
Abstract
Objective
Downregulation of miR-17-5p has been reported in several cancers, but whether and how miR-17-5p is downregulated in hepatocellular carcinoma (HCC) is unknown. Here, we examined whether miR-17-5p is downregulated in HCC and whether that affects expression of its target gene encoding transforming growth factor β receptor 2 (TGFβR).
Methods
We screened for potential microRNAs (miRNAs) involved in HCC by analyzing published transcriptomes from HCC patients. Expression of miR-17-5p was measured in HCC cell lines and in tissues from HCC patients using quantitative real-time PCR. The in vitro effects of miR-17-5p on HCC cells were assessed by EdU proliferation assay, CCK-8 cell proliferation assay, colony-formation assay, transwell migration/invasion assay, wound healing assay, and flow cytometry. Effects of miR-17-5p were evaluated in vivo using mice with subcutaneous tumors. Effects of the miRNA on the epithelial–mesenchymal transition (EMT) were assessed, while its effects on TGFβR2 expression were analyzed using bioinformatics and a dual luciferase reporter assay.
Results
Patients with low miR-17-5p expression showed lower rates of overall and recurrence-free survival than patients with high miR-17-5p expression, and multivariate Cox regression identified low miR-17-5p expression as an independent predictor of poor overall survival in HCC patients. In vitro, miR-17-5p significantly inhibited HCC cell proliferation, migration, invasion, and the EMT, while promoting apoptosis. In vivo, it slowed the development of tumors. These protective effects of miR-17-5p were associated with downregulation of TGFβR2.
Conclusion
The miRNA miR-17-5p can negatively regulate the expression of TGFβR2 and inhibit the EMT, thereby slowing tumor growth in HCC, suggesting a potential therapeutic approach against HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most prevalent cancer and third leading cause of cancer-related mortality globally. [1] The main risk factors for HCC are chronic infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) [2]. Because early symptoms of this disease are generally nonspecific, the majority of HCC patients are not diagnosed until a fairly advanced stage of the disease. [3] While there have been several therapeutic advances in recent years, especially immunotherapy, HCC patients still have generally poor clinical outcomes, and the 5-year survival rate is below 40%. [4] Thus, greater mechanistic insights into this disease are needed in order to develop better strategies to diagnose, treat, and even prevent the disease.
Numerous tumor suppressor genes and oncogenes have been shown to control the development of HCC [5, 6]. Among these genes are those encoding microRNAs (miRNAs), which are evolutionarily conserved, short, noncoding regulatory RNAs 20–24 nucleotides long [7, 8]. MicroRNAs suppress target gene expression by binding to specific sequences in the 3′-untranslated region (3′-UTR) of the mRNA, thereby disrupting its translation and/or promoting its degradation [9, 10]. While miRNAs always suppress their target genes, the result can be promotion or suppression of tumor development depending on function of the target gene [11, 12]. The dysregulation of normal miRNA expression has been closely linked to a number of processes vital to cancer progression, including proliferation, survival, angiogenesis, metastasis, and the epithelial–mesenchymal transition (EMT). [13,14,15]
Several studies have highlighted the potential miR-17-5p expression as a robust diagnostic and prognostic biomarker in gastric [16], pancreatic [17], breast [18], colon [19], and hematological cancers [20]. In previous work, we found that miR-17-5p may play a tumor suppressive role in HCC [21]. Therefore, in the present study, we aimed to evaluate the expression and functional relevance of miR-17-5p in HCC in order to determine whether it might be a prognostic marker and/or therapeutic target in this cancer.
The microRNA miR-17-5p has been shown to regulate TIMP2 [22], HSPB2, [23], and cyclin D1 [24] in various types of cancers. Bioinformatics predicts that it also binds to the 3′-UTR in the gene encoding transforming growth factor β receptor 2 (TGFβR2). [25, 26] This receptor recognizes cytokines that regulate proliferation, differentiation, adhesion, migration, and other functions in multiple cell types [27,28,29]. Whether miR-17-5p regulates TGFβR2 expression in HCC is unknown.
Here we explored whether miR-17-5p is downregulated in HCC and, if so, whether it affects tumor progression through TGFβR2. We also examined whether the miR-17-5p/TGFβR2 axis affects survival of HCC patients.
Materials and methods
Clinical samples
This study analyzed 74 pairs of human HCC tumor and corresponding adjacent tissues (para-cancerous tissues) that had been obtained from patients undergoing hepatectomy at Guangxi Medical University Affiliated Tumor Hospital (Nanning, China) between 5 January 2016 and 20 July 2020. After collection, all samples were stored at − 80 °C. Samples were included only if patients had pathology-confirmed HCC and if they had not received chemotherapy or radiotherapy before surgery.
This analysis of patient samples was approved by the Institutional Ethics Committee of the Guangxi Medical University Cancer Hospital (KY2020102), which waived the requirement for informed consent because patients, at the time of treatment, consented for their anonymized medical data to be analyzed and published for research purposes.
Cell culture and miR-17-5p transfection
MIHA human normal hepatocytes and six HCC cell lines (HepG2, Hep3B, Huh-7, PLC, 97H, and LM3) were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The HCC cell lines HepG2, Hep3B, Huh-7, 97H, and LM3 were grown in Dulbecco's modified Eagle medium (DMEM; Gibco, Waltham, MA, USA) containing 10% fetal bovine serum (FBS; Biological Industries, Kibbutz Beth Haemek, Israel) and 1% penicillin–streptomycin (Gibco). MIHA and PLC lines were grown in Roswell Park Memorial Institute (RPMI) 1640 (Gibco) containing 10% FBS (Biological Industries) and 1% penicillin–streptomycin (Gibco). Cells were cultured in humidified incubators at 37 ℃ in an atmosphere of 5% CO2.
Lentiviral plasmids encoding a miR-17-5p mimic (Lv-hsa-miR-17-5p mimic) or inhibitor (Lv-miR-17-5p-inhibitor), as well as negative controls for the mimic (Lv-mimic-NC) and inhibitor (Lv-inhibitor-NC), were synthesized by Genechem (Shanghai, China). Cells were cultured in 6-well plates (6 × 104 cells/well) for 24 h, then transfected for 12 h with lentiviral plasmids using transfection reagent (Genechem). Stably transfected cell lines were selected using 2 μg/mL puromycin (Solarbio, Beijing, China), and expression of miR-17-5p was checked using quantitative real-time PCR (see below).
Assessment of miRNA expression
Trizol (Invitrogen, Waltham, MA, USA) was used to extract total cellular RNA according to the manufacturer’s instructions. RNA (1 μg/μL) was used to prepare cDNA with the Mir-X miRNA First-Strand Synthesis Kit (Takara, Dalian, China) according to the manufacturer’s instructions. Quantitative PCR was performed using TB Green® Premix Ex Taq™ II (Tli RNaseH Plus, Takara) and the primers (Shangon Biotech, Shanghai, China) in Table 1, in the QuantStudio 5 real-time PCR detection system (Thermo Fisher, Waltham, MA, USA). Levels of miRNAs were calculated using the 2−ΔΔCt method relative to levels of U6 as an internal control.
Assessment of protein expression by western blotting
Samples were lysed in radio-immunoprecipitation assay (RIPA) buffer (Solarbio) and centrifuged. The supernatant was fractionated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Beyotime) and transferred onto polyvinylidene difluoride membranes (Millipore, San Diego, CA, USA). Blots were blocked using 5% bovine serum albumin (Beyotime) at 25 ºC for 2 h, followed by incubation for 16 h at 4 ºC with rabbit polyclonal antibodies against TGFβR2, E-cadherin, N-cadherin, Vimentin, or β-actin (1:1000; Cell Signaling Technology, MA, USA). Immunoblots were washed three times in Tris-buffered saline containing Tween-20 (TBST) and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG H&L (1:1,000; Beyotime). Antibody binding was visualized using an enhanced chemiluminescence detection kit (Beyotime).
Assessment of cell proliferation
Cell proliferation was assessed using a 5-ethynyl-2-deoxyuridine (EdU) assay kit (Beyotime). Briefly, cells were seeded in a 96-well plate (1 × 104 cells/well) and cultured for 24 h in DMEM (Gibco) medium containing 10% FBS (Biological Industries) and 1% penicillin–streptomycin (Gibco). On the following day, cells were incubated with 10 μM EdU solution for 2 h at 37 ºC, fixed in 4% paraformaldehyde (Solarbio) for 15 min at 25 ℃, permeabilized in 0.3% Triton X‐100 for 10 min at 25 ºC, and finally washed in phosphate-buffered saline (PBS; Solarbio) and stained in 100 μL Click reaction solution (Beyotime) in the dark for 30 min at 25 ºC. Nuclei were counter-stained by incubating cells in 100 μL Hoechst 33,342 in the dark for 10 min at 25 ºC. Images were captured under a fluorescence microscope (Thermo Fisher).
Cell proliferation and viability assay
Cell proliferation and viability were assessed using the Cell Counting Kit-8 (CCK-8) kit (Dojindo, Tokyo, Japan). Briefly, cells were seeded in a 96-well plate (2 × 103 cells/well) and incubated for 24 h. Then cells were stained in 10 μL CCK-8 solution at 24, 48, 72, or 96 h. Absorbance was measured at 450 nm using a microplate reader (Thermo Fisher).
Colony formation assay
Cells were seeded in 6-well plates (1 × 103 cells/well) and cultured in DMEM containing 10% FBS for 14 days, when cell colonies were visible. Colonies were fixed in 4% paraformaldehyde solution (Solarbio), stained in 0.1% crystal violet solution (Solarbio) and counted under a microscope (Olympus, Tokyo, Japan).
Wound healing assay
Cell migration was assessed using a wound healing assay. Briefly, cells were seeded in 6-well plates (5 × 105 cells/well) and cultured until 90% confluence in DMEM containing 10% FBS and 1% penicillin–streptomycin, then a 200 μL pipette tip was used to scratch across the cell monolayer. The wounded monolayer was washed with PBS to remove detached cells, then cultured in fresh DMEM containing 10% FBS and 1% penicillin–streptomycin. Monolayers were imaged immediately after washing (0 h) and 24 h later using an inverted microscope (Olympus). Wound width was measured using ImageJ (version 1.8.0, https://imagej.net/), and expressed as a percentage of the width at 0 h.
Transwell migration assay
The transwell migration assay was carried out to assess the migratory ability of tumor cells. Briefly, cells were seeded in the upper chamber of 24‐well transwell chambers (Millipore, San Diego, CA, USA) in serum-free DMEM (1 × 105 cells/well, 200 µL), then 500 μL DMEM supplemented with 20% FBS was added to the lower chamber as a chemoattractant, and cultures were incubated at 37 ℃. After 24 h of incubation, cells remaining in the upper layer of the chamber were removed with a cotton swab. Cells that migrated over the outer surface of the chamber of the insert were washed in PBS, fixed in 4% paraformaldehyde solution (Solarbio), and stained in 0.1% crystal violet solution (Solarbio) for 30 min. Finally, the number of migrated cells was counted under a light microscope (Olympus) at 100× magnification.
Transwell invasion assay
To assess tumor cell invasion, the invasion chamber of 24 well transwell chambers (Millipore) was loaded with 50 μL of Matrigel (Corning, Corning, NY, USA) and incubated for 2 h at 37 ºC. Cells were added to the upper chamber in serum-free DMEM (1 × 105 cells/well, 200 µL), then 500 μL DMEM supplemented with 20% FBS was added to the lower chamber as a chemoattractant, and cultures were incubated for 24 h at 37 ºC. Then cells remaining in the upper layer of the chamber were removed with a cotton swab. Cells that invaded the outer surface of the chamber of the culture insert were washed, fixed in 4% paraformaldehyde solution (Solarbio), and stained in 0.1% crystal violet (Solarbio) for 30 min. Finally, the number of invasive cells was counted under a light microscope (Olympus) at 100× magnification.
Assessment of apoptosis
Cells were processed using the Annexin V-APC/7-AAD kit (Multisciences Biotech, Hangzhou, China) as recommended by the manufacturer, then analyzed by flow cytometry (Beckman, Brea, CA, USA).
Animal experiments
Female BALB/c nude mice (4 weeks old) were purchased from the Animal Center of Guangxi Medical University and used for in vivo subcutaneous tumorigenesis experiments. All animal procedures were approved by the Animal Use and Care Committee of Guangxi Medical University (202,103,043).
LM3 cells (3 × 106) that had been transfected with Lv-hsa-miR-17-5p mimic, Lv-mimic-NC, Lv-inhibitor-NC, or Lv-miR-17-5p-inhibitor were suspended in 200 µL PBS, then injected subcutaneously into the right flank of the mice. Animals were randomly divided into four groups (5 mice/group): miR-17-5p mimic, mimic-NC, inhibitor, and inhibitor-NC. Every 4 days, tumor length and width were measured with a Vernier caliper and used to determine tumor volume according to the formula:
Animals were euthanized after 4 weeks.
Dual luciferase reporter assay
The 3′-UTR sequence of TGFβR2 containing a wild-type (WT) or mutated (MUT)-binding site for miR-17-5p was constructed by HanbioLab (Shanghai, China) and cloned into their PGL-3 luciferase reporter vector. HEK-293 T cells were incubated for 24 h in 24-well plates, then co-transfected with either the h-TGFβR2-3UTR-WT or h-TGFβR2-3UTR-MUT reporter plasmid, together with either the miR-17-5p mimic or miR-17-5p NC plasmid. Transfections were carried out using Lipofectamine 3000 (Invitrogen). Firefly and Renilla luciferase activities were evaluated using the Dual‐Luciferase Assay (Promega, Madison, WI, USA). The activity of firefly luciferase was normalized to that of the Renilla enzyme.
Bioinformatics analysis
We predicted mRNA targets of miR-17-5p using StarBase (version 2.0, https://starbase.sysu.edu.cn/), miRDB (https://mirdb.org/) and TargetScan (https://www.targetscan.org/vert_80/).
Statistical analysis
All statistical analyses were performed in SPSS 23.0 (IBM, Armonk, NY, USA) and GraphPad Prism version 5.0 (GraphPad, La Jolla, CA, USA). Results were presented as mean ± standard deviation (SD). Differences between two groups were assessed for significance using Student’s independent t test, while differences among three or more groups were assessed using one-way analysis of variance (ANOVA). Kaplan–Meier curves were generated to analyze the relationship between expression of miR-17-5p and 5-year overall survival (OS) or tumor-free survival of HCC patients. Differences in curves were assessed for significance using the log-rank test. Cox analysis was used to identify prognostic factors affecting overall survival of HCC patients. Levels of statistical significance were defined as *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
HCC is associated with miR-17-5p downregulation, and lower levels of this miRNA are associated with worse survival of patients
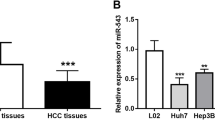
We detected the expression of miR-17-5p in normal hepatic epithelial cells (MIHA) and HCC cell lines (HepG2, Hep3B, Huh-7, PLC, 97H, and LM3) using RT-PCR. Compared with MIHA, miR-17-5p was significantly downregulated in all 6 HCC cell lines (Fig. 1A). Among HCC cell lines, the highest expression was observed in Hep3B cells and the lowest in LM3 cells. Analysis of matched tumor and para-cancerous tissues from HCC patients at our hospital confirmed miR-17-5p to be downregulated in the disease (Fig. 1B). In fact, the level of miR-17-5p was significantly associated with Edmondson grade 3–4, liver cirrhosis, microvascular invasion, Barcelona B/C stage, and incomplete tumor capsule in HCC patients (Table 2).

Expression of miR-17-5p and its association with prognosis in hepatocellular carcinoma (HCC). A Expression levels of miR-17-5p in normal hepatocytes and HCC cell lines. The expression of miR-17-5p was assessed by real-time quantitative PCR. B Expression of miR-17-5p in 74 pairs of HCC tissues (HCC) and normal para-cancerous tissues (PC). C Kaplan–Meier curves of overall survival of HCC patients after hepatectomy. Data are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001
Based on median miR-17-5p expression in our sample of 74 patients, 37 (50%) were categorized as showing low expression and 37 as showing high expression. In both univariate and multivariate analyses, a low level of miR-17-5p expression was independently associated with lower OS in patients with HCC after hepatectomy (Table 3). Kaplan–Meier survival analysis confirmed that patients expressing lower levels of miR-17-5p had worse OS than those expressing higher levels (Fig. 1C).
These results from cell lines and patients suggest that miR-17-5p is downregulated in HCC, and that lower levels of the miRNA are associated with worse prognosis.
miR-17-5p inhibits the proliferation, migration and invasion of HCC cells, while promoting apoptosis
We next explored the biological role(s) of miR-17-5p in HCC by expressing miR-17-5p mimic, mimic-NC, miR-17-5p inhibitor, or inhibitor-NC in Hep3B and LM3 cell lines (Fig. 2A). Overexpressing miR-17-5p impaired proliferation of both cell lines, while inhibiting miR-17-5p led to the opposite effects (Fig. 2B). Consistently, miR-17-5p overexpression significantly impaired migration (Fig. 2C) and invasion (Fig. 2D) of both cell lines, while inhibiting miR-17-5p led to the opposite effects. Overexpression of miR-17-5p also promoted the apoptosis of both cell lines (Fig. 2E), while the opposite was observed after inhibition of miR-17-5p. These findings suggest that miR-17-5p functions as a tumor suppressor in HCC cells.

miR-17-5p suppresses the proliferation, migration, invasion, and survival of HCC cells. A Expression of miR-17-5p in transfected HCC cells. B Representative images showing the viability of transfected HCC cells. Cell viability was assessed using 5-ethynyl-2-deoxyuridine (EdU), the Cell Counting kit-8 (CCK-8), and a colony-formation assay. Magnification, 200×. C Migration activity of transfected cells, as measured in wound healing assays (magnification, 100×) and transwell migration assays (magnification, 200X). D Invasion ability of stably transfected HCC cells assessed by transwell invasion assay. Magnification, 200×. (E) Proportion of apoptotic cells in stably transfected HCC cells. *p < 0.05, **p < 0.01, ***p < 0.001
miR-17-5p overexpression inhibits HCC tumor development in vivo
To verify whether miR-17-5p exerts anti-tumorigenic effects in vivo, we established a xenograft model in BALB/c nude mice by subcutaneously injecting stably transfected LM3 cells. Increased miR-17-5p expression suppressed tumorigenesis, whereas reduced miR-17-5p expression promoted tumor proliferation, based on tumor volume and weight (Fig. 3). The opposite effects were observed when mice were transfected with miR-17-5p inhibitors. These results in vivo are consistent with those obtained in vitro, indicating that miR-17-5p overexpression inhibits HCC tumor development.

miR-17-5p inhibits tumorigenesis in an HCC xenograft mouse model. A, B In vivo and harvested xenografts from mice injected with LM3 cells transfected with the indicated lentiviral constructs. C, D Tumor volume and weights. Data are mean ± SD. ***p < 0.001
miR-17-5p regulates expression of TGFβR2
To identify what downstream target genes of miR-17-5p might mediate its observed tumor suppressor activity, we utilized the miRDB, starbase, and TargetScan databases to predict potential target genes, and the gene encoding TGFβR2 emerged (Fig. 4A). Indeed, HCC cells expressing low levels of miR-17-5p showed upregulation of TGFβR2 (Fig. 4B). Consistently, TGFβR2 was expressed at significantly higher levels in HCC tumor tissue than in normal para-cancerous tissue from patients (Fig. 4C), and levels of TGFβR2 in HCC tissue samples showed a moderate negative correlation with levels of miRNA-17-5p expression (r = 0.5093, p = 0.0041; Fig. 4D). Overexpression of miR-17-5p in HCC downregulated TGFβR2 at the mRNA and protein levels, with miR-17-5p inhibition causing the opposite effects (Fig. 4E).

miR-17-5p binds to the 3’-untranslated region of the TGFβR2 gene and downregulates it in HCC. A Predicted interaction between miR-17-5p and the mRNA encoding TGFβR2. B Expression of TGFβR2 in normal hepatocytes and HCC cell lines. C Expression of TGFβR2 in 30 pairs of HCC and para-cancerous (PC) tissues. D Negative correlation between the expression of miR-17-5p and TGFβR2 in HCC tumors from 30 patients. Data are mean ± SD. ***p < 0.001. ns not significant. E Influence of miR-17-5p expression on the mRNA and protein levels of TGFβR2 in Hep3B and LM3 HCC cell lines. F Dual luciferase activity assay on cells expressing TGFβR2 mRNA containing a wild-type (WT) or mutated (MUT) predicted binding site for miR-17-5p in the 3’-untranslated region (UTR)
We verified that miR-17-5p targets the 3′-UTR of TGFβR2 mRNA by showing in a dual luciferase reporter assay that miR-17-5p mimic decreased the activity of luciferase encoded in mRNA carrying the wild-type, but not mutated, 3′-UTR of the TGFβR2 gene (Fig. 4F).
miR-17-5p inhibits the EMT by downregulating TGFβR2
TGF-β, which binds to TGFβR2, activates the EMT to promote various types of cancer [41]. Therefore, we examined whether miR-17-5p may influence the EMT in HCC via TGFβR2. Overexpression miR-17-5p markedly downregulated TGFβR2, vimentin, and N-cadherin while upregulating E-cadherin, indicating suppression of the EMT (Fig. 5). Knockdown of miR-17-5p had the opposite effects. These results strongly suggest that miR-17-5p suppresses the growth and metastasis of HCC cells by preventing TGFβR2 from activating the EMT.

miR-17-5p suppresses the epithelial–mesenchymal transition in HCC. Hep3B and LM3 HCC cell lines were transfected with the indicated lentiviral constructs. Representative Western blots are shown
Discussion
The identification of molecular pathways governing the development and progression of HCC can improve disease prevention, diagnosis, and treatment [30, 31]. Our experiments in HCC cell lines, patient samples and a mouse model suggest that HCC involves downregulation of miR‐17‐5p, which in turn leads to upregulation of TGFβR2, which stimulates the EMT, thereby driving tumor growth and metastasis. In this way, miR‐17‐5p appears to act as a tumor suppressor in HCC.
Our results add to the miRNAs whose dysregulation contributes to HCC. Another miRNA, miR-155-5p, promotes proliferation of HCC cells by activating the Wnt/β-Catenin signaling pathway [32]. Liu et al. reported that miR-103a promotes glycolytic metabolism and inhibits apoptosis of HCC cells by downregulating ATP11A and EIF5 [33]. Conversely, miR-199a downregulates HK2 and PKM2 to suppress proliferation of HCC cells [34]. All these miRNAs may be useful to pursue as biomarkers for diagnosing HCC and as targets for treating it.
Nevertheless, the limitations of this study should be acknowledged. Despite the significant downregulation of miR-17-5p characterized here in HCC along with previous reports among several types of cancers [35, 36], some studies have also reported that it can enhance the proliferation and metastasis of HCC [37] and non-small cell lung cancer [38]. Furthermore, Lou et al. found that the expression and the distribution of U6 exhibit a high degree of variability in HCC [39]. At present, the molecular mechanisms of HCC development are still not clearly revealed, and more explorations are needed to detect upstream or downstream effectors of miR-17-5p in HCC progression.
The EMT is a key stage in tumor progression in which epithelial cells undergo morphological and functional shifts toward a more motile, mesenchymal-like phenotype, thereby driving tumor metastasis [40, 41]. TGFβR2 can respond to signals from proximal stromal cells and tumor cells to drive the EMT. As a result, aberrant expression of TGFβR2 is associated with oncogenic activation of the EMT, the cell cycle, and tumor cell survival [42,43,44]. Our results illustrate how miR-17-5p helps keep such aberrant TGFβ signaling in check, thereby inhibiting cancer progression. Thus, the miR-17-5p/TGFβR2/EMT axis may be a potential therapeutic target in HCC.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Zhong JH, Ke Y, Gong WF, Xiang BD, Ma L, Ye XP, et al. Hepatic resection associated with good survival for selected patients with intermediate and advanced-stage hepatocellular carcinoma. Ann Surg. 2014;260(2):329–40.
Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604.
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6.
Komoll RM, Hu Q, Olarewaju O, von Döhlen L, Yuan Q, Xie Y, et al. MicroRNA-342-3p is a potent tumour suppressor in hepatocellular carcinoma. J Hepatol. 2021;74(1):122–34.
Liu ZK, Li C, Zhang RY, Wei D, Shang YK, Yong YL, et al. EYA2 suppresses the progression of hepatocellular carcinoma via SOCS3-mediated blockade of JAK/STAT signaling. Mol Cancer. 2021;20(1):79.
You C, Cui J, Wang H, Qi X, Kuo LY, Ma H, et al. Conservation and divergence of small RNA pathways and microRNAs in land plants. Genome Biol. 2017;18(1):158.
Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–37.
Oura K, Morishita A, Masaki T. Molecular and functional roles of microRNAs in the progression of hepatocellular carcinoma-a review. Int J Mol Sci. 2020;21(21):8362.
Han J, LaVigne CA, Jones BT, Zhang H, Gillett F, Mendell JT. A ubiquitin ligase mediates target-directed microRNA decay independently of tailing and trimming. Science. 2020;370(6523):eabc9546.
Sandbothe M, Buurman R, Reich N, Greiwe L, Vajen B, Gürlevik E, et al. The microRNA-449 family inhibits TGF-β-mediated liver cancer cell migration by targeting SOX4. J Hepatol. 2017;66(5):1012–21.
Boldrin E, Gaffo E, Niedermayer A, Boer JM, Zimmermann M, Weichenhan D, et al. MicroRNA-497/195 is tumor suppressive and cooperates with CDKN2A/B in pediatric acute lymphoblastic leukemia. Blood. 2021;138(20):1953–65.
Chen J, Liu A, Wang Z, Wang B, Chai X, Lu W, et al. LINC00173v.1 promotes angiogenesis and progression of lung squamous cell carcinoma by sponging miR-511-5p to regulate VEGFA expression. Mol Cancer. 2020;19(1):98.
Bhat AA, Younes SN, Raza SS, Zarif L, Nisar S, Ahmed I, et al. Role of non-coding RNA networks in leukemia progression, metastasis and drug resistance. Mol Cancer. 2020;19(1):57. https://doi.org/10.1186/s12943-020-01175-9. (Erratum in: Mol Cancer. 2020;19(1):174).
Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154(2):311–24.
Xie G, Dong P, Chen H, Xu L, Liu Y, Ma Y, et al. Decreased expression of ATF3, orchestrated by β-catenin/TCF3, miR-17-5p and HOXA11-AS, promoted gastric cancer progression via increased β-catenin and CEMIP. Exp Mol Med. 2021;53(11):1706–22.
Zhu Y, Gu J, Li Y, Peng C, Shi M, Wang X, et al. MiR-17-5p enhances pancreatic cancer proliferation by altering cell cycle profiles via disruption of RBL2/E2F4-repressing complexes. Cancer Lett. 2018;412:59–68.
Lee J, Kim HE, Song YS, Cho EY, Lee A. miR-106b-5p and miR-17-5p could predict recurrence and progression in breast ductal carcinoma in situ based on the transforming growth factor-beta pathway. Breast Cancer Res Treat. 2019;176(1):119–30.
Selven H, Andersen S, Pedersen MI, Lombardi APG, Busund LR, Kilvær TK. High expression of miR-17-5p and miR-20a-5p predicts favorable disease-specific survival in stage I-III colon cancer. Sci Rep. 2022;12(1):7080.
Hu L, Liu J, Meng Y, Zheng H, Ding C, Wang H, et al. Long non-coding RNA HOTAIR regulates myeloid differentiation through the upregulation of p21 via miR-17-5p in acute myeloid leukaemia. RNA Biol. 2021;18(10):1434–44.
Zhong JH, Xiang X, Wang YY, Liu X, Qi LN, Luo CP, et al. The lncRNA SNHG16 affects prognosis in hepatocellular carcinoma by regulating p62 expression. J Cell Physiol. 2020;235(2):1090–102.
Zou M, Zhang Q. miR-17-5p accelerates cervical cancer cells migration and invasion via the TIMP2/MMPs signaling cascade. Cytotechnology. 2021;73(4):619–27.
Yu W, Wang J, Li C, Xuan M, Han S, Zhang Y, et al. miR-17-5p promotes the invasion and migration of colorectal cancer by regulating HSPB2. J Cancer. 2022;13(3):918–31.
Kang H, Yu H, Zeng L, Ma H, Cao G. LncRNA Rian reduces cardiomyocyte pyroptosis and alleviates myocardial ischemia-reperfusion injury by regulating by the miR-17-5p/CCND1 axis. Hypertens Res. 2022;45(6):976–89.
Martinez-Hackert E, Sundan A, Holien T. Receptor binding competition: a paradigm for regulating TGF-β family action. Cytokine Growth Factor Rev. 2021;57:39–54.
Ma Y, He S, Gao A, Zhang Y, Zhu Q, Wang P, et al. Methylation silencing of TGF-β receptor type II is involved in malignant transformation of esophageal squamous cell carcinoma. Clin Epigenetics. 2020;12(1):25.
Clark DA, Coker R. Transforming growth factor-beta (TGF-beta). Int J Biochem Cell Biol. 1998;30(3):293–8.
Chen J, Gingold JA, Su X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends Mol Med. 2019;25(11):1010–23.
Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-β-induced epithelial–mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010;29(49):6485–98.
Yu M, Chen Z, Zhou Q, Zhang B, Huang J, Jin L, et al. PARG inhibition limits HCC progression and potentiates the efficacy of immune checkpoint therapy. J Hepatol. 2022;77(1):140–51.
Song Y, An O, Ren X, Chan THM, Tay DJT, Tang SJ, et al. RNA editing mediates the functional switch of COPA in a novel mechanism of hepatocarcinogenesis. J Hepatol. 2021;74(1):135–47.
Chen G, Wang D, Zhao X, Cao J, Zhao Y, Wang F, et al. miR-155-5p modulates malignant behaviors of hepatocellular carcinoma by directly targeting CTHRC1 and indirectly regulating GSK-3β-involved Wnt/β-catenin signaling. Cancer Cell Int. 2017;17:118.
Liu Y, Zhang Y, Xiao B, Tang N, Hu J, Liang S, et al. MiR-103a promotes tumour growth and influences glucose metabolism in hepatocellular carcinoma. Cell Death Dis. 2021;12(6):618.
Zhang LF, Lou JT, Lu MH, Gao C, Zhao S, Li B, et al. Suppression of miR-199a maturation by HuR is crucial for hypoxia-induced glycolytic switch in hepatocellular carcinoma. EMBO J. 2015;34(21):2671–85.
Li J, Lai Y, Ma J, Liu Y, Bi J, Zhang L, et al. miR-17-5p suppresses cell proliferation and invasion by targeting ETV1 in triple-negative breast cancer. BMC Cancer. 2017;17(1):745.
Liu DL, Lu LL, Dong LL, Liu Y, Bian XY, Lian BF, et al. miR-17-5p and miR-20a-5p suppress postoperative metastasis of hepatocellular carcinoma via blocking HGF/ERBB3-NF-κB positive feedback loop. Theranostics. 2020;10(8):3668–83.
Lu Z, Li X, Xu Y, Chen M, Chen W, Chen T, et al. microRNA-17 functions as an oncogene by downregulating Smad3 expression in hepatocellular carcinoma. Cell Death Dis. 2019;10(10):723.
Chen Y, Zhou X, Huang C, Li L, Qin Y, Tian Z, et al. LncRNA PART1 promotes cell proliferation and progression in non-small-cell lung cancer cells via sponging miR-17-5p. J Cell Biochem. 2021;122(3–4):315–25.
Lou G, Ma N, Xu Y, Jiang L, Yang J, Wang C, et al. Differential distribution of U6 (RNU6-1) expression in human carcinoma tissues demonstrates the requirement for caution in the internal control gene selection for microRNA quantification. Int J Mol Med. 2015;36(5):1400–8.
Pastushenko I, Mauri F, Song Y, de Cock F, Meeusen B, Swedlund B, et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature. 2021;589(7842):448–55.
Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18(2):128–34.
Bai F, Wang C, Liu X, Hollern D, Liu S, Fan C, et al. Loss of function of BRCA1 promotes EMT in mammary tumors through activation of TGFβR2 signaling pathway. Cell Death Dis. 2022;13(3):195.
Zhao Y, Sun H, Li X, Liu Q, Liu Y, Hou Y, et al. DGKZ promotes TGFβ signaling pathway and metastasis in triple-negative breast cancer by suppressing lipid raft-dependent endocytosis of TGFβR2. Cell Death Dis. 2022;13(2):105.
Cai Y, Ruan W, Ding J, Wei N, Wang J, Zhang H, et al. miR-93-5p regulates the occurrence and development of esophageal carcinoma epithelial cells by targeting TGFβR2. Int J Mol Med. 2021;47(3):3.
Funding
This work was supported by the Specific Research Project Grants of Guangxi for Research Bases and Talents (GuiKe AD22035057), the National Natural Science Foundation of China (82060510 and 82260569), the Guangxi Undergraduate Training Program for Innovation and Entrepreneurship (202110598178, 202110598073), the Key Laboratory of Early Prevention and Treatment for Regional High Frequency Tumors (Guangxi Medical University), and the Chinese Ministry of Education (GKE-ZZ202217).
Author information
Authors and Affiliations
Contributions
J-HZ conceived the study. H-TL, M-JJ, and Z-JD drafted the manuscript and analyzed data. All authors read and approved the final version of the manuscript to be published.
Corresponding authors
Ethics declarations
Conflict of interest
None.
Ethical approval, Research involving human participants and/or animals
The study protocol was approved by the Ethics Review Committee of the Guangxi Medical University Cancer Hospital (KY2020102) and the Animal Care & Welfare Committee(202103043)
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, HT., Luo, CP., Jiang, MJ. et al. miR-17-5p slows progression of hepatocellular carcinoma by downregulating TGFβR2. Clin Transl Oncol 25, 2960–2971 (2023). https://doi.org/10.1007/s12094-023-03164-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-023-03164-y