
Abstract
Ewing sarcoma is a rare tumor that arises in bones of children and teenagers but, in 15% of the patients it is presented as a primary soft tissue tumor. Balanced reciprocal chimeric translocation t(11;22)(q24;q12), which encodes an oncogenic protein fusion (EWSR1/FLI1), is the most generalized and characteristic molecular event. Using conventional treatments, (chemotherapy, surgery and radiotherapy) long-term overall survival rate is 30% for patients with disseminated disease and 65–75% for patients with localized tumors. Urgent new effective drug development is a challenge. This review summarizes the preclinical and clinical investigational knowledge about prognostic and targetable biomarkers in Ewing sarcoma, finally suggesting a workflow for precision medicine committees.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ewing sarcoma (ES) is morphologically a round blue cell tumor. Differential diagnosis of immature tumors as ES has been a challenge in pathology laboratories for decades [1]. WHO established new diagnosis criteria for ES in 2013 [2]. Since then, the presence of a typical translocation confirms diagnosis, independently of primary tumor location and peculiar morphological data. Balanced reciprocal chimeric translocation t(11;22)(q24;q12), which encodes an oncogenic protein fusion (EWSR1/FLI1), is the most generalized and characteristic molecular event. EWSR1 is a TET RNA-binding protein (like TLS and TAF15). FLI1 is a member of the ETS family transcription factors (like ERG, FEV, ETV1, E1AF). Other gene fusions affecting the TET/ETS family members have been identified in ES and have diagnostic value as well [3]. ES typically arises in children’s and teenagers’ bone, but in 15% of the patients it is presented as a primary soft tissue tumor [4, 5]. Using conventional treatments (chemotherapy, surgery and radiotherapy), long-term overall survival rate is 30% for patients with disseminated disease and 65–75% for patients with localized tumors. Results have not improved during the last decades [6, 7]. Therefore, urgent new effective drug development is a challenge for the mankind.
Gene fusion EWSR1/FLI1 acts as a transcription factor and has a main role in ES development, because it conditions an oncogenic transcriptional programme [8,9,10,11,12,13,14,15]. Moreover, it is suspected that EWSR1/FLI1 determines an epigenetic dysregulation which has a remarkable position in Ewing biology. Many current and previous innovative efforts have been directed to recover this disrupted epigenetic programme. In addition, chromosomal copy number variations (CNV) are well described in Ewing tumors. There are some common CNV alterations in ES: gain of chromosome 1q, 8, 12 and loss of 9p21 and 16q [16,17,18]. The pathogenic role of these chromosomal aberrations is not well understood. Furthermore, as far as mutational status is concerned, ES is one of the less mutated types of cancer [19, 20] and few mutations can be actionable with commercialized drugs.
Important personalized medicine projects for refractory tumors in pediatric oncology have been developed around the world in the past few years [21,22,23,24,25,26,27], aiming to transfer basic molecular information to the clinic. These investigational groups studied genomics and transcriptomics in a large patient series. Their interesting molecular data are enriching our previous knowledge about ES and other tumors in pediatric patients. Nevertheless, only few pediatric patients have benefited from this approximation, due to the fact that translation is a huge challenge in pediatric oncology, and many key points are still unknown [28]. In addition, a vast volume of data has been generated, and interpretation requires really important efforts.
This review summarizes the preclinical and clinical investigational knowledge about prognostic and targetable biomarkers in ES, finally suggesting a workflow for precision medicine committees.
Prognostic factors in ES
Clinical prognostic factors
The most important clinical prognostic factor at diagnosis is the presence of metastatic disease. This conditions a low survival rate [29, 30]. Other clinical prognostic factors have been described in the past few years: volume, tumor necrosis after neoadjuvant chemotherapy, age and axial location [29,30,31,32]. Comparing extraskeletal with skeletal located Ewing tumors, extraskeletal have better prognosis than those located in bone [33]. Patients that progressed while receiving initial therapy had worse prognosis than patients who relapsed later. Moreover, a short remission has bad prognosis [34, 35]. Nevertheless, this information is irrelevant when a patient affected by a refractory or relapsed ES tumor is forwarded to personalized medicine programmes [34, 35]. When patients are submitted to these programmes, general status, psychological burnout, and cumulative toxicity decrease the options of intensive interventions.
Biologic potential prognostic factors
Next to this hardly standarizable clinical data, biologic studies can enrich prognosis information at relapse. Some of them might be considered at diagnosis to stratify treatment (Table 1).
Translocation type
It was hypothesized that Ewing tumors with different translocations could have distinct prognosis in large cohorts [36, 37]. Fusion gene members have diagnostic importance, and must be taken into account when considering therapeutic alternatives. However, fusion type has not been demonstrated to be a prognostic marker [38,39,40].
Copy number variations (different to chr9p21, CDKN2A locus)
Some chromosomal CNV have been detected in Ewing tumors recurrently. These are gains of chromosome 1q, 8, 12 and loss of 9p21 and 16q. Chromosome 1q gain and chromosome 16q loss appear together because of an unbalanced translocation t(1;16) detected in this context.
It has been reported, in a short series, that patients with low copy number changes (≤ 3 copy number aberrations) have significantly better prognosis than patients with a high number of chromosomal alterations (in terms of event-free and overall survival) [41]. 1q gain and 16q loss have been associated with bad prognosis by several groups [17, 18, 42, 43]. No clear prognosis conclusions have been established about 8 and 12 chromosomal gains.
Copy number variations in 9p21 (CDKN2A locus)
CDKN2A (INK4A/ARF) homozygous deletion was first described by Kovar et al. in 1997 [44]. They observed CDKN2A deletions in 30% of ES tumors (8/27). This fact has been communicated later on several times [45,46,47,48]. Loss of this cell cycle arrest machinery component has been associated with worse prognosis [46, 47, 49]. Based on published data, the Children’s Oncology Group (COG) committee established CDKN2A loss as a negative prognostic marker [50]. However, in the last years, Lerman D et al. [51] demonstrated in a large cohort that CDKN2A leak is not a prognostic factor in localized Ewing tumors. The work of Tirade et al. does not support that hypothesis [52]. Therefore, CDKN2A must not be considered an adverse prognostic factor by itself.
TP53 mutations
TP53 mutations have been proposed as a bad prognosis biological marker in ES during the last decades. Several retrospective studies were concordant with this suspicion. Different investigations sustained this hypothesis [53,54,55,56]. Their work suggested worse overall survival in the presence of TP53 mutations. Recently, Lerman D et al. observed in a large series no significant differences in event-free survival of patients with TP53 mutations, in the presence or not of CDKN2A deletions. Consequently, TP53 mutations (present in around 10% ES patients) should not be considered as a bad prognosis factor when transferring personalized medicine study results to clinicians [51].
STAG2 (Stromal Antigen 2) mutations
STAG2 gene encodes for a component of cohesin complex (a complex required for sister chromatids cohesion after DNA replication) which is frequently mutated in ES (around 17% of tumors) [52]. STAG2 mutations and CDKN2A losses are considered mutually exclusive [52]. Tirode et al. observed bad prognosis when STAG2 and TP53 mutations coexist. Brohl et al., in another huge sequencing effort, observed STAG2 mutations in 21.5% of ES samples. They concluded that STAG2 mutations may be associated with more advanced disease and a modest decrease in overall survival, independently of TP53 status [20]. Crompton et al. described STAG2 loss in over 15% of ES tumors. It mostly occurs by point mutation, but also by rearrangement, and this suggests that it is likely conducted by non-genetic mechanisms. STAG2 losses were associated with metastatic disease, and therefore worse prognosis [57]. Taking into account the above, STAG2 status may be used as a prognosis marker.
RASSF2, NPTX2 and PHF11 methylation pattern
The Farida Latif group, in two related papers, described poor overall survival when whichever of these genes was methylated [58, 59]. RASSF2 (Ras Association Domain Family Member 2) codifies for one of the broad range of effector proteins that RAS protein family has [60]. RASSF2 protein interacts with KRAS [60]. RASSF2 and PAR4 (Proteinase-Activated Receptor 4) interact directly and this interaction is enhanced by activated KRAS [61]; PAR4–RASSF2 interaction allows tumor suppressor functions. When RASSF2 is methylated, this function is lost, and maybe this contributes towards tumor development, as shown in prostate cancer [61]. In ES, an equivalent effect might happen. NPTX2 (pentraxin II, neuronal) and PHF11 (PHD Finger Protein 11) have an undiscovered role in cancer.
CCL21 expression levels
Laurens G. L et al. reported that higher RNA expression levels of CCL21 condition an improved outcome. Therefore, it might be considered as a prognostic factor, although larger patient series are necessary to confirm this data [62].
PD1/PDL1 lymphocytes/ES expression
Kim et al. studied PD1 and PDL1 expression in a large cohort of ES patients. They concluded that patients with a PD1( +)/ PD-L1( +) pattern had the shortest survival time [63]. They also suggested that a PD1/PD-L1 positivity could be a criterion for a selection of patients susceptible of PD1-based immunotherapy.
Targetable biological markers
There are many molecular candidates postulated to become the best actionable target in Ewing sarcoma. However, there are very few that have gained importance (such as imatinib for GIST- or LMC-positive Philadelphia). In ES, there is still much to decide, and new candidates are being currently explored. These candidates are shown below.
EWSR1/FLI1 translocation
EWSR1/FLI1 fusion protein was described in 1992 [64]. The protein product results from t(11;22) and acts as an aberrant transcription factor [65]. During the last years, the oncogenic role of this fusion protein has been demonstrated in vitro and in vivo using different models [66,67,68,69]. It drives an oncogenic transcriptional programme, upregulating and downregulating thousands of genes [8, 9, 70]. Riggi et al. proposed the exact chromatin remodeling events leading to gene activation and repression [15], and Bilke and others reported the binding sites of EWSR1/FLI1 fusion protein as GGAA microsatellite regions [71,72,73,74,75].
Due to EWSR1/FLI1 suspected relevance in ES pathology, many therapeutic approaches have been directed to target the translocation or important proteins that present abnormal expression levels secondary to the fusion protein function.
A summary of them is presented.
Targeting EWSR1/FLI1 fusion protein
Targeting transcription factors directly has been a challenge for the scientific community until today. The most direct and specific approach to inactivate the EWSR1/FLI1 protein fusion is using oligonucleotides. They hybridize to specifically selected sequences and thereby break mRNA splicing and protein translation. Unfortunately, this strategy never reaches a complete absence of targeted proteins. Therefore, other strategies to block EWSR1/FLI1 activity have been developed.
Toretsky et al. described that RHA protein (RNA Helicase A) binds EWSR1/FLI1 and works in enhancing transcription signals in EWSR1/FLI1 target promoters [76]. Barber-Rotenberg et al. suggested that the small molecule YK-4-279 was able to disrupt the binding between EWSR1-FLI1 and RHA, blocking the transcriptional activity of EWSR1-FLI1 in vitro [77]. A more precise explanation for YK-4-279 mechanism of action was proposed by the same group: the disruption of EWSR1/FLI protein interactions within the spliceosome [78]. However, drug resistance was evident in some mouse cohorts [77, 79, 80]. The resistance mechanism has been studied by them, but no conclusive results have been obtained yet. Currently, clinical trials using this drug have not been registered.
Targeting downstream EWSR1/FLI1
EZH2 (enhancer of Zeste, Drosophila, Homolog 2)
EZH2 encodes a histone methyltransferase which methylates nucleosomal histone H3 at lysine-27 (H3-K27). It is part of the polycomb repressive complex-2 (PRC2). This complex initiates epigenetic silencing of genes involved in cell fate decisions [81, 82]. Richter et al. described an upregulation of EZH2 in ES cells in vitro and in vivo, since EWSR1/FLI binds EZH2 promoter to potentiate its expression. The high EZH2 activity in ES suggests an important role in oncogenicity and immaturity of this tumor [83] and therefore, it is a potential target [84]. Pankita et al. have studied (in vitro) if tazemetostat (highly selective EZH2 inhibitor) potentiates or not chemotherapy agents (etoposide and irinotecan). Their preclinical results, not published yet, are hopefully in this direction [85]. Currently, one pediatric MATCH clinical trial is recruiting ES patients that are EZH2 mutation carriers (NCT03155620) to receive tazemetostat, but it is not using EZH2 protein expression levels as a biomarker. In a small series, EZH2 high expression has been suggested as a bad prognosis factor too [86]. More preclinical studies are necessary. The combination of EZH2 inhibitors with chemotherapy agents should be studied in a trial on ES in the near future.
BET proteins
BET proteins (BRD2, BRD3, BRD4) are bromodomain (BRD)-containing proteins [87] that recognize acetylated lysine residues on the tails of histone proteins. EWSR1/FLI1 fusion protein deregulates epigenetic programme and can lead to the creation of specific epigenetic marks [15, 88]. On this basis, BET inhibitors were tested and have demonstrated downregulation of genes involved in ES pathogenesis [89]. BET inhibitors drive apoptosis in ES cell lines too and, interestingly, the association of these drugs with PI3K/mTOR inhibitor BEZ235 increases apoptosis [89]. Similar effects with EZH2 inhibitor combination are described for them. Several BET inhibitors have been tested in clinical trials, but none is recruiting ES patients yet.
LSD1 (lysine-specific demethylase 1)
Lysine-specific demethylase 1 was the first discovered histone lysine demethylase and can demethylate both H3K4me2/1 and H3K9me2/1 [90]. Due to the fact that ES fusion gene causes a deregulated epigenetic programme, it was hypothesized that this protein could be altered in ES tumors. Schildhaus et al. demonstrated high levels of LSD1 in different sarcoma types [91], and 1 year later Bennani-Baiti et al. confirmed the same phenomenon in ES, LSD1 inhibitors interfere with cell growth in ES (in vitro) [92]. Sankar et al. demonstrated that LSD1 inhibition with HCI2509 modifies the downstream oncogenic phenotypes driven by EWSR1/ETS fusions both in vitro and in vivo ES models [93]. Reversing epigenetic modifications using LSD1 inhibitors were reproduced for other groups [94]. Currently, two phase I trials are recruiting ES patients to test LSD1 inhibitors: SP-2577 (NCT03600649) and INCB059872 (NCT03514407).
NKX2.2 (NK2Homeobox2)
The protein encoded by this gene contains a homeobox domain and its physiologic function as a transcription factor is only partially understood. It may be involved in the morphogenesis of the central nervous system and develops a role in the maintenance of NEUROD1 expression in the endocrine pancreas cells. Curiously, Smith et al. discovered a high expression of NKX2.2 in ES tumors when this gene had not been related with cancer previously [95]. This work established NKX2.2 as a EWSR1/FLI target in ES. They showed that loss of NKX2.2 expression via RNAi results in a loss of oncogenic transformation [95]. Afterwards, Owen et al. concluded that NKX2.2 collaborates in oncogenic transformation via transcriptional repression (using repressor domains of NKX2.2), while the NKX2.2 transcriptional activation domain is dispensable [96]. Under these circumstances, NKX2.2 could also be proposed as a therapeutic target for Ewing sarcoma. Nevertheless, targeting transcription factors is difficult. Successfully, other therapeutic strategies have demonstrated NKX2.2 downregulation. In preclinical studies, treatment with HDAC inhibitor (vorinostat) depressed gene targets of EWSR1/FLI (NKX2.2, BCL11B). This effect is probably due to the fact that NKX2.2 recruits transcriptional corepressors and HDACs to gene promoters to develop its function [96]. Sampson et al. combined vorinostat with temozolomide, and irinotecan. Interestingly, they reported enhanced cytotoxicity in vitro when temozolomide is administered before vorinostat [97]. When targeting LSD1, similar effects to HDACs in ES models have been described [93].
Cyclin D1 gene (CCND1) and cyclin D4 gene (CDK4)
Starting from Riggi N. et al.’s study about chromatin remodeling mechanisms, Alyssa L. Kennedy et al. elaborated super-enhancer regions profiles. Super-enhancer regions of chromatin are regions of open chromatin with acetylated histones, transcription factors and transcriptional activators to promote transcription. Super-enhancer regions are corrupted in ES and they mark a tumor-specific gene expression programme [98]. They confirmed using this approach that ES was selectively dependent on cyclin D1 gene (CCND1) and cyclin D4 gene (CDK4) compared to other cancer cell lines. Moreover, they showed that ES cell lines are sensitive to pharmacological inhibition of CDK4/6, both in vitro and in vivo [98]. This study proposing CDK4/6 inhibitors is complementary to point 4 (see below).
PRKCB (protein kinase C beta)
Protein kinase C (PKC) family can be activated by calcium and second messenger diacylglycerol. PKC family members phosphorylate a wide variety of protein targets. Surdez et al. described that transcriptional activation of PRKCB (a member of PKC family) is directly regulated by the chimeric fusion EWSR1/FLI1. They detected that PRKCB phosphorylates histone H3T6 to permit global maintenance of H3K4 trimethylation at a variety of gene promoters involved in ES [99]. Nowadays, PKC inhibitors are in clinical trials, but none including ES patients.
HSP90 (heat shock 90 kDa protein)
HSP90 is a chaperone protein that promotes maturation, structural maintenance and proper regulation of specific protein targets involved in cell cycle control and signal transduction, among other functions. EWSR1/FLI1 and EWSR1/E1AF fusion genes activate telomerase and up-regulate TERT (reverse transcriptase telomerase) in ES cell lines [100]. Ambatia et al. described that TERT is a client protein of the HSP90 chaperone complex and inhibition of HSP90 led to decreased TERT expression in cell lines [101]. They hypothesized that the combination of HSP90 inhibitors and bortezomib may exert more potent inhibitory effects [101].
FOXO1 (Forkhead box O1)
FOXO1 is a transcription factor that regulates differentiation, proliferation, tumor suppression, autophagy, and cell death [102]. FOXO1 is transcriptionally repressed by EWSR1/FLI1 through direct promoter binding [102]. Interestingly, methylseleninic acid (MSA) increases FOXO1 expression in the presence of EWSR1/FLI1, and induces massive cell death [103, 104]. This drug is not currently used in clinical trials.
Non-specific downstream effective drugs
Trabectedin is a marine alkaloid isolated from the Caribbean tunicate Ecteinascidia turbinata [105]. This drug interferes with the activity of EWSR1/FLI1 [10]. Grohar et al. described that trabectedin reverses a gene signature of induced downstream targets of EWSR1/FLI1 in ES cell lines [106]. Until today, it has been approved for refractory adult sarcoma. Clinical trials with trabectedin in pediatric ES have been conducted. The trial of trabectedin in treating young patients with recurrent or refractory soft tissue sarcoma or Ewing's family of tumors (NCT00070109) has been completed. A number of eight patients received the drug: five progressed, two discontinued due to adverse events and one died. Amaral et al. published that trabectedin was able to strongly reduce EWSR1/FLI1 effects in vitro and in xenografts, but leading to IGF1R upregulation. The combination of trabectedin with IGFR1 inhibitors potentiates the efficacy of trabectedin in vitro and in vivo [107]. However, clinical trials are not underway yet. Lurbinectedin is a second-generation drug. Harlow et al. reported a complete reversal of EWSR1/FLI1 activity and elimination of established tumors in 30–70% of mice treated with this drug plus irinotecan [108]. This combination is being explored in clinical trials (NCT02611024).
Grohar et al. described ES preclinical sensitivity to mithramycin. They started a clinical trial but serious toxicities (hepatotoxicities) were detected and this limited its use [109]. The same group developed less toxic analogs EC-8105 and EC-8042. Both were less toxic than mithramycin in vivo but maintained suppression of EWS-FLI1 at similar concentrations [110]. None of them have been investigated in trials yet.
Midostaurin was also detected as a potential targeted therapy in ES during drug screening. This approach also found synergistic effect of simultaneous application of PKC412 (midostaurin) and IGF1R inhibitors [111].
IGF1/IGF1R
Autocrine and paracrine activated loop IGF1/IGF1R (insulin-like growth factor-I receptor) was postulated as a growing determinant in ES more than 20 years ago [112, 113], because EWSR1/FLI-1 gene fusion expression disrupts the IGFIR signaling pathway [114]. Therefore, it could be considered as a downstream element of ES fusion proteins. However, the importance of IGF1R in ES deserves an independent commentary. Kang et al. showed in vivo that IGF1R activation was independent of ES fusion protein type. They demonstrated IGF1-induced expression in the presence of EWSR1/FLI1, EWSR1/ERG and FUS/ERG fusion proteins [115]. IGF1R inhibitors were developed and tested in preclinical models [116,117,118]. Several difficulties have been detected during preclinical efforts and clinical trials. Due to the similarity of the structures of IGF1R and INSR (insulin receptor), synthesis of selective inhibitors of IGF1R is very complex. Monoclonal antibodies directed to IGF1R have been the main strategy developed. R1507 [119], cixutumumab and [120] figitumumab [121] were well-tolerated drugs, but modest responses in monotherapy were detected when treating patients. Moreover, detecting predictors of response is also difficult. There was not an apparent correlation between response to cixutumumab and tumor expression of IGF1, IGF2, or IGF1R [120]. However, a strong association between IGF-1 serum levels pretreatment and survival benefit was identified by Juergens et al. [121].
As in other drugs, acquired resistance to IGF1R blockage is a major problem when targeting IGFIR in ES [122]. Combination of IGF1R inhibitors and mTor inhibitors or ERK inhibitors has been proposed as alternative, after studying resistance mechanism in monotherapy responding patients [123, 124]. In this way, hopeful results were obtained trialing cixutumumab plus temsirolimus (mTor1 inhibitor) [125]. Ambatia et al. proposed an association of HSP90 inhibitors with IGF1R blocking drugs, because HSP90 was upregulated in treated patients [101]. Nowadays, there are no clinical trials with cixutumumab or figitumumab in monotherapy or combination, recruiting ES patients.
With some difficulties, small molecules targeting IGF1R have been developed too.
OSI-906 (IGF1R and insulin receptor (INSR) inhibitor) has been tested in combination with erlotinib in solid tumors [126]. ADW742 induces apoptosis in ES cell lines, and synergizes with imatinib [127, 128]. Nevertheless, these molecules are not currently in clinical recruiting trials.
Despite these limitations, addressing IGF1R in a combined way is still of interest in ES. Efforts aimed to establish predictive response markers are essential for our patients.
PARP (Poly-ADP-ribose polymerase)
PARP is an enzyme that collaborates in base excision repair (BER) pathway. It catalyzes the poly-ADP-ribosylation of some acceptor proteins involved in chromatin architecture and in DNA metabolism when DNA damage is present. It is an essential step leading to reparation of DNA strand breaks. Soldatenkov et al. described that wildtype ETS transcription factors regulate positively PARP levels, but surprisingly, changes in EWSR1/FLI1 expression do not cause changes in PARP expression level. Therefore, it was hypothesized that the fusion protein acted directly on DNA repair pathway and allowed tumor cells to avoid apoptosis, independently of PARP [129].
Although the exact biological reasons are not well understood, PARP presents high levels in ES tumors [130]. The DNA break repair is defective in ES and tumor responses were seen with PARP inhibitors. At present, this is one of the most expanding fields in new therapies for ES. The reasons of response to PARP inhibitors were studied by Gill et al. They concluded that effectiveness in ES is not caused by a defect in homologous DNA repair, but through hypersensitivity to trapped PARP1–DNA complexes. This drives accumulation of DNA damage during replication, ultimately leading to apoptosis [131]. Stewart et al. described in vivo cytotoxicity with olaparib (PARP inhibitor) alone and higher in combination with temozolomide or irinotecan [132]. Norris et al. did not demonstrate synergism in vivo [133]. However, Engert et al. again, suggested synergism between PARP inhibitors and temozolomide and also cytotoxicity with doxorubicin, etoposide or ifosfamide [134]. Other PARP inhibitors showed similar effects [135]. Based on these preclinical approximations, clinical trials have been conducted. Olaparib in monotherapy was trialed without objective responses (no selection of patients based in biological markers was done) [136]. At this moment, ES patients have been recruited in two clinical trials with olaparib monotherapy (NCT03233204) or combined (NCT01858168); talazoparib has one recruiting trial (NCT02116777) and there is an active trial with niraparib (NCT02044120).
Another interesting focus is PARP inhibitors in combination with trabectedin. Ordóñez et al. described relevant antitumor activity in preclinical models [137]. Smiths et al.’s proposal about synergic effects in apoptosis when associating radiotherapy with PARP inhibitors is also interesting [138].
Again, we are facing a huge problem: which biological markers should be used to select candidate patients for targeted therapy? Clinical trials and their published results are really required. We cannot personalize treatment in refractory patients without knowledge about these potential targetable biomarkers.
ATR (ataxia telangiectasia and Rad3-related protein)
ATR is an essential mediator of genomic integrity, replication-fork stability, cell cycle checkpoints, and DNA repair [139]. Activated oncogenes induce collapse of DNA replication forks, that generate replicative stress, double-strand breaks (DSBs) and therefore genomic instability [140]. High levels of replicative stress, facilitate tumoral cell death. ATR and CHEK1 are replicative stress response proteins. Targeting ATR leads to an increase in the levels of replicative stress that condition higher tumor toxicity. Nieto Soler et al. studied in ES cell lines that toxicity of ATR inhibitors correlated with CHEK1 and H2AFX expression levels (proportionally to the replicative stress) [141]. ATR inhibitors (VX-970, BAY1895344, M6620, AZD6738) are in clinical trials, but none are recruiting ES patients.
Homozygous CDKN2A deletion (9p21 deletion)
CDKN2A gene encodes 2 proteins: p16 (INK4), which is a cyclin-dependent kinase inhibitor, and p14 (ARF), which binds the p53-stabilizing protein MDM2 [142]. Wildtype p16 (INK4) arrests normal diploid cells in late G1 (p16-dependent cell cycle arrest is lost in cells lacking functional RB protein) [143, 144]. Promoting cell cycle arrest, p16 (INK4) interacts with CDK4 and inhibits its kinase activity [145]. Under these circumstances, CDK4 protein cannot phosphorylate RB protein, and this maintains cell cycle repression (RB mediated) [146].
Due to CDKN2A homozygous deletion in ES, the cell cycle is dysregulated and CDK4/CDK6 inhibitors could be useful targeted therapies, because they can mimic CDKN2A function at the cell cycle level. Marco Perez et al. investigated this in sarcomas (not exclusively ES) and their work supported the efficacy of CDK4 inhibitors against sarcomas displaying increased CDK4 levels, particularly in fibrosarcomas and MPNST (malignant peripheral nerve sheath tumor). High levels of p16 (INK4) indicated poor efficacy of CDK4 inhibitors [147]. This approximation hit depends on RB1 normal function. Unfortunately, RB1 is lost in around 10% ES tumors [48]. Moreover, Hu et al. described in ES cell cultures, that only hypophosphorylated forms of pRband p130 are detectable after knockdown of EWS/FLI1, thus indicating activation of the pRB pathway in EWSR1/FLI1-positive ES tumors [148]. Schwentner et al. hypothesized that EWSR1/FLI1 mediates cell cycle activation due to the replacement of E2F3/pRB by constitutively expressed repressive E2F4/p130 [149]. Summarizing, cell cycle oncogenic activation because of RB1 mutations and/or EWSR1/FLI1 effects could be a mechanism of CDK4/6 inhibitors resistance in ES.
Furthermore, IGF1R has been proposed as a gene whose overexpression promotes drug escape to CDK4/6 inhibitors in ES tumors. Guenther et al. found elevated levels of fosfo-IGF1R in resistant cells. They suggest that dual targeting of CDK4/6 and IGF1R provides a candidate synergistic drug combination in ES [150]. Murakami et al. evaluated the efficacy of inhibitors of CDK4/6 and IGF1R inhibitors on ES patient-derived orthotopic xenograft mouse. Results were hopeful in that personalized study, but the model came from a FUS/ERG-positive tumor, and therefore not EWSR1/FLI1, as it is commonly found [151].
Nowadays, some trials with CDK4/6 inhibitors in ES are in course trying to reach conclusions. NCI-COG Pediatric MATCH study has two clinical trials using palbociclib in ES. Besides, abemaciclib has one clinical trial registered in ES (NCT02644460). In spite of that, many questions are unsolved: would certain levels of CDK4, p16 (INK4) and wildtype pRB be predictive of a response to CDK4/6 inhibitors? Could association of CDK4/6 inhibitors and EWSR1/FLI1 targeted therapies be a therapeutic option? Will CDK4/6 inhibitors and IGF1R inhibitors association open new target therapies in ES? Can EWSR1/FLI1-negative ES tumors respond better to cyclin inhibitors?
Actionable mutated genes in ES
ES family tumors have few somatic mutations, but this low rate of mutations only exceptionally affects actionable genes [152]. Therefore, few pathogenic variants have been described in BRCA2 [20], KRAS (G13N), MET (T1010I and N375S), PIK3CA [153] or MLL2 [57]. Moreover, EZH2 is mutated in around 2.5% of ES tumors [52]. Exceptionally, mutations in BRAF (BRAF V600E) are present in ES [154]. Additional difficulties are detected when targeted drugs as vemurafenib (BRAF V600E inhibitor) do not achieve the same effect as in other tumors (melanoma, low-grade gliomas, etc.). Gouravan et al. studied BRAF V600E mutated ES cell lines, and detected reduced phosphorylation of ERK during treatment with vemurafenib, suggesting that the MAPK pathway is active under this circumstances. This could suggest a bypass pathway to be targeted in association with BRAF inhibitors [154].
Although ES has a low rate of actionable mutations, a small number of patients could benefit from this approximation. Therefore, next-generation sequencing should be a part of personalized focuses.
Tyrosine kinase receptors (c-KIT, PDGFR)
c-KIT and PDGFR are two class III receptor tyrosine kinases [155]. Scotlandi K et al. established that approximately 30% of patients expressed KIT in their primary ES tumor [156]. After that, Bozzi et al. provided evidence of constitutive KIT, PDGFRA, and PDGFRB expression/activation as a part of an autocrine/paracrine loop in ES (not activating mutations in tyrosine kinase domain of KIT, PDGFRA, and PDGFRB were detected) [157]. Both works propose imatinib (PDGFRA and KIT inhibitor) as a potential effective drug in this situation. Scotlandi et al. described that the drug acts additively with doxorubicin, inhibiting ES cell growth. Bozzi et al. combined imatinib with cisplatin in two chordoma patients and described a tumor response [157]. Yerushlami et al. studied that the effect of a combination of imatinib and cisplatin which produced a dose-dependent antiproliferative effect in ES cell lines, but there was no evidence of apoptosis [158].
On the basis of this preclinical information, imatinib has been investigated in ES clinical trials. Chao J et al. selected patients for tumor immunohistochemical expression equal or higher than 2 + /4 + . Only one patient with 3 + /4 + PDGFRA and 3 + /4 + KIT expression had a partial response. Other patients progressed under imatinib treatment [159]. Other concluded trials with imatinib in ES have not published their results yet. Imatinib–cisplatin and/or doxorubicin combinations have been scarcely investigated until today. Currently, clinical trials with regorafenib in monotherapy (NCT02048371) or combined with vincristine and irinotecan (NCT02085148) are ongoing. No other bimodal KIT and PDGFRA inhibitors (imatinib or pazopanib) are in trials.
VEGF/VEGFR(vascular endothelial growth factor and its receptor)
VEGF is highly expressed in ES tumors [160]. Endothelial development is mediated by VEGF-165 isoform [161]. Zhou et al. blocked VEGF receptor 2 (VEGFR-2) with antibodies and this suppressed tumor growth. This reduced tumor vessel formation in mice. The same group, based on previous knowledge about IGF1 function in VEGF-mediated angiogenesis, investigated a VEGFR2 inhibitor and a IGF1R inhibitor in mice with hopeful results [162, 163].
Cediranib (a pan-vascular endothelial growth factor receptor inhibitor) was used in 16 pediatric patients with solid tumors and one of three ES patients had a significant partial response [164]. Clinical trial NCT00516295 (with topotecan, vincristine, cyclophosphamide and bevacizumab) did not complete feasibility assessment. Multi-tyrosine kinase inhibitors (including VEGFR inhibitor) sorafenib and pazopanib have been tried and those trials were concluded. At present, regorafenib trials are ongoing (NCT02085148, NCT02048371, NCT02389244) (Table 2).
Based on adult tumors knowledge, some VEGF polymorphisms, VEGFR1 levels and VEGFR somatic mutations can be promising biomarkers for drug response in therapies targeting VEGF/VEGFR signaling [102].
Other interesting focuses
Winter et al. have published responses to tozasertib in ES cell lines. The combined inhibition of Aurora kinases A and B underlies its effect. Synergism with etoposide has also been proposed [165]. Alisertib did not complete phase I in ES patients [166].
STAT3 is a transcription factor that is activated by JAK proteins in the cytosol and upregulated in ES. It could be involved in sarcoma cell migration and invasion. Targeting it with a JAK-1/2 inhibitor, ruxolitinib might be an interesting investigation way in ES [167].
AKT phosphorylation level could serve as a biomarker of EGFR activity in ES and then it might be a biomarker of response to EGFR inhibitors. At the moment, this remains an untested hypothesis [168].
SIRT1 inhibitors are also under development. SIRT1 is overexpressed in ES tumors, participating in NOTCH pathway deregulation. ES patients may benefit from this approximation [169].
Targeting mutant p53 is a common goal for the scientist community. At the moment, APR-246 (PRIMA-1) is the most interesting drug. ES might benefit from clinical trials using this drug, but no data are available.
DKK2-SDF1α-CXCR4-Rac1 and ERBB4-PI3K-Akt-FAK-Rac1 pathways are potential drivers of metastasis in ES under exploration [170].
Immunotherapy
Targeted immunotherapy is in continuous development, and advances in this field must be taken into account when therapeutic decisions are being taken. Nevertheless, few biological markers can differentiate between potential responders and non-responders. About checkpoint inhibitors, phase I (nivolumab ± ipilimumab) studies in pediatric patients with ES have been completed. If PD1/PDL1 is a correct response marker, it is still under consideration [171]. Until more information is available, personalized programmes can use this immunohistochemical method to select immunotherapy candidate patients.
Discussion
Isolated advances in pediatric ES treatment arrived to clinic centers during the past 3 decades. Chemotherapy, radiotherapy and surgery are the main strategies to treat these patients [172]. Many recurrent or refractory ES patients are referred to personalized medicine programmes, in which therapeutic alternatives are studied and finally proposed. For this purpose, some biological data should be cautiously considered as potential biologic prognosis markers in ES (1q gain, 16q loss or STAG2 mutations). Moreover, some point mutations in specific genes (as in EZH2), CNVs at specific chromosomal locus (as 9p21), information from immunochemistry staining and expression level of certain proteins could be used to decide the most appropriate compassionate use therapy or the theoretically more convenient clinical trial for each individual patient.
What we have gathered in this work constitutes the nucleus of knowledge on which medical decisions should rest in front of an ES patient in relapse. First, looking for clinical trials must be the first effort of every medical doctor when a patient relapses and no other conventional therapies are available. Second, in a personalized way, biological studies must be conducted to enrich knowledge about the individual disease. These efforts, already developed in reference centers, must keep in mind the urgency of translational investigation and consideration of clinical and preclinical recent advances. Sadly, due to the limited number of biological markers to predict responses taking decisions from pediatric personalized medicine committees, good responses are very difficult. Thus, we consider that carrying out molecular studies in patients is necessary, at the best by massive analysis technologies (NGS), to find new potentially actionable molecular targets. Otherwise, we should search for potential response markers and obtain relevant conclusions, so that all these efforts do not become useless.
Through this review, we would also like to highlight that, for personalized medicine, it is important to squeeze positive responses happened in several clinical trials of small groups of patients. Likewise, many of the mentioned drugs are actively being used in clinical trials with adult patients, without more biological basis than in children. Therefore, we support pediatric patients should be more actively considered for clinical trials, regarding benefits of new targeted therapies. Indeed, running a certain degree of risk is the only option for many pediatric or young patients that do not have any other treatment options and a fatal outcome is expected.
Many of the approximations summarized in this review can only be used under compassionate use treatments and without enough molecular basis to choose the best option. Nevertheless, when clinical trials are not existing or recruiting and some biological markers suggest an alternative drug option, medical doctors should pursue target therapies in combination with conventional treatments, using preclinical data and recommendations of specialized precision medicine committees. Finally, as Vornicova et al. states, considering multimodal approximations is the unique way to get better results [173].
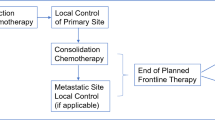
From our group and based on previous knowledge reported in this review, a proposal workflow for personalized medicine projects has been elaborated and is presented here (Fig. 1).

Ewing sarcoma proposal workflow within personalized medicine projects
References
Choi EY, Gardner JM, Lucas DR, McHugh JB, Patel RM. Ewing sarcoma. Semin Diagn Pathol. 2014;31(1):39–47.
Fletcher C, Bridge J, Hogendoorn P, Mertens F. Classification of tumours pathology and genetics of tumours of soft tissue and bone. In World Health Organization, 4thEd (Lyon: IARC Press); 2013: 306–309.
Savita S, Stephen L. Promiscuous Partnerships in Ewing’s Sarcoma. Cancer Genet. 2011;204(7):351–65.
Horowitz M, Malawer M, Woo S, et al. Ewing's sarcoma family of tumors: Ewing's sarcoma of bone and soft tissue and the peripheral primitive neuroectodermal tumors. Pizzo, PA.; Poplack, DG. (eds) Principles and practice of pediatric oncology. Philadelphia: Lippincott-Raven Publishers; 1997. p. 831–863.
Kimber C, Michalski A, Spitz L, Pierro A. Primitive neuroectodermal tumours: anatomic location, extentof surgery, and outcome. J Pediatr Surg. 1998;33:39–41.
Gaspar N, Hawkins DS, Dirksen U, Lewis IJ, Ferrari S, Le Deley MC, et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol. 2015;33(27):3036–46.
Gorlick R, Janeway K, Lessnick S, Randall RL, Marina N, Committee COGBT. Children’s Oncology Group’s 2013 blueprint for research: bone tumors. Pediatr Blood Cancer. 2013;60:1009–155.
Hancock JD, Lessnick SL. A transcriptional profiling meta-analysis reveals a core EWS-FLI gene expression signature. Cell Cycle. 2008;7:250–6.
Sankar S, Bell R, Stephens B, Zhuo R, Sharma S, Bearss DJ, et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 2013;32:5089–100.
Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu Rev Pathol. 2012;7:145–59.
Takigami I, Ohno T, Kitade Y, Hara A, Nagano A, Kawai G, et al. Synthetic siRNA targeting the breakpoint of EWS/Fli-1 inhibits growth of Ewing sarcoma xenografts in a mouse model. Int J Cancer. 2011;128:216–26.
Maksimenko A, Malvy C. Oncogene-targeted antisense oligonucleotides for the treatment of Ewing sarcoma. Expert Opin Ther Targets. 2005;9:825–30.
Mateo-Lozano S, Gokhale PC, Soldatenkov VA, Dritschilo A, Tirado OM, Notario V. Combined transcriptional and translational targeting of EWS/FLI-1 in Ewing's sarcoma. Clin Cancer Res. 2006;12:6781–90.
Stoll G, Surdez D, Tirode F, Laud K, Barillot E, Zinovyev A, et al. Systems biology of Ewing sarcoma: a network model of EWS-FLI1 effect on proliferation and apoptosis. Nucleic Acids Res. 2013;41(19):8853–71.
Riggi N, Knoechel B, Shawn M, Rheinbay E, Boulay G, Suvà M, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell. 2014;26(5):668–81.
Hattinger CM, Pötschger U, Tarkkanen M, Squire J, Zielenska M, Kiuru-Kuhlefelt S, et al. Prognostic impact of chromosomal aberrations in Ewing tumours. Br J Cancer. 2002;86:1763–9.
Mackintosh C, Ordonez JL, Garcia-Dominguez DJ, Sevillano V, Llombart-Bosch A, Szuhai K, et al. 1q gain and CDT2 overexpression underlie an aggressive and highly proliferative form of Ewing sarcoma. Oncogene. 2012;31:1287–98.
Roberts P, Burchill SA, Brownhill S, Cullinane CJ, Johnston C, Griffiths MJ, et al. Ploidy and karyotype complexity are powerful prognostic indicators in the Ewingʼs sarcoma family of tumors: A study by the united kingdom cancer cytogenetics and the childrenʼs cancer and leukaemia group. Genes Chromosomes Cancer. 2008;47:207–20.
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73.
Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10(7):e1004475.
Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314:913–25.
Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker E, et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the Individualized Cancer Therapy (iCat) study. JAMA Oncol. 2016;2:608–15.
Oberg JA, Glade Bender JL, Sulis ML, Pendrick D, Sireci AN, Hsiao SJ, et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med. 2016;8:133.
Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;2:616–24.
Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag A, et al. Next-generation personalised medicine for high-risk paediatric cancer patients the INFORM pilot study. Eur J Cancer. 2016;65:91–101.
Harttrampf AC, Lacroix L, Deloger M, Deschamps F, Puget S, Auger N, et al. MOlecular Screening for CAncerTreatment Optimization (MOSCATO-01) in pediatric patients: a single institutional prospective molecular stratification trial. Clin Cancer Res. 2017;23:6101–12.
Pincez T, Clement N, Lapouble E, Pierron G, Kamal M, Bieche I, et al. Feasibility and clinical integration of molecular profiling for target identification in pediatric solid tumors. Pediatr Blood Cancer. 2017;64:e26365.
Glade Bender J, Verma A, Schiffman JD. Translating genomic discoveries to the clinic in pediatric oncology. Curr Opin Pediatr. 2015;27(1):34–433.
Cotterill SJ, Ahrens S, Paulussen M, Jürgens HF, Voûte PA, Gadner H, et al. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol. 2000;18(17):3108–14.
Rodríguez-Galindo C, Navid F, Liu T, Billups CA, Rao BN, Krasin MJ. Prognostic factors for local and distant control in Ewing sarcoma family of tumors. Ann Oncol. 2008;19(4):814–20.
Sauer R, Jurgens H, Burgers JM, Dunst J, Hawlicek R, Michaelis J. Prognostic factors in the treatment of Ewing’ssarcoma The Ewing’s Sarcoma Study Group of the German Society of Paediatric Oncology CESS 81. Radiother Oncol. 1987;10:101–10.
Lee J, Hoang BH, Ziogas A, Zell JA. Analysis of prognostic factors in Ewing sarcoma using a population based cancer registry. Cancer. 2010;116:1964–73.
Cash T, McIlvaine E, Krailo MD, Lessnick SL, Lawlor ER, Laack N, et al. Comparison of clinical features and outcomes in patients with extraskeletal versus skeletal localized Ewing sarcoma: a report from the children’s oncology group. Pediatr Blood Cancer. 2016;63(10):1771–9.
Shankar AG, Ashley S, Craft AW, Pinkerton CR. Outcome after relapse in an unselected cohort of children and adolescents with Ewing sarcoma. Med Pediatr Oncol. 2003;40:141–7.
Barker LM, Pendergrass TW, Sanders JE, Hawkins DS. Survival after recurrence of Ewing’s sarcoma. J Clin Oncol. 2005;23:4354–62.
De Alava E, Kawai A, Healey JH, Fligman I, Meyers PA, Huvos AG, et al. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol. 1998;16:1248–55.
Zoubek A, Dockhorn-Dworniczak B, Delattre O, Christiansen H, Niggli F, Gatterer-Menz I, et al. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol. 1996;14:1245–51.
Le Deley MC, Delattre O, Schaefer KL, Burchill SA, Koehler G, Hogendoorn PC, et al. Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro- EWING 99 trial. J Clin Oncol. 2010;28:1982–8.
Barr FG, Meyer WH. Role of fusion subtype in Ewing sarcoma. J Clin Oncol. 2010;28:1973–4.
Van Doorninck JA, Ji L, Schaub B, Shimada H, Wing MR, Krailo MD, et al. Current treatment protocol shave eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2010;28:1989–94.
Savola S, Klami A, Tripathi A, Niini T, Serra M, Picci P, et al. Combined use of expression and cgh arrays pin points novel candidate genes in Ewing sarcoma family of tumors. BMC Cancer. 2009;9:17.
Brisset S, Schleiermacher G, Peter M, Mairal A, Oberlin O, Delattre O, et al. CGH analysis of secondary genetic changes in Ewing tumors: correlation with metastatic disease in a series of 43 cases. Cancer Genet Cytogenet. 2001;130(1):57–61.
Sannino G, Orth M, Grünewald T. Next steps in Ewing sarcoma (epi-) Genomics. Future Oncol. 2017;13(14):1207–11.
Kovar H, Jug G, Aryee DN, Zoubek A, Ambros P, Gruber B, et al. Among genes involved in the RB dependent cell cycle regulatory cascade, the p16 tumor suppressor gene is fre quently lost in the Ewing family of tumors. Oncogene. 1997;15:2225–32.
Brownhill SC, Taylor C, Burchill SA. Chromosome 9p21 gene copy number and prognostic significance of p16 in ESFT. Br J Cancer. 2007;96:1914–23.
Wei G, Antonescu CR, De Alava E, Leung D, Huvos AG, Meyers PA, et al. Prognostic impact of INK4A deletion in Ewing sarcoma. Cancer. 2000;89:793–9.
Tsuchiya T, Sekine K, Hinohara S, Namiki T, Nobori T, Kaneko Y. Analysis of the p16 INK4, p14 ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet Cytogenet. 2000;120:91–8.
Maitra A, Roberts H, Weinberg AG, Geradts J. Aberrant expression of tumor suppressor proteins in the Ewing family of tumors. Arch Pathol Lab Med. 2001;125:1207–12.
Honoki K, Stojanovski E, Mcevoy M, Fujii H, Tsujiuchi T, Kido A, et al. Prognostic significance of p16 INK4 alteration for Ewing sarcoma:a meta-analysis. Cancer. 2007;110:1351–60.
Shukla N, Schiffman JD, Reed D, Davis IJ, Womer RB, Lessnick SL, et al. Biomarkers in Ewing sarcoma: the promise and challenge of personalized medicine A report from the Children’s Oncology Group. Front Oncol. 2013;3:141.
Lerman DM, Monument MJ, McIlvaine E, Liu X, Huang ML, et al. Tumoral TP53 and/or CDKN2A alterations are not reliable prognostic biomarkers in patients with localized Ewing sarcoma: a report from the children’s oncology group. Pediatr Blood Cancer. 2015;62(5):759–65.
Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of stag2 and tp53 mutations. Cancer Discov. 2014;4:1342–53.
Abudu A, Mangham DC, Reynolds GM, Pynsent PB, Tillman RM, Carter SR, et al. Overexpression of p53 protein in primary Ewing’s sarcoma of bone: relationship to tumour stage, response and prognosis. Br J Cancer. 1999;79:1185–9.
de Alava E, Antonescu CR, Panizo A, Leung D, Meyers PA, Huvos AG, et al. Prognostic impact of P53 status in Ewing sarcoma. Cancer. 2000;89:783–92.
Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH, et al. Ewing sarcomas with p53 mutation or p16/p14 ARF homozygous deletion: a highly lethal subset associated with poor chemo response. J Clin Oncol. 2005;23:548–58.
Lopez-Guerrero JA, Machado I, Scotlandi K, Noguera R, Pellin A, Navarro S, et al. Clinicopathological significance of cell cycle regulation markers in a large series of genetically confirmed Ewing’s sarcoma family of tumors. Int J Cancer. 2011;128:1139–50.
Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326–41.
Alholle A, Brini AT, Gharanei S, Vaiyapuri S, Arrigoni E, Dallo A, et al. Functional epigenetic approach identifies frequently methylated genes in Ewing sarcoma. Epigenetics. 2013;8(11):1198–204.
Gharanei S, Brini AT, Vaiyapuri S, Alholle A, Dallo A, Arrigoni E, et al. RASSF2 methylation is a strong prognostic markerin younger age patients with Ewing sarcoma. Epigenetics. 2013;8(9):893–8.
Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J. Biol Chem. 2003;278:28045–51.
Donninger H, Hesson L, Vos M, Beebe K, Gordon L, Sidransky D, et al. The Ras effector RASSF2 controls the PAR-4 tumor suppressor. Molec Cell Biol. 2010;30:2608–20.
Sand LG, Berghuis D, Szuhai K, Hogendoorn PC. Expression of CCL21 in Ewing sarcoma shows an inverse correlation with metastases and is a candidate target for immunotherapy. Cancer Immunol Immunother. 2016;65:995–1002.
Kim JR, Moon YJ, Kwon KS, Bae JS, Wagle S, Kim KM, et al. Tumor infiltrating PD1-positive lymphocytes and the expression of PD-L1 predict poor prognosis of soft tissue sarcomas. PLoS ONE. 2013;8(12):e82870.
Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992;359:162–5.
May WA, Gishizky ML, Lessnick SL, Lunsford LB, Lewis BC, Delattre O, et al. Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proc Natl Acad Sci. 1993;90:5752–6.
Castillero-Trejo Y, Eliazer S, Xiang L, Richardson JA, Ilaria RL. Expression of the EWS/FLI-1 oncogene in murine primary bone-derived cells Results in EWS/FLI-1 dependent, Ewing sarcoma-like tumors. Cancer Res. 2005;65:8698–705.
Riggi N, Cironi L, Provero P, Suva ML, Kaloulis K, Garcia-Echeverria C, et al. Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Res. 2005;65:11459–68.
Tanaka K, Iwakuma T, Harimaya K, Sato H, Iwamoto Y. EWS-Fli1 antisense oligodeoxynucleotide inhibits proliferation of human Ewing’s sarcoma and primitive neuroectodermal tumor cells. J Clin Invest. 1997;99:239–47.
Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted, nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005;65:8984–92.
Herrero-Martín D, Osuna D, Ordóñez JL, Sevillano V, Martins AS, Mackintosh C, et al. Stable interference of EWS–FLI1 in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signalling and reveals TOPK as a new target. Br J Cancer. 2009;101:80–90.
Bilke S, Schwentner R, Yang F, Kauer M, Jug G, Walker RL, et al. Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res. 2013;23:1797–809.
Gangwal K, Close D, Enriquez CA, Hill CP, Lessnick SL. Emergent properties of EWS/FLI regulation via GGAA microsatellites in Ewing's sarcoma. Genes Cancer. 2010;1:177–87.
Gangwal K, Sankar S, Hollenhorst PC, Kinsey M, Haroldsen SC, Shah AA, et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proc Natl Acad Sci USA. 2008;105:10149–54.
Guillon N, Tirode F, Boeva V, Zynovyev A, Barillot E, Delattre O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS ONE. 2009;4:e4932.
Patel M, Simon JM, Iglesia MD, Wu SB, McFadden AW, Lieb JD, Davis IJ. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res. 2012;22:259–70.
Toretsky JA, Erkizan V, Levenson A, Abaan OD, Parvin JD, Cripe TP, et al. Oncoprotein EWS-FLI1 Activity Is Enhanced by RNA Helicase A. Cancer Res. 2006;66(11):5574–81.
Hong SH, Youbi SE, Hong SP, Kallakury B, Monroe P, Erkizan HV, et al. Pharmacokinetic modeling optimizes inhibition of the 'undruggable' EWS-FLI1 transcription factor in Ewing Sarcoma. Oncotarget. 2014;5(2):338–50.
Selvanathan SP, Graham GT, Erkizan HV, Dirksen U, Natarajan TG, Dakic A, et al. Oncogenic fusion protein EWS-FLI1 is a network hub that regulates alternative splicing. Proc Natl Acad Sci USA. 2015;112(11):E1307–E13161316.
Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nat Med. 2009;15(7):750–6.
Lamhamedi-Cherradi SE, Menegaz BA, Ramamoorthy V, Aiyer RA, Maywald RL, Buford AS, et al. An oral formulation of YK-4-279: preclinical efficacy and acquired resistance patterns in Ewing sarcoma. Mol Cancer Ther. 2015;14(7):1591–604.
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298:1039–43.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nature Genet. 2010;42:722–6.
Richter GH, Plehm S, Fasan A, Rössler S, Unland R, Bennani-Baiti IM, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endotelial and neuro-ectodermal differentiation. Proc Natl Acad Sci USA. 2009;106(13):5324–9.
Ciarapica R, Miele L, Giordano A, Locatelli F. Rota R Enhancer of zeste homolog 2 (EZH2) in pediatric soft tissue sarcomas: first implications. BMC Med. 2011;9:63.
Pandya PH, Bailey B, Elmi AE, Bates HB, Hemenway CN, Sinn AL, et al. Preclinical validation of EZH2 as a therapeutic target in pediatric Ewing's sarcoma [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2018; Cancer Res. 2018; 78(13 Suppl): Abstract nr 3180.
Ramaglia M, D'Angelo V, Iannotta A, Di Pinto D, Pota E, Affinita MC, et al. High EZH2 expression is correlated to metastatic disease in pediatric soft tissue sarcomas. Cancer Cell Int. 2016;16:59.
Maruyama T, Farina A, Dey A, Cheong J, Bermudez VP, Tamura T, et al. A Mammalian bromodomain protein, brd4, interacts with replication factor C and inhibits progression to S phase. Mol Cell Biol. 2002;22:6509–20.
Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLLfusion leukaemia. Nature. 2011;478:529–33.
Hensel T, Giorgi C, Schmidt O, Calzada-Wack J, Neff F, Buch T, et al. Targeting the EWS-ETS transcriptional program by BET. Oncotarget. 2016;7(2):1451–63.
Shao GB, Chen JC, Zhang LP, Huang P, Lu HY, Jin J, et al. Dynamic patterns of histone H3 lysine 4 methyltransferases and demethylases during mouse preimplantation development. In Vitro Cell Dev Biol Anim. 2014;50:603–13.
Schildhaus HU, Riegel R, Hartmann W, Steiner S, Wardelmann E, Merkelbach-Bruse S, et al. Lysine-specific demethylase 1 is highly expressed in solitary fibrous tumors, synovial sarcomas, rhabdomyosarcomas, desmoplastic small round cell tumors, and malignant peripheral nerve sheath tumors. Hum Pathol. 2011;42(11):1667–755.
Bennani-Baiti IM, Machado I, Llombart-Bosch A, Kovar H. Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing's sarcoma, osteosarcoma, and rhabdomyosarcoma. Hum Pathol. 2012;43(8):1300–7.
Sankar S, Theisen ER, Bearss J, Mulvihill T, Hoffman LM, Sorna V, et al. Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth. Clin Cancer Res. 2014;20(17):4584–97.
Theisen ER, Pishas KI, Saund RS, Lessnick SL. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget. 2016;7(14):17616–30.
Smith R, Owen LA, Trem DJ, Wong JS, Whangbo JS, Golub TR, et al. Expression profiling of EWS/FLI identifies NKX22 as a critical target gene in Ewing’s sarcoma. Cancer Cell. 2006;9(5):405–16.
Owen LA, Kowalewski AA, Lessnick SL. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing’s sarcoma. PLoS One.1965; 3: 1965.
Sampson VB, Vetter NS, Kamara DF, Collier AB, Gresh RC, Kolb EA. Vorinostat enhances cytotoxicity of SN-38 and temozolomide in Ewing sarcoma cells and activates STAT3/AKT/MAPK pathways. PLoS ONE. 2015;10(11):e0142704.
Kennedy AL, Vallurupalli M, Chen L, Crompton B, Cowley G, Vazquez F, et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget. 2015;6(30):30178–93.
Surdez D, Benetkiewicz M, Perrin V, Han ZY, Pierron G, Ballet S, et al. Targeting the EWSR1-FLI1 oncogene-induced protein kinase PKC-beta abolishes Ewing sarcoma growth. Can Res. 2012;72:4494–503.
Takahashi A, Higashino F, Aoyagi M, Yoshida K, Itoh M, Kyo S, et al. EWS/ ETS fusions activate telomerase in Ewing’s tumors. Cancer Res. 2003;63(23):8338–444.
Ambatia SR, Lopes EC, Kosugi K, Mony U, Zehir A, Shah SK, et al. Moore Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol Oncol. 2014;8(2):323–36.
Yu H, Ge Y, Guo L, Huang L. Potential approaches to the treatment of Ewing’s sarcoma. Oncotarget. 2017;8(3):5523–39.
Yang L, Hu HM, Zielinska-Kwiatkowska A, Chansky HA. FOXO1 is a direct target of EWS-Fli1 oncogenic fusion protein in Ewing’s sarcoma cells. Biochem Biophys Res Commun. 2010;402:129–34.
Niedan S, Kauer M, Aryee DN, Kofler R, Schwentner R, Meier A, et al. Suppression of FOXO1 is responsible for a growth regulatory repressive transcriptional sub-signature of EWS-FLI1 in Ewing sarcoma. Oncogene. 2014;33:3927–38.
Pommier Y, Kohlhagen G, Bailly C, Waring M, Mazumder A, Kohn KW. DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry. 1996;35:13303–9.
Grohar PJ, Griffin LB, Yeung C, Chen QR, Pommier Y, Khanna C, et al. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia. 2011;13:145–53.
Amaral AT, Garofalo C, Frapolli R, Manara MC, Mancarella C, Uboldi S, et al. Trabectedin efficacy in Ewing sarcoma is greatly increased by combination with Anti-IGF signaling agents. Clin Cancer Res. 2015;21(6):1373–82.
Harlow ML, Maloney N, Roland J, Guillen Navarro MJ, Easton MK, Kitchen-Goosen SM, et al. Lurbinectedin inactivates the Ewing sarcoma oncoprotein EWS-FLI1 by redistributing it within the nucleus. Cancer Res. 2016;76(22):6657–68.
Grohar PJ, Glod J, Peer CJ, Sissung TM, Arnaldez FI, Long L, et al. A phase I/II trial and pharmacokineticstudy of mithramycin in children and adults with refractory Ewing sarcoma and EWS-FLI1 fusion transcript. Cancer Chemother Pharmacol. 2017;80(3):645–52.
Osgood CL, Maloney N, Kidd CG, Kitchen-Goosen S, Segars L, Gebregiorgis M. Identification of mithramycin analogs with improved targeting of the EWS-FLI1 transcription factor. Clin Cancer Res. 2016;22(16):4105–18.
Radic-Sarikas B, Tsafou KP, Emdal KB, Papamarkou T, Huber KV, Mutz C, et al. Combinatorial drug screening identifies Ewing sarcoma–specific sensitivities. Mol Cancer Ther. 2017;16(1):88–101.
Scotlandi K, Benini S, Sarti M, Serra M, Lollini PL, Maurici D, et al. Insulin-like growth factor I receptor-mediated circuit in Ewing's sarcoma peripheral neuroectodermal tumor: a possible therapeutic target. Cancer Res. 1996;56(20):4570–4.
Scotlandi K, Benini S, Nanni P, Lollini PL, Nicoletti G, Landuzzi L, et al. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing's sarcoma in athymic mice. Cancer Res. 1998;58(18):4127–31.
Toretsky JA, Kalebic T, Blakesley V, LeRoith D, Helman LJ. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J Biol Chem. 1997;272(49):30822–7.
Kang HG, Jenabi JM, Liu XF, Reynolds CP, Triche TJ, Sorensen PH. Inhibition of the insulin-like growth factor i receptor by epigallocatechin gallate blocks proliferation and induces the death of Ewing tumor cells. Mol Cancer Ther. 2010;9(5):1396–407.
Kolb EA, Gorlick R, Lock R, Carol H, Morton CL, Keir ST, et al. Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2011;56(4):595–603.
Houghton PJ, Morton CL, Gorlick R, Kolb EA, Keir ST, Reynolds CP, et al. Initial testing of a monoclonal antibody (IMC-A12) against IGF-1R by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2010;54(7):921–6.
Manara MC, Landuzzi L, Nanni P, Nicoletti G, Zambelli D. Lollini PL preclinical invivo studyof new insulin-like growth factor-I receptor specific inhibitor in Ewing’s sarcoma. Clin Cancer Res. 2007;13(4):1322–30.
Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. 2014;120(16):2448–566.
Malempati S, Weigel B, Ingle AM, Ahern CH, Carroll JM, Roberts CT, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol. 2012;30(3):256–62.
Juergens H, Daw NC, Geoerger B, Ferrari S, Villarroel M, Aerts I, et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29(34):4534–40.
Arnaldez FI, Helman LJ. New strategies in ewing sarcoma: lost in translation? Clin Cancer Res. 2014;20(12):3050–6.
Subbiah V, Naing A, Brown RE, Chen H, Doyle L, LoRusso P. Targeted morphoproteomic profiling of Ewing's sarcoma treated with insulin-like growth factor 1 receptor (IGF1R) Inhibitors: response/resistance signatures. PLoS ONE. 2011;6(4):e18424.
Garofalo C, Mancarella C, Grilli A, Manara MC, Astolfi A, Marino MT, et al. Identification of common and distinctive mechanisms of resistance to different anti-IGF-IR agents in Ewing's sarcoma. Mol Endocrinol. 2012;26:1603–16.
Naing A, LoRusso P, Fu S, Hong DS, Anderson P, Benjamin RS, et al. Insulin growth factor receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res. 2012;18:2625–31.
Macaulay VM, Middleton MR, Eckhardt SG, Juergens RA, Stephens AW, Poondru S, McCarthy SP, Gadgeel SM. Phase I study of OSI-906, dual tyrosine kinase inhibitor of insulinlike growth factor-1 receptor (IGF-1R) and insulin receptor (IR) in combination with erlotinib (E) in patients with advanced solid tumors. J Clin Oncol. 2011;29:3098.
Kolb EA, et al. Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2011;56:595–603.
Martins AS, Mackintosh C, Martín DH, Campos M, Hernández T, Ordóñez JL, et al. Insulin-like growth factor I receptor pathway inhibition by ADW742, alone or in combination with imatinib, doxorubicin, or vincristine, is a novel therapeutic approach in Ewing tumor. Clin Cancer Res. 2006;12:3532–40.
Soldatenkov VA, Trofimova IN, Rouzaut A, McDermott F, Dritschilo A, Notario V. Differential regulation of the response to DNA damage in Ewing's sarcoma cells by ETS1 and EWS/FLI-1. Oncogene. 2002;21:2890–5.
Prasad SC, Thraves PJ, Bhatia KG, Smulson ME, Dritschilo A. Enhanced poly (adenosine diphosphate ribose) polymerase activity and gene expression in Ewing's sarcoma cells. Cancer Res. 1990;50:38–433.
Gill SJ, Travers J, Pshenichnaya I, Kogera FA, Barthorpe S, Mironenko T, et al. Combinations of PARP inhibitors with temozolomide drive PARP1 trapping and apoptosis in Ewing's sarcoma. PLoS ONE. 2015;10(10):e0140988.
Stewart E, Goshorn R, Bradley C, Griffiths LM, Benavente C, Twarog NR, et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep. 2014;9(3):829–41.
Norris RE, Adamson PC, Nguyen VT, Fox E. Preclinical evaluation of the PARP inhibitor, olaparib, in combination with cytotoxic chemotherapy in pediatric solid tumors. Pediatr Blood Cancer. 2014;61(1):145–60.
Engert F, Schneider C, Weiβ LM, Probst M, Fulda S. PARP inhibitors sensitize Ewing sarcoma cells to Temozolomide-induced apoptosis via the mitocondrial pathway. Mol Cancer Ther. 2015;14(12):2818–30.
Smith MA, Reynolds CP, Kang MH, Kolb EA, Gorlick R, Carol H, et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin Cancer Res. 2015;21(4):819–32.
Choy E, Butrynski JE, Harmon DC, Morgan JA, George S, Wagner AJ, et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer. 2014;14:813.
Ordóñez JL, Amaral AT, Carcaboso AM, Herrero-Martín D, del Carmen G-M, Sevillano V. The PARP inhibitor olaparib enhances the sensitivity of Ewing sarcoma to trabectedin. Oncotarget. 2015;6(22):18875–90.
Lee HJ, Yoon C, Schmidt B, Park DJ, Zhang AY, Erkizan HV, et al. Combining poly(ADP-ribose) polymerase 1 (PARP-1) inhibition and radiation in Ewing sarcoma results in lethal DNA damage. Mol Cancer Ther. 2013;12(11):2591–600.
Tanaka A, Weinel S, Nagy N, O'Driscoll M, Lai-Cheong JE, Kulp-Shorten CL, et al. Germline mutation in ATR in autosomal-dominant oropharyngeal cancer syndrome. Am J Hum Genet. 2012;90:511–7.
Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene induced DNA damage model for cancer development. Science. 2008;319(5868):1352–5.
Nieto-Soler M, Morgado-Palacin I, Lafarga V, Lecona E, Murga M, Callen E. Efficacy of ATR inhibitors as single agents in Ewing sarcoma. Oncotarget. 2016;7(37):58759–67.
Robertson KD, Jones PA. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene. 1999;18:3810–20.
Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, et al. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. 1995;375:503–6.
Koh J, Enders GH, Dynlacht BD, Harlow E. Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. 1995;375:506–10.
Lin YC, Diccianni MB, Kim Y, Lin HH, Lee CH, Lin RJ, et al. Human p16-gamma, a novel transcriptional variant of p16 (INK4A), coexpresses with p16(INK4A) in cancer cells and inhibits cell-cycle progression. Oncogene. 2007;26:7017–27.
Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–69.
Perez M, Muñoz-Galván S, Jiménez-García MP, Marín JJ, Carnero A. Efficacy of CDK4 inhibition against sarcomas depends on their levels of CDK4 and p16ink4 mRNA. Oncotarget. 2015;6(38):40557–74.
Hu HM, Zielinska-Kwiatkowska A, Munro K, Wilcox J, Wu DY, Yang L, Chansky HA, et al. EWS/FLI1 suppresses retinoblastoma protein function and senescence in Ewing’s sarcoma cells. J Orthop Res. 2008;26(6):886–93.
Schwentner R, Papamarkou T, Kauer MO, Stathopoulos V, Yang F, Bilke S. EWS-FLI1 employs an E2F switch to drive target gene Expression. Nucleic Acids Res. 2015;43(5):2780–9.
Lillian M. Guenther, Neekesh V. Dharia, Linda Ross, Amy S. Conway, Amanda L. Robichaud, Alanna J. Church, RajarshiGuha, Mindy I. Davis, Gabriela Alexe, Jaume Mora, Federica Piccioni and Kimberly Stegmaier. Abstract 1629: Targeting resistance mechanisms to CDK4/6 inhibitors in Ewing sarcoma with an IGF1R inhibitor drug combination strategy. AACR Annual Meeting 2018; April 14–18, 2018; Chicago, IL.
Murakami T, Singh AS, Kiyuna T, Dry SM, Li Y, James AW, et al. Effective molecular targeting of CDK4/6 and IGF-1R in a rare FUS-ERG fusion CDKN2A-deletion doxorubicin-resistant Ewing’s sarcoma patient-derived orthotopicxenograft (PDOX) nude-mouse model. Oncotarget. 2016;7(30):47556–64.
Sand LG, Szuhai K, Hogendoorn PC. Sequencing overview of Ewing sarcoma: a journey across genomic, epigenomic and transcriptomic landscapes. Int J Mol Sci. 2015;16:16176–215.
Jiang Y, Subbiah V, Janku F, Ludwig JA, Naing A, Benjamin RS, et al. Novel secondary somatic mutations in Ewingʼs sarcoma and desmoplastic small round cell tumors. PLoS ONE. 2014;9(8):e93676.
Gouravan S, Meza-Zepeda LA, Myklebost O, Stratford EW, Munthe E. Preclinical evaluation of vemurafenib as therapy for BRAFV600E mutated sarcomas. Int J Mol Sci. 2018;19:969.
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78.
Scotlandi K, Manara MC, Strammiello R, Landuzzi L, Benini S, Perdichizzi S. C-kit receptor expression in Ewing's sarcoma: lack of prognostic value but therapeutic targeting opportunities in appropriate conditions. J ClinOncol. 2003;21(10):1952–60.
Bozzi F, Tamborini E, Negri T, Pastore E, Ferrari A, Luksch R, et al. Evidence for activation of KIT, PDGFRa, and PDGFRb receptors in the Ewing sarcoma family of tumors. Cancer. 2007;109(8):1638–45.
Yerushalmi R, Nordenberg J, Beery E, Uziel O, Lahav M, Luria D, Fenig E. Combined antiproliferative activity of imatinibmesylate (STI-571) with radiation or cisplatin in vitro. Exp Oncol. 2007;29(2):126–31.
Chao J, Budd GT, Chu P, Frankel P, Garcia D, Junqueira M, Loera S, Somlo G, Sato J, Chow WA. Phase II clinical trial of imatinibmesylate in therapy of KIT and/or PDGFRalpha-expressing Ewing sarcoma family of tumors and desmoplastic small round cell tumors. Anticancer Res. 2010;30(2):547–52.
Kumar R, Sankineani S, Rastogi S, Prakash S, Bakhshi S, Sharma MC, et al. Expression of vascular endothelial growth factor in Ewing’s sarcoma. Int Orthop. 2012;36:1669–722.
Reddy K, Cao Y, Zhou Z, Yu L, Jia SF, Kleinerman ES. VEGF165 expression in the tumor microenvironment influences the differentiation of bone marrow-derived pericytes that contribute to the Ewing’s sarcoma vasculature. Angiogenesis. 2008;11:257–67.
Zhou Z, Bolontrade MF, Reddy K, Duan X, Guan H, Yu L, et al. Suppression of Ewing’s sarcoma tumor growth, tumor vessel formation, and vasculogenesis following anti vascular endothelial growth factor receptor-2 therapy. Clin Cancer Res. 2007;13:4867–73.
Ackermann M, Morse BA, Delventhal V, Carvajal IM, Konerding MA. Anti-VEGFR2 and anti-IGF-1R-Adnectins inhibit Ewing’s sarcoma A673-xenograft growth and normalize tumor vascular architecture. Angiogenesis. 2012;15(4):685–95.
Fox E, et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J Clin Oncol. 2010;28:5174–81.
Winter GE, Rix U, Lissat A, Stukalov A, Müllner MK, Bennett KL, et al. An integrated chemical biology approach identifies specific vulnerability of Ewing's sarcoma to combined inhibition of aurora kinases A and B. Mol Cancer Ther. 2011;10(10):1846–56.
Mossé YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B, et al. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children’s Oncology Group Phase I Consortium study. Clin Cancer Res. 2012;18:6058–64.
Seethalakshmi Hariharan, Doris A. Phelps, Peter JH. Role of STAT3 in pediatric sarcoma cell lines. In: Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; 2016 Apr 16–20; New Orleans, LA. Philadelphia (PA): AACR; Cancer Res 2016; 76: 1128.
Jiang Y, Ludwig J, Janku F. Targeted therapies for advanced Ewing sarcoma family of tumors. Cancer Treat Rev. 2015;41(5):391–400.
Ban J, Aryee DN, Fourtouna A, van der Ent W, Kauer M, Niedan S, et al. Suppression of deacetylase SIRT1 mediates tumor suppressive NOTCH response and offers a novel treatment option in metastatic Ewing sarcoma. Cancer Res. 2014;74(22):6578–88.
Lawlor ER, Sorensen PH. Twenty years on what do we really know about Ewing sarcoma and what is the path forward. Crit Rev Oncog. 2015;20:155–71.
Davis KL, Fox E, Reid JM, Liu X, Minard CG, Weigel B, et al. ADVL1412: Initial results of a phase I/II study of nivolumab and ipilimumab in pediatric patients with relapsed/refractory solid tumors a COG study. J Clin Oncolournal Clin Oncol. 2017;35(15):10526–10526.
Ozaki T. Diagnosis and treatment of Ewing sarcoma of the bone: a review article. J Orthop Sci. 2015;20(2):250–63.
Vornicova O, Bar-Sela G. Investigational therapies for Ewing sarcoma: a search without a clear finding. Expert Opin Investig Drugs. 2016;25(6):679–86.
Author information
Authors and Affiliations
Contributions
All authors have participated in the research, but especially in revising it critically and all authors have approved the final article.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, formal consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Referees: Papo AS: alberto.pappo@stjude.org. Choy E: echoy@mgh.harvard.edu. Fox E: foxe@e-mail.chop.edu. All three are leaderships in translational medicine, and particularly in Ewing Sarcoma. They are from a different geographical area and we didn't meet them.
Rights and permissions
About this article

Cite this article
Gargallo, P., Juan, A., Yáñez, Y. et al. Precision medicine in Ewing sarcoma: a translational point of view. Clin Transl Oncol 22, 1440–1454 (2020). https://doi.org/10.1007/s12094-020-02298-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-020-02298-7