
Abstract
Background
Acute myeloid leukemia (AML) is the most common type of acute leukemia and biologically heterogeneous diseases with poor prognosis. Thus, we aimed to identify prognostic markers to effectively predict the prognosis of AML patients and eventually guide treatment.
Methods
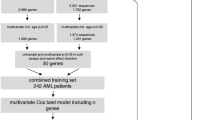
Prognosis-associated genes were determined by Kaplan–Meier and multivariate analyses using the expression and clinical data of 173 AML patients from The Cancer Genome Atlas database and validated in an independent Oregon Health and Science University dataset. A prognostic risk score was computed based on a linear combination of 5-gene expression levels using the regression coefficients derived from the multivariate logistic regression model. The classification of AML was established by unsupervised hierarchical clustering of CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 expression levels.
Results
High FCHO2 and LRCH4 expression was related to decreased mortality. While high CALCRL, DOCK1, PLA2G4A expression was associated with increased mortality. The risk score was predictive of increased mortality rate in AML patients. Hierarchical clustering analysis of the five genes discovered three clusters of AML patients. The cluster1 AML patients were associated with lower cytogenetics risk than cluster2 or 3 patients, and better prognosis than cluster3 patients (P values < 0.05 for all cases, fisher exact test or log-rank test).
Conclusion
The gene panel comprising CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 as well as the risk score may offer novel prognostic biomarkers and classification of AML patients to significantly improve outcome prediction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Acute myeloid leukemia (AML) is a heterogeneous disease with a relatively poor prognosis [1]. The incidence of AML ranges from three to five cases per 100,000 individuals in US. An estimated 20,830 new cases were diagnosed, and more than 10,000 patients died from the disease in 2015 alone [2]. Though considerable progress in the treatment of AML has significantly improved clinical outcomes for younger patients, prognosis remains poor for the elderly [3]. As high as 70% of patients above 65 years old die from AML within 1 year of diagnosis [4].
Cytogenetic abnormalities have been well established to serve as diagnostic and prognostic markers for AML patients, suggesting that they may play a critical role in leukemogenesis [5]. For example, genetic markers, such as translocation (8;21), inversion (16)/translocation (16;16), are associated with favorable outcome. In contrast, inversion (3)/translocation (3;3) may indicate poor prognosis in AML patients [5]. However, a fraction of AML genomes are lack of structural abnormalities; thus, the prediction of prognosis may not be possible for this subset of AML patients [6, 7]. In recent years, the prognostic value of somatic mutations has been systematically evaluated. Patel et al. found internal tandem duplication in Fms-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD), mixed-lineage leukemia-partial tandem duplication (MLL-PTD), ASXL Transcriptional Regulator 1(ASXL1) and Tumor protein p53 (TP53) mutations were associated with inferior prognosis. While, mutations in CCAAT Enhancer Binding Protein Alpha (CEBPA) and isocitrate dehydrogenase 2 (IDH2) were associated with improved prognosis. DNA methyltransferase 3A (DNMT3A), Nucleophosmin 1 (NPM1) mutations and MLL translocations showed to improve risk stratification for AML patients with normal karyotype [8]. These findings suggest that mutational profiling could potentially be used for risk stratification and to inform prognosis in AML. However, mutation profiling may be not applicable to those AML patients without DNMT3A, NPM1 mutations and mixed-lineage leukemia translocations [8]. Therefore, the current prognostic predictors might not meet the clinical demand, new biomarkers are needed for better prognostic classification, ultimately, better therapeutic targets.
In this study, we performed Kaplan–Meier and multivariate analyses to screen for prognosis-associated genes using the expression and clinical data of 173 AML patients from The Cancer Genome Atlas (TCGA) database [9] and validated the results in an independent Oregon Health and Science University (OHSU) dataset [10]. We established a prognostic risk score based on a linear combination of 5-gene expression levels to effectively predict the overall survival (OS) of AML patients. Lastly, we utilized unsupervised hierarchical clustering of five genes and defined AML genomic subgroups and their relevance to clinical outcomes. The completion of our study paves the way for developing molecular markers in prognostication and treatment decision-making for AML patients.
Materials and methods
Data acquisition
RNA-seq expression data of 20,531 genes, and clinicopathologic characteristics of 173 AML patients were obtained from the TCGA database [9]. Genes which have expression values in less than 10% AML samples were removed from the study. 18,366 genes were included in the study. Clinicopathologic characteristics analysed in the study included patients’ age, gender, percent of bone marrow blast cells (PBMBC), European Leukemia Net (ELN) classification, isocitrate dehydrogenase 1 (IDH1), IDH2, DNMT3A, NPM1, FLT3, CEBPA, TP53, ASXL1, Runt-related transcription factor 1 (RUNX1) mutation status, OS and neoadjuvant therapy. To verify the associations of gene expression with OS, gene expression and clinical data of 405 AML patients were downloaded from the Tyner’s study (OHSU dataset) [10]. Clinicopathologic characteristics included patients’ age, gender, PBMBC, cytogenetic risk, FLT3-IDT, IDH1, CEBPA, DNMT3A, NPM1, TP53, ASXL1, RUNX1 mutation, OS, chemotherapy, bone marrow transplant and targeted therapy data in the OHSU dataset. Mutation data of the gene panel comprising Calcitonin Receptor Like Receptor (CALCRL), Dedicator Of Cytokinesis 1 (DOCK1), Phospholipase A2 Group IVA (PLA2G4A), FCH Domain Only 2 (FCHO2) and Leucine Rich Repeats And Calponin Homology Domain Containing 4 (LRCH4) were obtained from the cbioportal database and visualized with the cbioportal online tools [11].
Survival analyses
To investigate the association of gene expression or risk score with OS in the TCGA and OHSU datasets, a prognostic risk score formula was established based on a linear combination of expression levels weighted with the regression coefficients derived from the multivariate logistic regression analysis. Risk score = expression of gene 1 × β1 + expression of gene 2 × β2 + ⋯ + expression of gene n × βn. β values are the regression coefficients derived from the multivariate logistic regression analysis of the TCGA dataset. AML patients were split into high-risk and low-risk subgroups based on the median expression values and median risk score. We used Kaplan–Meier curves and log-rank methods to study the prognostic importance of gene expression and the risk score using the survival package [12, 13]. Multivariate survival analyses were performed to confirm whether gene expression and risk score are independent prognostic biomarkers after adjustment of the prognosis‑related risk factors using logistic regression model. Receiver operating characteristic (ROC) curve analysis was conducted by the R package of pROC to further validate the prognostic importance of risk score [14]. Area under curve (AUC) values were computed accordingly by the R package of pROC for the risk score. P < 0.05 was considered statistically significant.
Diagnostic analyses of five genes
Transcripts Per Million (TPM) expression data of 173 AML patients came from the TCGA database. TPM expression data of 70 bone marrow tissues were obtained from The Genotype-Tissue Expression (GTEx) project [15]. Gene expression difference of CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 was compared by the Wilcoxon sum-rank test between 173 AML patients and 70 bone marrow tissues. ROC curve analysis was conducted by the R package of pROC to determine the diagnostic values of the five genes [14]. AUC values were computed accordingly by the R package of pROC for the five genes.
Unsupervised hierarchical clustering analysis
Unsupervised hierarchical clustering of CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 was conducted using the function Pheatmap of the R package of pheatmap [16]. Difference in quantitative clinical factors was compared by the Wilcoxon sum-rank test between subgroups of patients. Count data were compared by Fisher’s exact test among the three subgroups of AML patients. Kaplan–Meier curves were plotted using the R package of survival [12], and survival rates were compared among the three clusters using the log-rank test. P < 0.05 was predefined as statistically significant.
Results
Characteristics of AML patients
Detailed clinical information of the 173 AML patients of the TCGA dataset is shown in Table 1. Patient’s age, ELN classification and TP53 expression were found to be negatively associated with OS (P < 0.05 for all cases, Student’s t-test or Fisher’s exact test, Table 1). In the OHSU database, older patient’s age and higher ELN classification and TP53 expression were negatively associated with inferior OS (P < 0.05 for all cases, Student’s t-test or Fisher’s exact test, Supplementary Table 1). Chemotherapy, bone marrow transplant and targeted therapy were positively correlated with improved OS (P < 0.05 for all cases, Fisher’s exact test, Supplementary Table 1). The other characteristics did not exhibit a significant association with OS in the TCGA and OHSU datasets (P values > 0.05 for all cases, Student’s t-test or Fisher’s exact test, Table 1 and Supplementary Table 1).
Survival analyses between patient mortality and gene expression in AML
To evaluate the predictive capability of gene expression for patients’ OS, the 173 AML patients in the TCGA dataset were divided into low and high expression groups based on median values. Kaplan–Meier survival analysis showed that high expression levels of 1352 genes and 1099 genes were associated with favourable or poor prognosis, respectively, such as CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 (P < 0.05 for all cases, log-rank test, Fig. 1, Supplementary Fig. 1). Then, multivariate analyses were performed between patients’ OS and the mortality-associated features, including patients’ age, ELN classification, and 2451 gene expression levels. Multivariate survival analyses confirmed that high expression of 337 genes was associated with decreased mortality, such as FCHO2 and LRCH4 (P = 0.01, OR: 0.36, 95% CI: 0.17–0.75; P = 0.01, OR: 0.38, 95% CI: 0.18–0.79; respectively, Supplementary Table 3). While high expression of 165 genes was associated with increased mortality, such as CALCRL, DOCK1, PLA2G4A (P = 0.03, OR: 2.32, 95% CI: 1.09–5.03, P = 0.03, OR: 2.23, 95% CI: 1.08–4.70; P < 0.001, OR: 3.88, 95% CI: 1.83–8.50; respectively, Supplementary Table 2, Supplementary Fig. 1).

Kaplan–Meier survival analysis of patients’ OS with CALCRL (a), DOCK1 (b), FCHO2 (c) and LRCH4 (d) and PLA2G4A (e) expression levels in 173 AML patients of the TCGA dataset
Validation of survival analyses
In order to validate the findings above, the association between 502 gene expression and mortality was evaluated in 405 AML samples of the OHSU dataset. Of 502 prognosis-associated genes, Kaplan–Meier survival analysis confirmed that high expression levels of 22 genes were associated with a favorable prognosis in AML. In contrast, high expression of 19 genes was associated with a poor prognosis (P < 0.05 for all cases, log-rank test, Supplementary Fig. 1 and Supplementary Fig. 2). Then, multivariate analyses were performed between patients’ OS and the mortality-associated features, including patients’ age, ELN classification, chemotherapy, bone marrow transplant, targeted therapy and 41 gene expression levels. Multivariate survival analyses confirmed that high expression of FCHO2 and LRCH4 was associated with decreased mortality (P < 0.001, OR: 0.47, 95%CI: 0.28–0.77; P = 0.02, OR:0.56, 95%CI: 0.34–0.90, respectively, Supplementary Fig. 1, Supplementary Table 3) and high expression of CALCRL, DOCK1, PLA2G4A was associated with increased mortality (P = 0.03, OR: 1.75, 95% CI: 1.07–2.88; P = 0.03, OR: 1.69, 95% CI: 1.04–2.75; P < 0.001, OR: 2.26, 95% CI: 1.40–3.72, respectively, Supplementary Fig. 1, Supplementary Table 3).
Risk score is a negative prognostic factor in AML
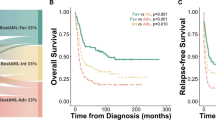
A prognostic risk score formula was established based on a linear combination of the expression levels weighted with the regression coefficients derived from multivariate logistic regression analysis: Risk score = 2.32 × expression of CALCRL + 2.23 × expression of DOCK1 + 0.36 × expression of LRCH4 + 0.38 × expression of FCHO2 + 3.88 × expression of PLA2G4A. Risk scores were computed for AML patient and then they were divided into high and low risk groups based on the median risk score. Kaplan–Meier survival analysis showed the patients with high-risk scores showed higher mortality rates than those with low-risk scores (P < 0.001, Fig. 2a). Following adjustment of prognostic risk factors, multivariate analysis confirmed that the risk score was associated with increased mortality rate in AML patients (P < 0.001, OR: 3.36, 95% CI: 1.57–7.48, Table 2). With respect to the associations of risk score with known prognostic biomarkers, the five patients with double CEBPA mutations were predicted to have low risk scores, 3 out of 15 core binding factor, 6 out of 22 NPM1-mutated/FLT3-wild type AML patients were classified as high-risk score patients. Of the 103 and 32 ELN intermediate and favorable patients, 53, 4 AML patients were predicted high-risk, respectively, risk score was a negative factor for overall survival in the ELN intermediate and favorable groups of the TCGA dataset (Supplementary Table 4). To validate the findings above, risk score was calculated following the formula in the TCGA dataset. The negative correlation was validated between OS and risk score in the OHSU dataset (Table 2 and Fig. 2b). The 25 patients with double CEBPA mutations were predicted to have high-risk scores, 32 core binding factor, 17 out of 53 NPM1-mutated/FLT3-wild type AML patients were classified as high-risk score patients. Among the ELN intermediate and favorable groups, 68 in 142, 38 in 117 AML patients were predicted high-risk, risk score was a negative factor for overall survival in the intermediate and favorable groups of the OHSU dataset following adjustment of prognosis-associated features (Supplementary Table 4). The ROC curve analysis scores were 0.74 and 0.64 for TCGA and OHSU datasets, respectively (Fig. 2c), indicating the good sensitivity and specificity of the risk score in predicting OS in AML patients.

Risk score is negative prognostic biomarker in AML. a Kaplan–Meier survival analysis of patients’ OS with risk score in the TCGA dataset, b Kaplan–Meier survival analysis of patients’ OS with risk score in the OHSU dataset. c The ROC curves of the risk scores in the TCGA and OHSU datasets
Assessment of diagnostic value
Then, the cBioPortal database was used to analyze the genomics alternations of CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 from the TCGA and OHSU datasets. The results showed that DOCK1, FCHO2 and PLA2G4A had mutations in 1%, 0.5% and 0.5% patients in the TCGA dataset. DOCK1 displayed a mutation frequency of 0.38% in the OHSU dataset (Supplementary Fig. 3). By comparing expression levels of CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 between 173 AML samples and 70 bone marrow tissues, CALCRL, DOCK1, PLA2G4A and LRCH4 were found to be up-regulated, while FCHO2 was down-regulated in AML samples (P < 0.05 for all cases, Wilcoxon sum-rank test, Fig. 3a). ROC curves were constructed to further explore the diagnostic values of the five genes. CALCRL, DOCK1, PLA2G4A and LRCH4 in particular exhibited high accuracy in differentiating AML tissues from bone marrow tissues (Fig. 3b, P values < 0.05, AUC > 0.85 for all cases).

Diagnostic value of the gene panel. a Expression difference of CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 between 173 AML samples and 70 bone marrow tissues. b The ROC curves of the five genes in the TCGA dataset
Unsupervised hierarchical clustering analysis
Hierarchical clustering analysis of the five genes revealed three subgroups of AML patients in the TCGA dataset (Supplementary Fig. 4). The cluster1 AML patients were associated with lower cytogenetics risk than cluster2 or 3 tumors, and more favorable OS than cluster3 patients (P values < 0.05 for all cases, fisher exact test or log-rank test, Fig. 4a, b). The remaining factors NPM1, DNMT3A, IDH1, IDH2, FLT3, CEBPA, TP53, ASXL1 and RUNX1 mutations, gender and neoadjuvant treatment did not exhibit significant difference between subgroups of AML patients (P values > 0.05 for all cases, Fisher’s exact test). To validate the findings, we performed the classification of 405 AML patients using the gene panel and found three clusters of AML patients in the OHSU dataset (Supplementary Fig. 5). Cluster1 tumors were significantly associated with lower cytogenetics risk, higher frequencies of TP53, RUNX1 and targeted therapy than those in cluster 2, lower frequency of FLT3-ITD mutation than cluster3 tumors, higher frequency of FLT3-ITD mutations than cluster2 tumors and more favourable OS than cluster2, 3 tumors (P values < 0.05 for all cases, Wilcoxon sum-rank test, Fisher’s exact test or log-rank test, Fig. 4c, d and Supplementary Fig. 6).

The three clusters of AML patients (1–3) showed significant differences in cytogenetic risk (a), OS (b) in the TCGA dataset, cytogenetic risk (c) and OS (d) in the OHSU dataset
Discussion
Acute myeloid leukemia (AML) is the most common type of acute leukemia and biologically heterogeneous diseases with poor prognosis. Accurate assessment of prognosis is central to the management of AML for genomics researchers and physicians. The 2017 ELN guidelines are widely used for the evaluation of prognostic risk and classifying patients into “favorable,” “intermediate,” and “adverse” subgroups on the basis of leukemia cell cytogenetics and somatic mutations in several key driver genes [17]. Papaemmanuil et al. [18] developed a Bayesian statistical model to compartmentalize AML into mutually exclusive subtypes based on patterns of co-mutation and defined 11 classes of AML, each with distinct diagnostic features and clinical outcomes. Ciftciler et al. [19] demonstrated that pre-transplant bone marrow blast percentage is a positive prognostic factor for patients with AML, with patients with pre-transplant bone marrow blast cells < 5 showing more favourable survival than those with pre-transplant bone marrow blast cells 5–10%.
In recent years, an increasing number of mRNAs have been demonstrated to be potential prognostic biomarkers in AML. Lee [20] reported that elevated expression of DOCK1 confers poor prognosis in acute myeloid leukemia. Angenendt [21] revealed increasing expression levels of CALCRL were associated with decreasing complete remission rates, 5-year overall, and event-free survival. Despite significant advances in the risk classification of AML, a single gene might be an inaccurate predictor, because various factors can affect a single gene. There are few reports on a gene signature comprising various genes to predict cancer outcomes.
In this study, we performed Kaplan–Meier and multivariate analyses using the mRNA expression data of two independent datasets and found CALCRL, DOCK1, PLA2G4A, FCHO2 and LRCH4 expression levels could predict the OS of AML patients. Furthermore, we computed a risk score using a linear combination of 5-gene expression levels and β-values from subsequently multivariate logistic regression models. The risk score remained significantly associated with poor OS after adjusting for established prognosticators. The five genes play diverse roles in the tumorigeneses of cancers. For instance, the PLA2G4A gene encodes a member of the cytosolic phospholipase A2 group IV family. The enzyme catalyzes the hydrolysis of membrane phospholipids to release arachidonic acid which is subsequently metabolized into eicosanoids. Eicosanoids, including prostaglandins and leukotrienes, are lipid-based cellular hormones that regulate hemodynamics, inflammatory responses, and other intracellular pathways [22]. PLA2G4A is up-regulated in glioblastoma [23], AML [24], lung cancer [25] and colon cancer [26]. PLA2G4A depletion moderately inhibited glioblastoma proliferation and survival but remarkably sensitized chemo-resistant glioblastoma cells to several chemotherapeutic agents through suppressing the PI3K/Akt/mTOR pathway in glioblastoma cells [23]. Similarly, reduction in PLA2G4A activity caused decreased growth of A549 and H460 lung cancer cells [25] and reduced both basal and the leukotriene D4 -induced proliferation, the effects being most pronounced in Caco-2 tumor cells [26]. These results combined with our study suggest PLA2G4A may serve as oncogene in cancers.
Furthermore, the 5-gene expression signature effectively stratified AML patients into three subgroups with different survival probabilities. Given the 5-gene expression signature is independent of known prognosis-associated mutations in NPM1, DNMT3A, IDH1, IDH2 and CEBPA, the 5-gene expression signature may have prognostic values for the faction of AMLs who harbor normal or risk-indeterminate karyotypes. In addition to prognostic value, the five genes also showed diagnostic value for AML patients. Our study revealed that CALCRL, DOCK1, PLA2G4A and LRCH4 differentiated AML tissues from bone marrow tissues with high accuracy. Lastly, the five genes may also pave the way for developing targeted therapies for AML patients. For instance CRISPR-Cas9-mediated knockout of CALCRL significantly inhibited colony formation in human myeloid leukemia cell lines [21]. Selective inhibition of DOCK1 ablated cellular invasion in Ras-transformed cells and suppressed cancer metastasis and growth in vivo in mice [27].
Conclusion
Taken together, this study is the first to report a 5-gene risk signature that has prognostic and diagnostic values and successfully stratifies AML patients. A higher risk score indicates a poorer prognosis. These findings will help researchers identify new treatments for AML and to provide more therapeutic targets to cure AML patients in the future.
Availability of data and material
The datasets generated and/or analysed during the current study are available upon reasonable request.
References
Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–907.
Siegel R, Miller K, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:29. https://onlinelibrary.wiley.com/doi/10.3322/caac.21254/pdf
Shah A, Andersson TML, Rachet B, Björkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971–2006: a population-based study. Br J Haematol. 2013;162:509–16.
Meyers J, Yu Y, Kaye JA, Davis KL. Medicare fee-for-service enrollees with primary acute myeloid leukemia: an analysis of treatment patterns, survival, and healthcare resource utilization and costs. Appl Health Econ Health Policy. 2013;11:275–86. https://doi.org/10.1007/s40258-013-0032-2.
Mrózek K, Heerema NA, Bloomfield CD. Cytogenetics in acute leukemia. Blood Rev. 2004;18:115–36.
Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA. 2009;106:12950–5. https://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2716381&tool=pmcentrez&rendertype=abstract
Bullinger L, Krönke J, Schön C, Radtke I, Urlbauer K, Botzenhardt U, et al. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia. 2010;24:438–49.
Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. https://doi.org/10.1056/NEJMoa1112304 (Massachusetts Medical Society).
Network TCGAR. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med. 2013;368:2059–74. https://www.nejm.org/doi/10.1056/NEJMoa1301689
Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526–31. https://doi.org/10.1038/s41586-018-0623-z.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1–pl1. https://pubmed.ncbi.nlm.nih.gov/23550210
Therneau T. Survival Analysis. Cran [Internet]. 2016; https://cran.r-project.org/web/packages/survival/survival.pdf
Fox J. Cox Proportional-Hazards Regression for Survival Data The Cox Proportional-Hazards Model. Most. 2002;2008:1–18. https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.110.2264&rep=rep1&type=pdf
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. BioMed Central Ltd; 2011;12:77. https://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-12-77
Consortium Gte. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–5. https://pubmed.ncbi.nlm.nih.gov/23715323
Warnes G, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, et al. gplots: Various R programming tools for plotting data. R Packag. version. 2005.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. https://pubmed.ncbi.nlm.nih.gov/27895058 (American Society of Hematology)
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Ciftciler R, Goker H, Buyukasık Y, Malkan UY, Saglam EA. Impact of pre-transplant bone marrow blast percentage on survival in acute myeloid leukemia patients. Int J Clin Exp Med. 2019;12:9288–94.
Lee S, Chiu Y, Li Y, Lin C, Hou H, Chou W, et al. High Expression of dedicator of Cytokinesis 1 ( DOCK1) Confers Poor Prognosis in Acute Myeloid Leukemia. Oncotarget. 2017;8:72250–9.
Angenendt L, Bormann E, Pabst C, Alla V, Görlich D, Braun L, et al. The neuropeptide receptor calcitonin receptor-like (CALCRL) is a potential therapeutic target in acute myeloid leukemia. Leukemia. 2019;33:2830–41. https://doi.org/10.1038/s41375-019-0505-x.
Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–5. https://nar.oxfordjournals.org/lookup/doi/10.1093/nar/gkl842
Yang L, Zhang H. Expression of cytosolic phospholipase A2 alpha in glioblastoma is associated with resistance to chemotherapy. Am J Med Sci. 2018;356:391–8. https://doi.org/10.1016/j.amjms.2018.06.019.
Runarsson G, Feltenmark S, Forsell PKA, Sjöberg J, Björkholm M, Claesson H-E. The expression of cytosolic phospholipase A2 and biosynthesis of leukotriene B4 in acute myeloid leukemia cells. Eur J Haematol. 2007;79:468–76. https://doi.org/10.1111/j.1600-0609.2007.00967.x.
Sundarraj S, Kannan S, Thangam R, Gunasekaran P. Effects of the inhibition of cytosolic phospholipase A2α in non-small cell lung cancer cells. J Cancer Res Clin Oncol. 2012;138:827–35. https://doi.org/10.1007/s00432-012-1157-7.
Parhamifar L, Jeppsson B, Sjölander A. Activation of cPLA2 is required for leukotriene D4-induced proliferation in colon cancer cells. Carcinogenesis. 2005;26:1988–98.
Tajiri H, Uruno T, Shirai T, Takaya D, Matsunaga S, Setoyama D, et al. Targeting ras-driven cancer cell survival and invasion through selective inhibition of DOCK1. Cell Rep. 2017;19:969–80.
Funding
None.
Author information
Authors and Affiliations
Contributions
KS designed the study. RP, KS, LC downloaded somatic mutation, RNA-seq data and clinical data from the TCGA and OHSU databases. YL, PZ, RP, XS performed the survival analyses and differentially expressed gene analysis. ZF, LC conducted unsupervised hierarchical clustering analysis. KS prepared the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
The study was approved by the Ethics Committee of the Department and conformed to the ethical guidelines of the Helsinki Declaration (as revised in Tykyo 2004).
Informed consent
All participants have provided written informed consent in this study.
Code availability
The codes in the current study are available upon reasonable request.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12094_2020_2460_MOESM2_ESM.tif
Supplementary file2 Supplementary Figure 1. The overlap of prognosis-associated genes between TCGA and OHSU datasets. A. The overlap of protective type genes between TCGA and OHSU datasets, A. The overlap of risk type genes between TCGA and OHSU datasets. (TIF 1959 kb)
12094_2020_2460_MOESM3_ESM.tif
Supplementary file3 Supplementary Figure 2. Kaplan-Meier survival analysis of patients’ OS with CALCRL (A), DOCK1 (B), FCHO2 (C) and LRCH4 (D) and PLA2G4A (E) expression levels in 405 AML patients of the OHSU dataset. (TIF 1232 kb)
12094_2020_2460_MOESM4_ESM.tif
Supplementary file4 Supplementary Figure 3. The mutation frequencies of DOCK1 (A), FCHO2 (B) and PLA2G4A (C) in the TCGA and OHSU datasets. (TIF 1898 kb)

12094_2020_2460_MOESM5_ESM.jpg
Supplementary file5 Supplementary Figure 4. Unsupervised hierarchical clustering of the 5-gene panel uncovered three classes of AML patients in the TCGA dataset. (JPEG 1663 kb)

12094_2020_2460_MOESM6_ESM.jpg
Supplementary file6 Supplementary Figure 5. Unsupervised hierarchical clustering of the 5-gene panel uncovered three classes of AML patients in the OHSU dataset. (JPEG 978 kb)
12094_2020_2460_MOESM7_ESM.tif
Supplementary file7 Supplementary Figure 6. The three clusters of AML patients exhibited significant differences in FLT3-ITD mutation in the OHSU dataset. (TIF 1847 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Sha, K., Lu, Y., Zhang, P. et al. Identifying a novel 5-gene signature predicting clinical outcomes in acute myeloid leukemia. Clin Transl Oncol 23, 648–656 (2021). https://doi.org/10.1007/s12094-020-02460-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-020-02460-1