible on the CancerSEA website (http://biocc.hrbmu.edu.cn/CancerSEA/home.jsp).
Drug sensitivity analysis
In this study, we investigated the association between TMEM92 expression and the half-maximal inhibitory concentrations (IC50) of 192 therapeutic compounds sourced from the GDSC2 database (https://www.cancerrxgene.org/) using Spearman’s correlation analysis.
Statistical analysis
Gene expression data underwent normalization via a log2 transformation. The relationship between two variables was ascertained either through Spearman’s or Pearson’s correlation tests. To compare differences between two distinct groups, either the Student’s t-test or the Wilcoxon rank sum test was employed. For comparisons involving more than two experimental groups, one-way analysis of variance (ANOVA) was used. For the assessment of survival outcomes, both the Kaplan–Meier method and univariate Cox regression analysis were employed. The statistical evaluations of the bioinformatics results were carried out with R software (Version 3.9.1). A P value of < 0.05 was deemed statistically significant. The significance levels were denoted as follows: *, P < 0.05; **, P < 0.01; and ***, P < 0.001.
Results
Pan-cancer analysis of TMEM92 expression
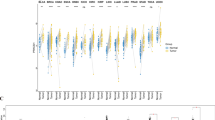
Using the GTEx datasets, we initiated an assessment of the physiologic gene profiles of TMEM92 across an array of normal tissues, subsequently categorizing the expression from low to high. As delineated in Fig. 1A, the small intestine manifested the most pronounced expression of TMEM92, while the majority of other standard samples predominantly displayed diminished levels of TMEM92. Furthermore, an exploration of TMEM92 expression across diverse tumors, as indexed in the TCGA database, revealed heightened expression in LUAD, PAAD, and STAD, yet diminished in KICH (Fig. 1B). Given the limited array of normal samples in the TCGA, a comprehensive analysis was facilitated by amalgamating data from both the GTEx and TCGA datasets. This enlarged dataset facilitated an investigation into the expression across 33 tumor types, revealing that TMEM92 mRNA expression was significantly augmented in 21 distinct tumors, inclusive of BLCA, BRCA, CESC, COAD, ESCA, GBM, HNSC, KIRC, KIRP, LAML, LGG, LUAD, LUSC, OV, PAAD, READ, SKCM, TGCT, THCA, UCEC, UCS (Fig. 1C). Contrarily, a decrease in TMEM92 was observed in DLBC, KICH, and PRAD (Fig. 1C). Proceeding with data from the CCLE database, it was discerned that TMEM92 expression levels were predominantly elevated across tumor cell lines, reaching a zenith in KIRC (Fig. 1D). Drawing from the ExoRBase 2.0 data, it was evident that TMEM92 expression levels in EVs (The sources are blood; urine; CSF and bile) were generally subdued in a majority of tumor cell lines, albeit an upsurge was noted in ESCC and MEL (Fig. 1E).

Profiling TMEM92 expression in cancers. (A) TMEM92 expression pattern in 31 types of normal samples from the GTEx database. (B, C) TMEM92 mRNA expression in pan-cancer based on TCGA(B) and GTEx-TCGA database (C). (D) TMEM92 expression pattern in 30 kinds of tumor cell lines from CCLE database. (E) Expression profiles of TMEM92 in extracellular vesicles (EVs) from ExoRBase 2.0. (F–L) Differential expression of TMEM92 in paired tumors and adjacent normal tissues from TCGA database. (M–Q) The correlation between TTMEM92 expression and the pathological stages of cancers using GEPIA. (R–U) The protein expression of TMEM92 in immunohistochemical images of normal (left) and tumor (right) groups from the HPA database. The tumors examined are as follows: (R) LIHC, (S) KIRC, (T) OV, and (U) BLCA. (V) Immunofluorescence staining of the microtubules, endoplasmic reticulum (ER), and nucleus for the subcellular distribution of TMEM92 expression in A549, CACO-2, and UOS cells based on the HPA database
In a subsequent analysis of paired tumors from the TCGA dataset, TMEM92 displayed an elevated expression in BRCA, HNSC, KIRC, KIRP, LUAD, and THCA, whereas a decreased expression was observed in KICH when juxtaposed with their respective matched normal samples (Fig. 1F–L). Concurrently, box plots demonstrated significant disparities in TMEM92 expression between normal and tumor specimens across 12 distinct cancer categories, as sourced from the TCGA dataset (Fig. S1A–L). A deeper probe into the data revealed heterogeneous TMEM92 expression across various stages of BRCA, KICH, KIRC, PAAD, and THCA (Fig. 1M–Q). Nevertheless, a meaningful correlation between TMEM92 expression and disease stage was not discernible for other cancer types (Fig. S2A–S). Focusing on protein expression, the HPA database was used to assess TMEM92 protein levels. The interrogation revealed that in normal tissues, including liver, kidney, ovary, urinary bladder (Fig. 1R–U), cerebral cortex, endometrium, lung, and skin (Fig. S1M–P), TMEM92 was either not detected or only faintly visible. In stark contrast, the corresponding tumor specimens exhibited moderate to pronounced staining for TMEM92. Concurrently, immunofluorescence (IF) imagery for the TMEM92 protein, as sourced from the HPA, indicated its primary localization within the nucleoplasm in A549, CACO-2, and U2OS cancer cell lines (Fig. 1V). Cumulatively, these observations underscore that the upregulation of TMEM92 is pervasive at both mRNA and protein levels in a majority of the evaluated malignancies.
Prognostic significance of TMEM92
We explored the prognostic potential of TMEM92 across a spectrum of cancers. Our investigations revealed that heightened TMEM92 expression corresponded with reduced survival durations in LUSC, LGG, PAAD, HNSC, ACC, BRCA, CESC, and CHOL, as evidenced by Cox regression survival analysis (Fig. 2A). This observation was congruent with findings from the Kaplan–Meier survival analysis (Fig. 2B–H). Consequently, TMEM92 appears to hold prognostic relevance across diverse malignancies.

Correlation between TMEM92 expression and OS. (A) Correlation between TMEM92 expression and OS of patients using the Cox regression survival analysis. (B–H) Kaplan–Meier survival curves revealed that TMEM92 expression was highly correlated with clinical outcomes in different cancers
The genetic alteration landscape and methylation analysis of TMEM92 in pan-cancer
By using the cBioPortal platform for genomic analysis, we observed that the predominant genetic modification related to TMEM92 was characterized as “amplification.” This pattern was discernible across virtually all TCGA cancer cases. Notably, breast invasive carcinoma exhibited the most pronounced alteration frequency, registering at 6.55% (Fig. 3A).

Mutation feature of TMEM92 in different tumors of TCGA. The alteration frequency with mutation type (A) and mutation site (B) displayed using the cBioPortal tool. The mutation site with the highest alteration frequency (P138Lfs*86) in the 3D structure of TMEM92 (C)
The cumulative somatic mutation frequency for TMEM92 was registered at 0.3%. Within this, missense mutations constituted the primary category (Fig. 3B). The specific alteration of P138Lfs*86 was identified in multiple instances: 3 cases of UCEC, 2 of STAD, 1 of COAD, and 1 of HNSC. The P138 locus was conspicuously evident in the 3D structural representation of the TMEM92 protein (Fig. 3C). Within the altered group, several genes, namely ALOX12P1, C9ORF85, RUN6-69P, RPS15, LINC00971, and PLK5, demonstrated a propensity for genetic modification (Fig. S3A).
Subsequently, we conducted the Pearson’s correlation between TMEM92 CNV and its mRNA expression. As depicted in Fig. 4A, a positive correlation between CNV and TMEM92 expression was evident across a substantial number of tumor types. Contrarily, in PAAD and THCA, an inverse correlation was observed (Fig. 4A). Figure 4B–G presents the six tumor types exhibiting the most pronounced correlation scores.

CNV and methylation of TMEM92. (A) The relationship between TMEM92 expression and CNV. (B–G) The top 6 positively correlated. (H) The correlation between TMEM92 expression and methylation. (I–N) The top 6 negatively correlated. (O–W) Kaplan–Meier curves illustrate the relationships between TMEM92 methylation levels with OS (O–Q)
In the broader context of pan-cancer analysis, we mapped the DNA methylation landscape of TMEM92. As depicted in Fig. 4H, the lollipop chart elucidates a salient correlation between TMEM92 expression and its methylation across 33 distinct tumor types. Predominantly, the data suggests an inverse relationship between TMEM92 expression and its gene promoter methylation as quantified by the Pearson’s correlation coefficient. Figure 4I–N depicts the six tumor types manifesting the most pronounced correlation metrics. To deepen the understanding on the interplay between promoter methylation and prognostic implications, we conducted a Kaplan–Meier survival analysis spanning various cancers. Notably, for patients diagnosed with BLCA and UVM, a trend towards compromised OS was observed in the presence of promoter hypermethylation, as visualized in Fig. 4O–P. Conversely, for patients with LGG, promoter hypermethylation appeared to be associated with an enhanced survival prognosis, as illustrated in Fig. 4Q.
Correlation analysis between TME and TMEM92 expression
The TME plays a pivotal role in modulating tumor progression, metastatic potential, and therapeutic responsiveness [18]. In light of this, we endeavored to elucidate the relationship between TMEM92 expression and various facets of the TME. Figure 5A, showcasing a heatmap, depicts the degree of correlation between TMEM92 expression and specific TME parameters. Notably, EMT2, EMT3, Pan_F_TBRs, EMT1, and antigen_processing_machinery exhibited a positive correlation with TMEM92 expression.

The role of TMEM92 in the tumor microenvironment. (A) Heatmap presenting the correlation strength between TMEM92 expression and TME-related terms in pan-cancer. (B–D) Lollipop plots displaying the correlations between TMEM92 expression and ESTIMATEScore (B), ImmuneScore (C), and StromalScore (D) in pan-cancer, respectively. The top five tumors with the most significant correlation between the degree of immune infiltration and TMEM92 expression for ESTIMATEScore (E); for ImmuneScore (F); for StromalScore (G), solely
To bolster the evidence supporting the involvement of TMEM92 within the TME, we analyzed the ESTIMATEScore, ImmuneScore, and StromalScore across 33 cancer types, deriving these scores from expression data profiles (Fig. 5B–D). Insights garnered from the ESTIMATE algorithm revealed that in cancers such as THCA, KICH, KIRC, GBM, ACC, KIRP, LIHC, BRCA, PAAD, LUSC, and PRAD, TMEM92 expression exhibited a positive correlation with ESTIMATEScore, ImmuneScore, and StromalScore.
Furthermore, when ranked by the magnitude of correlation, the top quintet of cancers displaying a pronounced association between TMEM92 expression and TME-associated scores were KICH, THCA, KIRC, GBM, and PCPG as per ESTIMATEScore (Fig. 5E); THCA, KICH, KIRC, ACC, and KIRP as per ImmuneScore (Fig. 5F); and KICH, THYM, DLBC, THCA, and PCPG according to StromalScore (Fig. 5G).
Subsequent analyses discerning the correlation between TMEM92 expression levels and a spectrum of immune-related genes—encompassing those involved in immune activation (Fig. S4A), immunosuppression (Fig. S4B), chemokine synthesis (Fig. S4C), chemokine receptor expression (Fig. S4D), as well as MHC (Fig. S4E)—were executed across various cancer types. Intriguingly, the majority of these immune-related genes manifested co-expression patterns with TMEM92, with the exception being in DLBC. This robust association underscores the profound involvement of TMEM92 in the TIME.
The relevance of TMEM92 expression and immune cell infiltrates in pan-cancer
Immune cells, encompassing components of both the adaptive and innate immune systems, permeate the TME and exert considerable influence over the trajectory of tumor evolution [19]. In our effort to delineate the nexus between immune cell infiltration and TMEM92 expression at a pan-cancer scale, we leveraged data concerning immune cell infiltration sourced from an array of databases, subsequently undertaking correlation analyses. It was discerned that the expression of TMEM92 exhibited a positive correlation with the infiltration intensities of NKTs, monocytes, Th17 cells, nTregs, and macrophages. Conversely, a negative correlation was identified in relation to CD8 + T cells, CD8 + naive T cells, T cells gamma delta, and B cells across a majority of cancer types (Fig. 6A). Upon executing sequence analyses via the TIMER2.0 portal, a marked positive association was evident between TMEM92 expression and the infiltration of macrophages. Simultaneously, a conspicuous negative correlation surfaced between TMEM92 and the infiltration degrees of CD8 + T cells (Fig. 6B). Correlations with additional immune cell varieties are depicted in Fig. S3B. Along with these findings, data sourced from “The Immune Landscape of Cancer” reaffirmed that TMEM92 expression correlates positively with macrophage levels (Fig. 6C) and negatively with CD8 + Tcell infiltration (Fig. 6D). Synthesizing the aforementioned data, it can be inferred that TMEM92 may significantly influence its oncogenic potential in a plethora of tumor types by modulating the infiltration dynamics of immune cells.

Relationship between TMEM92 expression and immune cell infiltration in different cancers. (A) Correlations between TMEM92 expression level and immune cells in pan-cancer using the ImmuCellAI database. (B) Correlation between TMEM92 expression and the infiltration of macrophages and CD8+ T cell, utilizing the TIMER 2.0 database. (C, D) Scatter plots of macrophages (C) and CD8+ T cell (D) infiltration levels related to TMEM92 expression are presented
The relationships between TMEM92 expression and TMB, MSI, and MMR
Alterations in TMB, MSI, and MMR often culminate in the accumulation of neo-antigens, which in turn augments the likelihood of T cell recognition [20]. In light of this, we undertook an investigation to elucidate the interrelationship between TMEM92 expression and the parameters of TMB, MSI, and MMR. Analysis revealed a positive correlation between TMEM92 expression and TMB in both TGCT and ESCA. Conversely, a negative association was identified in BLCA, LUAD, and READ (Fig. 7B). Furthermore, significant associations between TMEM92 expression and MSI were observed in six cancer types: ESCA, TGCT, MESO, COAD, OV, and UCS. Notably, patients with OV and UCS demonstrated a negative correlation with TMEM92 expression, whereas the remaining cancer types manifested an inverse correlation (Fig. 7C). Detailed scatter plots for each of the aforementioned tumors are presented in Fig. S5A, B.

Expression of TMEM92 is correlated with TMB, MSI, and MMR. (A) The heatmap displays the correlations between TMEM92 and MMR genes in pan-cancer. Radar maps of correlations between TMEM92 expression and TMB (B) and MSI (C)
Subsequent to discerning a link between TMEM92 expression and the mutation indicators TMB and MSI, we delved deeper to ascertain the relationship between TMEM92 expression and deficits in MMR. Our analysis revealed prominent associations across 33 cancer types, with the exceptions being READ, UVM, GBM, DLBC, MESO, LAML, BRCA, PCPG, and LIHC (Fig. 7A). Cumulatively, these results suggest that TMEM92 could serve as a prospective biomarker for genome stability across most cancerous conditions.
Functional enrichment analysis of TMEM92 in multiple cancers
In an endeavor to discern the biological processes or signaling cascades intertwined with TMEM92 in oncological contexts, we utilized GSEA and GSVA (Fig. 8). Through GO terms, our findings underscored the profound involvement of TMEM92 in immune regulatory pathways across pan-cancer, prominently in domains such as adaptive immune response, immune effector processes, activation of multiple cells within the immune response, and both positive and negative modulations of the immune system process. This also extended to the production and regulation of cytokines (Fig. 8A–F). Concurrently, KEGG analysis signified a prominent connection between TMEM92 and pathways like cytokine-cytokine receptor interaction and ECM-receptor interaction, among other established pathways. Additionally, metabolic pathway associations were unearthed specifically within DLBC, ESCA, and LUAD contexts.

GSEA and GSVA of TMEM92 in pan-cancer. GO annotations of TMEM92 in the indicated six types of tumors, including ACC (A), CESC (B), DLBC (C), ESCA (D), LUAD (E), and SKCM (F). KEGG annotations of TMEM92 in the indicated six types of tumors, including ACC (G), CESC (H), DLBC (I), ESCA (J), LUAD (K), and SKCM (L). The top 20 GSEA terms are displayed. (M) Heatmap for different hallmark pathway enrichment scores with TMEM92 expression level. (N) GSVA data of TMEM92 in the indicated six types of cancers
A visualization of GSVA for assorted hallmark pathway enrichment scores with TMEM92 expression levels is illustrated as a heatmap (Fig. 8M), leveraging data from the MsigDB database. Our analysis pinpointed a positive correlation of TMEM92 with several oncogenic pathways, notably hypoxia, EMT, and angiogenesis, alongside pathways pertinent to substance metabolism. The cellular immune factor inflammatory responses, encompassing factors like interferon-gamma, interferon-alpha, IL6, and TGF-beta, manifested a pronounced positive association with TMEM92 across a multitude of tumors. Such findings are congruently echoed in Fig. 8N.
The functional states of TMEM92 across different cancers in scRNA-Seq datasets
To explore the potential pathophysiological mechanisms underlying the role of TMEM92 in oncology, we examined the interrelation between TMEM92 and the functional states of various cancers at the single-cell resolution, leveraging the CancerSEA database. Our analysis unveiled a positive correlation between TMEM92 expression and several cellular processes, notably angiogenesis, metastasis, quiescence, differentiation, and hypoxia. Conversely, TMEM92 expression exhibited a pronounced negative correlation with processes such as the cell cycle, DNA damage, DNA repair, and proliferation (Fig. 9A). The intricacies of this correlation between TMEM92 and the functional states across eight distinct cancer types are delineated in Fig. 9B–I. It is particularly noteworthy that the relationship between TMEM92 and functional states may exhibit a stronger correlation in lung adenocarcinoma, a subtype of non-small cell lung cancer, compared to other pathological types of lung cancer (Fig. 9B, C).

CancerSEA was used for single-cell analysis to determine the functions of TMEM92. (A) Average correlations between TMEM92 and functional states in different cancers from cancerSEA. (B–I) Functional relevance of TMEM92 in LUAD (B), NSCLC (C), RCC (D), MEL (E), RB (F), UM (G), HNSCC (H), and BRCA (I) from cancerSEA. Red plots suggest a positive correlation while blue plots suggest a negative correlation
Correlation of sensitivity to drug (IC50) with TMEM92 expression
Gene modifications play a pivotal role in determining clinical treatment responses [21]. In light of this, an investigation was conducted to discern the relationship between mRNA expression levels of TMEM92 and IC50 values relative to antineoplastic agents as sourced from the GDSC database. Our analysis covered 192 drugs, and we ultimately determined that 151 drugs were statistically correlated with TMEM92. Notably, 144 drugs exhibited a positive correlation while 7 manifested a negative correlations. Drugs displaying the most robust positive correlations comprised AZD5991, BIBR-1532, EPZ004777, Sabutoclax, Venetoclax, Pyridostain, PCI-34051, and Telomerase Inhibitor IX. Comprehensive results from the drug sensitivity evaluation are documented in the Supplementary Table S1. It is pertinent to highlight that widely prescribed antineoplastic agents in carcinoma management—namely cisplatin, gemcitabine, sorafenib, paclitaxel, docetaxel, crizotinib, oxaliplatin, and tamoxifen—presented elevated IC50 metrics in patients manifesting heightened TMEM92 expression levels.
Discussion
Carcinogenesis is fundamentally underpinned by the ubiquitous presence of driver mutations and epigenetic aberrations even within ostensibly normal tissues [22]. In our present work, a comprehensive pan-cancer assessment of TMEM92 was conducted. The genetic alterations landscape of TMEM92 across diverse cancer types was unveiled, with amplification emerging as the predominant alteration. DNA methylation, an epigenetic mechanism pivotal in modulating gene expression, exhibits aberrant patterns strongly linked to oncogenesis [23]. We discerned a pronounced inverse correlation between TMEM92 expression and DNA methylation. Furthermore, survival analyses across cancers indicated that hypermethylation of the TMEM92 promoter was associated with improved OS in patients diagnosed with LGG, conversely being detrimental for patients diagnosed with UVM and BLCA. This directed our hypothesis towards the possible involvement of the mutational and epigenetic states of TMEM92 in tumorigenesis.
The TME is not a mere passive entity; rather, it actively propels cancer progression [24]. Our findings underscored a positive correlation between TMEM92 expression across cancers and TME, particularly in relation to EMT markers—processes known to augment tumor initiation and metastatic capacity [25]. It is empirically supported that TMEM92 modulates EMT-associated proteins, bolstering breast cancer cell viability [13]. Aligning with this, our dataset indicates that TMEM92 may influence the EMT cascade and instigate carcinogenic processes. Moreover, evaluations of StromalScore and ImmuneScore gauged the degree of stromal and immune cell infiltration, with higher scores and diminished tumor purity correlating with advanced tumor stage and poor OS [26].
Anti-tumor immunity is primarily orchestrated by immune cells capable of potent immune reactions [27, 28]. Nevertheless, malignant cells have evolved intricate mechanisms, such as augmenting negative regulatory pathways, to elude immune scrutiny [19]. The presence of immunosuppressive cells is intricately linked with resistance to immunotherapy [29]. In our study, we analyzed the abundance of immune cells across 33 cancer types. The data revealed a positive correlation between TMEM92 expression and macrophage levels, but a negative correlation with CD8 + T cell infiltration. Such patterns suggest that TMEM92 may interact with immune cells across a spectrum of cancers, potentially serving as an indicator of immune responsiveness and efficacy of immunotherapies.
MSI testing and TMB stand as genomic indicators, identifying patients poised to benefit from immune checkpoint inhibitors [30]. TMB is regarded as a prognostic marker for pembrolizumab monotherapy in cases of advanced tumors, while detection of high MSI via liquid biopsy has been associated with sustained immunotherapy responses in pancreatic cancer [31, 32]. Our findings linked TMEM92 expression with TMB and MSI across multiple malignancies. At the same time, a significant correlation was observed between TMEM92 expression and mutation rates of MMR genes, underscoring the potential of TMEM92 as a therapeutic guide in oncology.
Building upon the established association between TMEM92 and TME, we utilized GSEA and GSVA to elucidate the regulatory mechanism of TMEM92 in a pan-cancer context. Our results highlighted positive correlations of TMEM92 expression with angiogenesis, metastasis, cellular quiescence, differentiation, and hypoxia. This establishes theoretical evidence of the intricate interplay with immunity and cancer metabolism in TMEM92.
In terms of pharmacological sensitivity, 144 out of 192 evaluated anti-cancer drugs, encompassing numerous conventional drugs, demonstrated diminished efficacy in the high-expression TMEM92 cohort. This suggests that there is a potential involvement of TMEM92 in drug resistance. Focal adhesion kinase (FAK), a pivotal regulator of cellular adhesion and migration, is implicated in resistance to cancer therapeutics. Current preclinical evidence suggests enhanced utility of FAK inhibitors when combined in specific patient populations [33]. Collectively, our analyses point to TMEM92 as a potential therapeutic target to some extent.
While our investigation pioneers a pan-cancer analysis of TMEM92 from multifaceted viewpoints, it is not devoid of limitations. The intricacies of the role of TMEM92 in cancer largely hinge upon bioinformatics analyses, with limited direct experimental validation currently available. Consequently, we aim to explore the multifarious roles, mechanistic underpinnings, and therapeutic potential of TMEM92 across diverse cancers via a slew of experimental designs.
Conclusion
In the present study, we elucidated the tumorigenic significance and prognostic implications of TMEM92 across a spectrum of cancers. Our findings highlighted associations between TMEM92 expression and multiple oncogenic factors, including DNA methylation, TME, immune cell infiltration, immune-associated gene expression, MSI, TMB, MMR genes, and drug susceptibility. These findings augment the understanding on the multifaceted roles of TMEM92 in cancer initiation and progression, laying the groundwork for potential clinical interventions targeting TMEM92 in forthcoming therapeutic strategies.
Data availability
All data generated or analysed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Abbreviations
- ACC:
-
Adrenocortical carcinoma
- BLCA:
-
Bladder urothelial carcinoma
- BRCA:
-
Breast invasive carcinoma
- CESC:
-
Cervical squamous cell carcinoma and endocervical adenocarcinoma
- CHOL:
-
Cholangio carcinoma
- COAD:
-
Colon adenocarcinoma
- DLBC:
-
Lymphoid neoplasm diffuse large B-cell lymphoma
- ESCA:
-
Esophageal carcinoma
- ESCC:
-
Esophageal squamous cell carcinoma
- GBM:
-
Glioblastoma multiforme
- HNSC:
-
Head and neck squamous cell carcinoma
- KICH:
-
Kidney chromophobe
- KIRC:
-
Kidney renal clear cell carcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- LAML:
-
Acute myeloid leukemia
- LGG:
-
Brain lower grade glioma
- LIHC:
-
Liver hepatocellular carcinoma
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- MEL:
-
Melanoma
- MESO:
-
Mesothelioma
- OV:
-
Ovarian serous cystadenocarcinoma
- PAAD:
-
Pancreatic adenocarcinoma
- PCPG:
-
Pheochromocytoma and paraganglioma
- PRAD:
-
Prostate adenocarcinoma
- READ:
-
Rectum adenocarcinoma
- SKCM:
-
Skin cutaneous melanoma
- TGCT:
-
Testicular germ cell tumors
- THCA:
-
Thyroid carcinoma
- THYM:
-
Thymoma
- UCEC:
-
Uterine corpus endometrial carcinoma
- UCS:
-
Uterine carcinosarcoma
- UVM:
-
Uveal melanoma
References
Global Burden of Disease Cancer C, Kocarnik JM, Compton K, Dean FE, Fu W, Gaw BL, et al. Cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life years for 29 cancer groups from 2010 to 2019: a systematic analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022;8(3):420–44. https://doi.org/10.1001/jamaoncol.2021.6987.
Saw PE, Chen J, Song E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. 2022;8(7):527–55. https://doi.org/10.1016/j.trecan.2022.03.001.
Schmit K, Michiels C. TMEM proteins in cancer: a review. Front Pharmacol. 2018;9:1345. https://doi.org/10.3389/fphar.2018.01345.
Zhao Y, Zhang K, Pan H, Wang Y, Zhou X, Xiang Y, et al. Genetic analysis of six transmembrane protein family genes in Parkinson’s disease in a large Chinese cohort. Front Aging Neurosci. 2022;14: 889057. https://doi.org/10.3389/fnagi.2022.889057.
Hayez A, Malaisse J, Roegiers E, Reynier M, Renard C, Haftek M, et al. High TMEM45A expression is correlated to epidermal keratinization. Exp Dermatol. 2014;23(5):339–44. https://doi.org/10.1111/exd.12403.
Lipinski P, Stepien KM, Ciara E, Tylki-Szymanska A, Jezela-Stanek A. Skeletal and bone mineral density features, genetic profile in congenital disorders of glycosylation: review. Diagnostics (Basel). 2021;11(8):1438. https://doi.org/10.3390/diagnostics11081438.
Deng H, Li T, Wei F, Han W, Xu X, Zhang Y. High expression of TMEM200A is associated with a poor prognosis and immune infiltration in gastric cancer. Pathol Oncol Res. 2023;29:1610893. https://doi.org/10.3389/pore.2023.1610893.
Rao J, Wu X, Zhou X, Deng R, Ma Y. TMEM205 Is an independent prognostic factor and is associated with immune cell infiltrates in hepatocellular carcinoma. Front Genet. 2020;11: 575776. https://doi.org/10.3389/fgene.2020.575776.
Du Y, Zeng X, Yu W, Xie W. A transmembrane protein family gene signature for overall survival prediction in osteosarcoma. Front Genet. 2022;13: 937300. https://doi.org/10.3389/fgene.2022.937300.
Pei J, Grishin NV. Unexpected diversity in Shisa-like proteins suggests the importance of their roles as transmembrane adaptors. Cell Signal. 2012;24(3):758–69. https://doi.org/10.1016/j.cellsig.2011.11.011.
Listing H, Mardin WA, Wohlfromm S, Mees ST, Haier J. MiR-23a/-24-induced gene silencing results in mesothelial cell integration of pancreatic cancer. Br J Cancer. 2015;112(1):131–9. https://doi.org/10.1038/bjc.2014.587.
Zhang S, Wan X, Lv M, Li C, Chu Q, Wang G. TMEM92 acts as an immune-resistance and prognostic marker in pancreatic cancer from the perspective of predictive, preventive, and personalized medicine. EPMA J. 2022;13(3):519–34. https://doi.org/10.1007/s13167-022-00287-0.
Lin MZ, Teng LL, Sun XL, Zhang LP, Chen F, Yu LJ. Transmembrane protein 92 performs a tumor-promoting function in breast carcinoma by contributing to the cell growth, invasion, migration and epithelial-mesenchymal transition. Tissue Cell. 2020;67: 101415. https://doi.org/10.1016/j.tice.2020.101415.
Zeng D, Li M, Zhou R, Zhang J, Sun H, Shi M, et al. Tumor microenvironment characterization in gastric cancer identifies prognostic and immunotherapeutically relevant gene signatures. Cancer Immunol Res. 2019;7(5):737–50. https://doi.org/10.1158/2326-6066.CIR-18-0436.
Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. https://doi.org/10.1038/ncomms3612.
Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–30e14. https://doi.org/10.1016/j.immuni.2018.03.023.
Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen HZ, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. 2017;2017: PO.17.00073. https://doi.org/10.1200/PO.17.00073.
Bilotta MT, Antignani A, Fitzgerald DJ. Managing the TME to improve the efficacy of cancer therapy. Front Immunol. 2022;13: 954992. https://doi.org/10.3389/fimmu.2022.954992.
Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–21. https://doi.org/10.1038/s41423-020-0488-6.
He Y, Zhang L, Zhou R, Wang Y, Chen H. The role of DNA mismatch repair in immunotherapy of human cancer. Int J Biol Sci. 2022;18(7):2821–32. https://doi.org/10.7150/ijbs.71714.
Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):D955–61. https://doi.org/10.1093/nar/gks1111.
Ushijima T, Clark SJ, Tan P. Mapping genomic and epigenomic evolution in cancer ecosystems. Science. 2021;373(6562):1474–9. https://doi.org/10.1126/science.abh1645.
Huang W, Li H, Yu Q, Xiao W, Wang DO. LncRNA-mediated DNA methylation: an emerging mechanism in cancer and beyond. J Exp Clin Cancer Res. 2022;41(1):100. https://doi.org/10.1186/s13046-022-02319-z.
Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30(16):R921–5. https://doi.org/10.1016/j.cub.2020.06.081.
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84. https://doi.org/10.1038/s41580-018-0080-4.
Wang Y, Li W, Jin X, Jiang X, Guo S, Xu F, et al. Identification of prognostic immune-related gene signature associated with tumor microenvironment of colorectal cancer. BMC Cancer. 2021;21(1):905. https://doi.org/10.1186/s12885-021-08629-3.
Halle S, Halle O, Forster R. Mechanisms and dynamics of T cell-mediated cytotoxicity in vivo. Trends Immunol. 2017;38(6):432–43. https://doi.org/10.1016/j.it.2017.04.002.
Oh DY, Fong L. Cytotoxic CD4(+) T cells in cancer: expanding the immune effector toolbox. Immunity. 2021;54(12):2701–11. https://doi.org/10.1016/j.immuni.2021.11.015.
Tie Y, Tang F, Wei YQ, Wei XW. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. 2022;15(1):61. https://doi.org/10.1186/s13045-022-01282-8.
Palmeri M, Mehnert J, Silk AW, Jabbour SK, Ganesan S, Popli P, et al. Real-world application of tumor mutational burden-high (TMB-high) and microsatellite instability (MSI) confirms their utility as immunotherapy biomarkers. ESMO Open. 2022;7(1): 100336. https://doi.org/10.1016/j.esmoop.2021.100336.
Cristescu R, Aurora-Garg D, Albright A, Xu L, Liu XQ, Loboda A, et al. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: a pan-tumor retrospective analysis of participants with advanced solid tumors. J Immunother Cancer. 2022;10(1): e003091. https://doi.org/10.1136/jitc-2021-003091.
Chakrabarti S, Bucheit L, Starr JS, Innis-Shelton R, Shergill A, Dada H, et al. Detection of microsatellite instability-high (MSI-H) by liquid biopsy predicts robust and durable response to immunotherapy in patients with pancreatic cancer. J Immunother Cancer. 2022;10(6): e004485. https://doi.org/10.1136/jitc-2021-004485.
Dawson JC, Serrels A, Stupack DG, Schlaepfer DD, Frame MC. Targeting FAK in anticancer combination therapies. Nat Rev Cancer. 2021;21(5):313–24. https://doi.org/10.1038/s41568-021-00340-6.
Acknowledgements
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Funding
This study was funded by Natural Science Foundation of Hebei Province (No. H2022206221) Medical science research project of Hebei Province (No. 20240095)
Author information
Authors and Affiliations
Contributions
Conception and design of the research: Wu Z, Wang ZY. Acquisition of data: Zhang YH, Guo SH, Liu Y. Analysis and interpretation of the data: Liu Y, Zhang L, Li W. Statistical analysis: Guo SH, Liu Y. Obtaining financing: Wu Z. Writing of the manuscript: Wu Z, Pan T. Critical revision of the manuscript for intellectual content: Li W, Zhang YH, Wang ZY. All authors read and approved the final draft.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, Z., Pan, T., Li, W. et al. Comprehensive pan-cancer analysis reveals prognostic implications of TMEM92 in the tumor immune microenvironment. Clin Transl Oncol 26, 2701–2717 (2024). https://doi.org/10.1007/s12094-024-03477-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-024-03477-6