
Abstract
Purpose
The fusion gene BCR-ABL has an important role to the progression of chronic myeloid leukemia (CML) and several signaling pathways have been characterized as responsible for the terminal blastic phase (BP). However, the initial phase, the chronic phase (CP), is long lasting and there is much yet to be understood about the critical role of BCR-ABL in this phase. This study aims to evaluate transcriptional deregulation in CD34+ hematopoietic cells (CD34+ cells) from patients with untreated newly diagnosed CML compared with CD34+HC from healthy controls.
Methods
Gene expression profiling in CML-CD34 cells and CD34 cells from healthy controls were used for this purpose with emphasis on five main pathways important for enhanced proliferation/survival, enhanced self-renewal and block of myeloid differentiation.
Results
We found 835 genes with changed expression levels (fold change ≥ ±2) in CML-CD34 cells compared with CD34 cells. These include genes belonging to PI3K/AKT, WNT/b-catenin, SHH, NOTCH and MAPK signaling pathways. Four of these pathways converge to MYC activation. We also identified five transcripts upregulated in CD34-CML patients named OSBPL9, MEK2, p90RSK, TCF4 and FZD7 that can be potential biomarkers in CD34-CML-CP.
Conclusion
We show several mRNAs up- or downregulated in CD34-CML during the chronic phase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic myeloid leukemia (CML) originates in multipotent hematopoietic cells (HSCs) and is characterized by the presence of the Philadelphia chromosome and fusion gene, BCR–ABL. The natural course of CML has three phases: chronic phase (CP), accelerated phase (AP), and blastic phase. BCR–ABL codes a chimeric protein p21BCR–ABL with elevated tyrosine kinase activity, considered to be responsible for the disease progression [1]. The initial phase is characterized by massive expansion of the granulocytic cell lineage and mature cells in bone marrow, peripheral blood, and extramedullary sites [2]. Therefore, better understanding of the transcriptional deregulation in CD34+ hematopoietic cells (CD34-HC) in this phase will allow understanding several signaling pathways responsible for their aberrantly self-renew capacity acquisition, which leads to leukemia stem cell (LSC) generation and blastic crisis transformation.
In CML patients, gene expression profiling has been used for better characterization of BCR–ABL effects on mononuclear cells (MNC) from CML patients compared with cells from healthy individuals [3], as well as, on highly enriched CD34-HSC from patients with untreated newly diagnosed CML [4, 5]. Several overexpressed genes were identified; most of them were components of pathways downstream of BCR–ABL and related to several pathways such as phosphatidylinositol 3-kinase (PI3K/AKT) pathway, mitogen-activated protein kinase (MAPK) pathway and p38 MAPK pathway. This suggests that in CML-CD34+ cells, increased signaling is not only based on phosphorylation but also on transcriptional activation of signaling molecules.
Gene expression profiling has also been used to detect tyrosine kinase inhibitor treatment (TKI)-resistant mutations [6], as well as major DNA methylation changes, which occur during CML progression [7]. Sonic hedgehog (SHH), WNT/b-catenin and NOTCH are another interesting group of pathways, which might be involved in haematopoietic stem cell self-renewal and maintenance, are associated with different forms of human cancer and are probably crucial to the tumorigenicity of cancer stem cells [8]. These three pathways in CML progression to terminal blast crisis have been previously studied [9]. However, these pathways have not been studied in CD34 cells of CP-CML leaving open their oncogenic or tumor-suppressing role in CP-CML. Therefore, by means of cDNA array technology and hierarchical cluster analysis, we investigate the gene expression pattern of CP-CD34 cells from patients with untreated newly diagnosed CML.
There is much yet to be understood about the biological and molecular roles of non-coding RNAs, such as microRNAs (miRNAs), on various aspects of cancer and metastasis, including BCR–ABL effects. Previous studies identified miR-17/92 cluster on chromosome 13q31.1 as both a tumor suppressor and an oncogene depending on the tumor context [10]. In a clinical setting, miR-17/92 polycistronic pri-miRNA transcripts and mature miRNAs (mi-17, miR-18a, miR-19a, miR-20a, miR-19b-1, miR-92a-1) are overexpressed in chronic phase (CP) compared with healthy donors, but not in blast crisis (BC) compared with healthy donors [11, 12]. The miR-17/92 cluster is regulated by BCR–ABL and cMyc and may exert an oncogenic function by down-regulating the tumor suppressor PTEN, a protein involved in the PTEN/PI3K pathway and in CML stem cells [10]. As has been demonstrated in older patients with heart failure [13] members of miR-17/92 (miR-18 and miR-19) target the extracellular matrix proteins such as connective tissue growth factor (CTGF). Moreover, a reduction of CTGF levels in Myc-transformed neoplasms has been shown [14]. This suggests that it may be worthwhile studying the correlation between members of the miR-17/92 cluster with PTEN and CTGF in chronic phase CML.
We also explore whether differences in miRNA expression and target gene could further explain the distinct feature of CP-CML samples and healthy donors leading to differences in transcriptome regulation.
Patients, materials and methods
Patients and cells
Eleven Bone marrow (BM) and four peripheral blood (PB) samples were obtained from newly diagnosed, non-treated patients showing Ph+ CML and in chronic phase. The diagnosis was based on clinical, hematological, cytogenetic and molecular analyses and Sokal’s index [15] (Table 1). BM and PB mononuclear cells from all samples were separated by Ficoll-Hypaque as previously described [16]. BM CD34+HCs from CML and healthy controls with over 97% purity were used for microarray experiments. The purity of other samples used for validation by real time PCR varied from 80 to 100%.
As a control, CD34+ cells were obtained from healthy donors (22 BM and 6 PB samples). The HC-PB donors were mobilized with G-CSF (10 mcg/kg/day) and collected with COBE spectra, gambro BCT (Lakewood, CO).
RNA isolation and microarray
Isolation of total RNA from CD34+HC was performed using the Trizol LS reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). Gene expression analysis was performed with Amersham CodeLink UniSet Human I BioArrays (Amersham Biosciences), containing 10,000 probes. Biotin-labelled cRNA was generated and hybridized to arrays and detected with a Cy5-Streptavidin conjugate. Fluorescence images were acquired using a GenePix 4000B scanner and the software GenePix Pro 6.0 (Axon Instruments) and analyzed with the CODELINK expression analysis software (CodeLink EXP v4.1, Amersham Biosciences). Experiments were run in duplicates, in such way that six hybridizations were performed for the three CML samples and four hybridizations for the two control samples.
Analysis of data
Data analysis of the microarray hybridizations was performed with the SAM [17]. Twofold or more differentially expressed genes were selected. A delta value of 2.34 was used to select a set of genes with a median FDR of 0.001.
Hierarchical clustering analysis based on euclidian distance was performed using UPGMA as implemented in cluster and Treeview software. The ontology analysis was performed using the gene ontology (http://www.ebi.ac.uk/GOA/). KEGG pathway database (http://www.genome.jp/kegg/pathway.html) and the pathway assist 3.0 software (Strategene) were used to identify signaling pathways associated with aberrant gene expression pattern in CML.
Real-time quantitative reverse transcription-PCR
Ten differentially expressed genes were selected to validate our microarray data by real-time PCR, using CML BCR/ABL CD34+ (BM = 10 and PB = 4) and normal CD34+ samples (BM = 22 and PB = 5). Reverse transcription was performed using high-capacity cDNA archive kit following the manufacturer’s protocol (Applied Biosystems). Normalization of quantitative data was based on the expression of GAPDH. All oligonucleotides used are listed in Table 2. All reactions were carried out duplicate and relative expression levels were calculated using the 2−∆∆Ct method with the mean of the healthy controls used as calibrator. The Mann–Whitney U test and the Kruskal–Wallis test were used, followed by Dunn’s post test. GraphPad prism 4.0 software was used for statistic analysis.
For quantitative analysis of miRNA expression, standard TaqMan assays primers and probes (applied biosystems) were used. The reverse transcription was using high-capacity cDNA archive kit, according to manufacturer’s instructions. Total RNA input was normalized based on the Ct values for RNU24 (PN4373379) endogenous controls. The fold change was calculated using 2−∆∆Ct method and the healthy controls as calibrator [18].
R (version 3.1.3) was used to analyse the statistical significance of pairwise correlation values between two mRNAs and two miRNAs. We calculated the Pearson correlation coefficient for patients and controls separately to check for any difference in correlation coefficients in both groups. To assess the significance of the correlations, bootstrapping was used. We also perform joint logistic regression of cell types (patient or control) against expression and microRNA expression.
Results
We compared gene expression profiles of hematopoietic cells CD34+ (CD34-HC) from two healthy volunteers and three non-treated, newly diagnosed, CP-CML. The total number of differentially expressed genes was 835 (8.3%, fold changes ≥2.0 or ≤0.5). Of those, 306 genes were upregulated and 529 were downregulated (shown in supplementary tables S1A and S1B). We performed gene ontology analysis to identify the different categories and biological processes expressed by leukemic and healthy CD34+HC. We found several genes involved with leukemogenesis (Fig. 1). Hierarchical clustering analysis was performed. The CML group from the normal group was separated, as well as the replicates that were clustered together.

Genes and pathways deregulated in leukemic CD34+ cells. Heat-map showing 129 differentially in leukemic and normal CD34+ cells. We selected and classified eight groups based on criteria involved in leukemogenesis (adhesion, cell cycle, cell death and apoptosis, cell motility, differentiation, cell division, DNA repair and cell proliferation). Microarray gene expression profiling was performed in two normal CD34+ cells and three leukemic CD34+ cells in duplicate
Among all genes with at least twofold changes in expression between CD34+HC from CP-CML compared with healthy individuals, we focused on genes present in five main pathways. The following genes were upregulated: CRKL; AKT1, MYC and also RAS, RAB10, MEK1, MEK2, p38GAMMA and p90RSK. p90RSK is the highest overexpressed gene (14-fold) in the CML group and is also a transcriptional target of MAPK pathway (Fig. 2, in red). However, other genes from PI3 kinase pathway were downregulated, for example, the BAD gene (2, zerofold downregulated).

Transcripts network in CD34 chronic myeloid leukemia cells. Transcripts of five pathways are aberrantly expressed in leukemic cells. Four of these pathways converge on myc oncogene upregulation that activates miR-19a and miR-18a from the polycistron miR-17/92 which directly inhibit the tumor suppressor gene Pten. In red are the upregulated genes, in blue the downregulated genes and in gray some transcripts that do not show differential expression in CML CD34+ cells
The other three pathways, which have been implicated in HSC self-renewal and maintenance, SHH, WNT and NOTCH were deregulated in CML patients (Fig. 2, in blue). Two inhibitors of SHH pathway, HHIP and Gli-3 genes, were downregulated in HSC CML, suggesting an upregulation of this pathway in these cells. As regards the Wnt/b-catenin pathway, we could observe an upregulation of FZD7 (fold change = 2), which is a receptor for Wnt signaling proteins. The antagonist of FZD7, FRZB was downregulated (fold-change = −2.6). The major effect of Wnt pathway is the stabilization of cytoplasmic beta-catenin protein, which did not show differential expression in HSC CML (Fig. 2, in gray). The third pathway associated with HSC self-renewal, NOTCH, seems to be inhibited in CML HSC. Deltex (DTX1), a modulator of the Notch-signaling pathway, was downregulated (fold change = −17.5), as well some targets of Notch: CD28, CHI3L2, CDKN1A, CLEC4A and CDKN2A (Fig. 2, in blue).
To validate the microarray data, we selected eight genes (AKT1, FAS, CASP3, HDAC1, CTGF, DTX1, HMOX1 and BAD) and compared gene expression values by qPCR in CD34+HC samples from 10 CML patients and 22 samples from healthy volunteers. The analysis showed that all of these eight genes are differentially expressed. We observed that AKT1, FAS, CASP3 and HDAC1 are 2.1–5-fold upregulated in CD34+HC-BM CML (Fig. 3a–d). On the other hand, CTGF, DTX1, HMOX1 and BAD are 2–100-fold downregulated in CD34+ (Fig. 3e–h). The differences observed between the CD34+HC from normal bone marrow and HC-BM CML were significant (P < 0.001) in all the genes tested. In addition, for CTGF, we also performed qPCR using peripheral blood (PB) CD34+HCs from four CML patients and six healthy volunteers. We found that there is no significant difference between CTGF expression levels in CD34+HC-PB CML. However, the difference between CTGF expression levels in CD34+HC from PB-CML patients and BM-CML is significant (Fig. 3e).

Quantitative validation of the microarray results by real time PCR for eight selected genes. AKT1, FAS, CASP3, HDAC1 are enhanced and BAD, CTGF, DTX1 eHMOX1 are repressed in CD34+HC of patients with CML. The gene expression levels from quantitative PCR are expressed in fold with respect to the median of the expression levels obtained in CD34+ of bone marrow from healthy controls. The differences observed between the CD34+HC from normal bone marrow and CML were significant (P < 0.001) in all the genes tested. In addition, we also quantified the expression of CTGF gene in CD34+HC isolated from peripheral blood of patients with CML and healthy controls and we found that there is no significant difference (α = 0.05). We also evaluated the CTGF expression level in CD34+HC from PB-CML and healthy BM and we found that the difference is significant
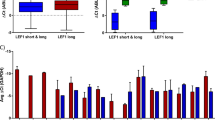
We also investigated the expression levels of PTEN and two miRNAs from the miRNA-17/92 cluster, miR-18a and miR-19a. qPCR showed no significant difference of these protein-coding and non-coding RNAs between CD34+HC from CML and from healthy controls (Fig. 4a–c).

a–c The difference in fold change between the expression levels of PTEN, miR-19a and mir-18a between patients and controls. d, e Shows the correlation between gene expression and microRNA levels in patients and controls. L denotes patients and H healthy control
For each of these two pairs protein-coding and non-coding RNAs, we studied the correlation between CML patients and healthy controls. In both Fig. 4d, e it appears that the pattern of correlation between gene expression and microRNA level is quite different in patients and healthy controls. To check this, we first computed the Spearman correlation between the levels of miR-19a and PTEN in patients and controls separately. For healthy controls the value of the sample Spearman correlation coefficient is 0.23 while for patients it is 0.024. However, for both patients and controls the p values are not significant (0.47 and 0.97). This calculation is repeated for the levels of miR-18a and CTGF, again for patients and controls separately. Once again the sample values are very different, 0.17 for controls and −0.51 for patients but the p values are not significant (0.6 and 0.2, respectively).
From Fig. 4 it appears that the joint analysis of PTEN and miR-19a levels may separate the two cell types. A similar separation may be possible considering miR-18a and CTGF. As a check, we perform logistic regression with the two cell types as dependent variables. In one logistic regression, the independent variables are PTEN and miR-19a. For this joint analysis, PTEN is significant (p value = 0.033) even after controlling for miR-19a. A similar joint analysis was performed using miR-18a and CTCF as predictors. However, the results were not statistically significant.
Discussion
In our study, the comparison of transcript levels from newly diagnosed CML patients and healthy controls of components of these pathways that have been implicated in several biological processes such as enhanced proliferation/survival (for example PI3K/AKT), enhanced self-renewal (for example WNT/b-catenin, SHH and NOTCH) and block of myeloid differentiation (for example, MAPK pathway) in highly enriched CD34+ cells showed that they are deregulated in initial CP of CML patients, suggesting that might contribute in the pathogenesis of CML.
Microarray analysis showed upregulation of OSBPL9, LYN, RAS, MEK1, MEK2, p90RSK (RPS6KA1), MYC, CRKL, AKT1, TCF4 and FZD7 (Fig. 2). Some of these mRNAs are upregulated in cancer stem cells (CSC) of other tumors. For example, LYN have been shown to be upregulated in CSC in pancreatic adenocarcinoma [19]; MEK1 in CSC in hepatocellular carcinoma [20] and AKT1 in CSC in breast cancer [21]. We also performed quantitative real time PCR of AKT1 and demonstrated that this transcript is significantly upregulated in CD34 of CML patients (Fig. 3). Similar to our study [5], have revealed upregulation of LYN, KRAS, NRAS, MEK1, MYC, CRKL and AKT1 in HSC from CLM patients in CP by microarray analysis [5]. However, overexpression of OSBPL9, MEK2, p90RSK, TCF4 and FZD7 has not been previously reported in HSC in CML in CP.
Microarray analysis also revealed downregulation of HHIP, GLI3, SP1, PTEN, BAD, CTGF, LEF1, FRZB, AXUD1 (CSRNP1), DTX1, CD28, CHI3L2, CDKN1A, CLEC4A and CDKN2A (Fig. 2). Of note, PTEN loss or scarcely expression of PTEN has been shown to have a role in CSC in several tumors such as: prostate, breast, colorectal, lung and melanoma [22,23,24,25]. On the other hand, inversely to our study inhibition of the transcription factor SP1 suppresses colon cancer stem cell growth [26] and overexpression of DTX1 was observed in liver CSC through in vitro sphere-forming cell approach of hepatoma cell lines [27]. We also performed quantitative real time PCR of BAD and DTX1 and demonstrated that these transcripts are significantly downregulated in CD34 of CML patients (Fig. 3). This is also contrary to a previous study of [5] which showed that BAD transcript is not differentially expressed in HSC of CML in CP [5]. Moreover, downexpression of all the other transcripts have not been reported in HSC in CML patients compared with HSC healthy controls.
MYC gene activated in various cancers is crucial for the BCR/ABL mediated transformation [28]. Our data showed that in CD34+HC-CML-CP deregulation of SHH, MAPK, PI3K and WNT pathways converge to MYC activation (Fig. 2). Also, MYC activates the miR-17/92 cluster which has characterized in a variety of solid tumor type, as well as hematopoietic malignancies including CML. In CD34+HC-CML-CP qPCR of two components of this cluster (miR-19a and miR-18a, Fig. 4) showed a tendency but not significant increase in expression levels compared with healthy CD34+HC. Interestingly, Fallah et al. (2015) showed significant upregulation of two other microRNAs from this cluster named miR-20a and miR-92a-1 in CML-CP [29].
Moreover, we performed qPCR of two transcripts target of miR-18a and miR-19 named CTGF and PTEN, respectively. We found that CTGF is significantly downregulated in CD34+HC in CML-CP compared with CD34+HC in healthy control, but not PTEN (Figs. 3, 4a–c).
There are accumulated studies suggesting that the changes in PTEN expression might have a role in CSC and tumorigenesis; CTGF deregulation has been associated with stem-like properties during cancer progression in head and neck cancer cells [25, 30]. Considering two microRNA targets of these RNAs, we studied the correlation between these RNAs of CD34+HC-CML and healthy controls. Our results suggest a difference, which could show statistical significance in a larger sample.
To conclude, in this study we demonstrated several mRNAs up- or downregulated in CD34+HC CML during chronic phase. Several tyrosine kinase inhibitors (TKIs) have been used to target BCR–ABL1 and eradicate HSC in CP. However, recurrence has been the main challenge in the management of CML, suggesting the need to identify alternative targets and biomarkers. The pathways here analyzed are indeed conserved through evolution, and we identify potential biomarkers in CD34+HC CML-CP for eradicating CML-CD34+HC. For example, we have shown that OSBPL9, MEK2, p90RSK, TCF4 and FZD7 are overexpressed in the CP and this result needs to be confirmed in an independent study to better understand the biology of CD34+HC in chronic phase and the transition from the chronic phase to the blastic phase.
References
Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7(6):441–53. doi:10.1038/nrc2147.
Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103(11):4010–22. doi:10.1182/blood-2003-12-4111.
Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T, Skorski T. Chronic myelogenous leukemia molecular signature. Oncogene. 2003;22(25):3952–63. doi:10.1038/sj.onc.1206620.
Kronenwett R, Butterweck U, Steidl U, Kliszewski S, Neumann F, Bork S, et al. Distinct molecular phenotype of malignant CD34+ hematopoietic stem and progenitor cells in chronic myelogenous leukemia. Oncogene. 2005;24(34):5313–24. doi:10.1038/sj.onc.1208596.
Diaz-Blanco E, Bruns I, Neumann F, Fischer JC, Graef T, Rosskopf M, et al. Molecular signature of CD34+ hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia. 2007;21(3):494–504. doi:10.1038/sj.leu.2404549.
Machova Polakova K, Kulvait V, Benesova A, Linhartova J, Klamova H, Jaruskova M, et al. Next-generation deep sequencing improves detection of BCR-ABL1 kinase domain mutations emerging under tyrosine kinase inhibitor treatment of chronic myeloid leukemia patients in chronic phase. J Cancer Res Clin Oncol. 2015;141(5):887–99. doi:10.1007/s00432-014-1845-6.
Heller G, Topakian T, Altenberger C, Cerny-Reiterer S, Herndlhofer S, Ziegler B, et al. Next-generation sequencing identifies major DNA methylation changes during progression of Ph+ chronic myeloid leukemia. Leukemia. 2016;30(9):1861–8. doi:10.1038/leu.2016.143.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12(8):445–64. doi:10.1038/nrclinonc.2015.61.
Sengupta A, Banerjee D, Chandra S, Banerji SK, Ghosh R, Roy R, et al. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and Notch signaling in chronic myeloid leukemia progression. Leukemia. 2007;21(5):949–55. doi:10.1038/sj.leu.2404657.
Yeh CH, Moles R, Nicot C. Clinical significance of microRNAs in chronic and acute human leukemia. Mol Cancer. 2016;15(1):37. doi:10.1186/s12943-016-0518-2.
Yong AS, Melo JV. The impact of gene profiling in chronic myeloid leukaemia. Best Pract Res Clin Haematol. 2009;22(2):181–90. doi:10.1016/j.beha.2009.04.002.
Venturini L, Battmer K, Castoldi M, Schultheis B, Hochhaus A, Muckenthaler MU, et al. Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood. 2007;109(10):4399–405. doi:10.1182/blood-2006-09-045104.
van Almen GC, Verhesen W, van Leeuwen RE, van de Vrie M, Eurlings C, Schellings MW, et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. 2011;10(5):769–79. doi:10.1111/j.1474-9726.2011.00714.x.
Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–5. doi:10.1038/ng1855.
Sokal JE, Cox EB, Baccarani M, Tura S, Gomez GA, Robertson JE, et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood. 1984;63(4):789–99.
Tognon R, Gasparotto EP, Leroy JM, Oliveira GL, Neves RP, Carrara Rde C, et al. Differential expression of apoptosis-related genes from death receptor pathway in chronic myeloproliferative diseases. J Clin Pathol. 2011;64(1):75–82. doi:10.1136/jcp.2010.080895.
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98(9):5116–21. doi:10.1073/pnas.091062498.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45.
Skoda J, Hermanova M, Loja T, Nemec P, Neradil J, Karasek P, et al. Co-expression of cancer stem cell markers corresponds to a pro-tumorigenic expression profile in pancreatic adenocarcinoma. Plos one. 2016;11(7):e0159255. doi:10.1371/journal.pone.0159255.
Cheng J, Liu C, Liu L, Chen X, Shan J, Shen J, et al. MEK1 signaling promotes self-renewal and tumorigenicity of liver cancer stem cells via maintaining SIRT1 protein stabilization. Oncotarget. 2016;7(15):20597–611. doi:10.18632/oncotarget.7972.
Gargini R, Cerliani JP, Escoll M, Anton IM, Wandosell F. Cancer stem cell-like phenotype and survival are coordinately regulated by Akt/FoxO/Bim pathway. Stem Cells. 2015;33(3):646–60. doi:10.1002/stem.1904.
Hill R, Wu H. PTEN, stem cells, and cancer stem cells. J Biol Chem. 2009;284(18):11755–9. doi:10.1074/jbc.R800071200.
Lombardo Y, Scopelliti A, Cammareri P, Todaro M, Iovino F, Ricci-Vitiani L, et al. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology. 2011;140(1):297–309. doi:10.1053/j.gastro.2010.10.005.
Sette G, Fecchi K, Salvati V, Lotti F, Pilozzi E, Duranti E, et al. Mek inhibition results in marked antitumor activity against metastatic melanoma patient-derived melanospheres and in melanosphere-generated xenografts. J Exp Clin Cancer Res. 2013;32:91. doi:10.1186/1756-9966-32-91.
Ciuffreda L, Falcone I, Incani UC, Del Curatolo A, Conciatori F, Matteoni S, et al. PTEN expression and function in adult cancer stem cells and prospects for therapeutic targeting. Adv Biol Regul. 2014;56:66–80. doi:10.1016/j.jbior.2014.07.002.
Zhao Y, Zhang W, Guo Z, Ma F, Wu Y, Bai Y, et al. Inhibition of the transcription factor sp1 suppresses colon cancer stem cell growth and induces apoptosis in vitro and in nude mouse xenografts. Oncol Rep. 2013;30(4):1782–92. doi:10.3892/or.2013.2627.
Cao L, Zhou Y, Zhai B, Liao J, Xu W, Zhang R, et al. Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol. 2011;11:71. doi:10.1186/1471-230X-11-71.
Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. doi:10.1016/j.cell.2012.03.003.
Fallah P, Amirizadeh N, Poopak B, Toogeh G, Arefian E, Kohram F, et al. Expression pattern of key microRNAs in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Int J Lab Hematol. 2015;37(4):560–8. doi:10.1111/ijlh.12351.
Chang CC, Hsu WH, Wang CC, Chou CH, Kuo MY, Lin BR, et al. Connective tissue growth factor activates pluripotency genes and mesenchymal-epithelial transition in head and neck cancer cells. Cancer Res. 2013;73(13):4147–57. doi:10.1158/0008-5472.CAN-12-4085.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest.
Human participants
All procedures performed in studies involving human participants were in accordance with the ethical standards of the FMRP-USP Hospital and with the 1964 Helsinki declaration and its later amendments.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
de Cássia Viu Carrara, R., Fontes, A.M., Abraham, K.J. et al. Expression differences of genes in the PI3K/AKT, WNT/b-catenin, SHH, NOTCH and MAPK signaling pathways in CD34+ hematopoietic cells obtained from chronic phase patients with chronic myeloid leukemia and from healthy controls. Clin Transl Oncol 20, 542–549 (2018). https://doi.org/10.1007/s12094-017-1751-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-017-1751-x