
Abstract
Prostaglandin signaling controls a wide range of biological processes from blood pressure homeostasis to inflammation and resolution thereof to the perception of pain to cell survival. Disruption of normal prostanoid signaling is implicated in numerous disease states. Prostaglandin signaling is facilitated by G-protein-coupled, prostanoid-specific receptors and the array of associated G-proteins. This review focuses on the expression, characterization, regulation, and mechanism of action of prostanoid receptors with particular emphasis on human isoforms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Prostaglandins are amphipathic, bioactive signaling molecules derived from the oxidation of arachidonic acid. They are involved in a wide range of roles from homeostasis of blood pressure and blood flow, the initiation and resolution of inflammation, the perception of pain, cell survival, and the progression of numerous disease states. These biomolecules act most often as autocrine or paracrine signaling agents and most have relatively short half-lives. Prostanoid signaling is accomplished through specific G-protein coupled receptors (Figure 1). With the exception of a few isoforms with unknown biological function, all known biologically competent receptors are heptahelical, multi-pass membrane proteins, members of the G-protein coupled receptor 1 family (GPCR) and are among the most abundant membrane proteins (Binda et al. 2004). The diversity of prostanoid action is not only defined by specific receptors and their regulated expression, but also to the diversity of G-proteins that most receptors are able to couple to, leading to actuation of different signaling pathways by the same receptor. Although the actions of these receptors are diverse, many show commonalities in their regulation. Each display agonist-induced desensitization that is usually found to be associated with receptor phosphorylation by various protein kinases. Phosphorylation by G-protein receptor kinases (GRK) can lead to arrestin binding that promotes receptor uptake into clatherin-coated pits leading to sequestration into punctate vesicles. This review focusses primarily on human receptors but discusses receptors from other species when information about the human receptors is lacking.

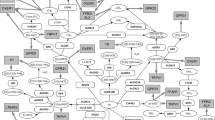
Prostanoid metabolic pathway. Gene designations are given for the participating enzymes (rounded boxes), accepted acronyms given for metabolites (ovals), and accepted acronyms for receptors (grey boxes). Receptor binding only indicated for metabolites with an EC50 in the nM range. Enzymes: AKR1B1, aldo-keto reductase 1B1; AKR1C3, aldo-keto reductase 1C3; CBR1, carbonyl reductase 1; FAM213B, prostamide/prostaglandin F synthase; HPGDS, Hematopoietic prostaglandin D synthase; HSA, human serum albumin; PTGDS, prostaglandin D2 synthase; PTGES, prostaglandin E synthase; PTGES2, prostaglandin E synthase-2; PTGES3, prostaglandin E synthase-3; PTGIS, prostacyclin (PGI2) synthase; PTGS1, Prostaglandin G/H Synthase 1; PTGS2, Prostaglandin G/H Synthase 2; TBXAS1, Thromboxane A Synthase 1. Note: Figure 1 is in color
Prostaglandin D2 Receptors
Introduction
There are two distinct types of prostaglandin D2 receptors found in humans, the prostanoid DP1 receptor (PGD receptor, PGD2 receptor, Gene: PTGDR) and the DP2 receptor (CRTH2, G-protein coupled receptor 44, CD294, Gene: PTGDR2). Both are coupled to G-proteins and upon binding to PGD2 or its PGJ2 series metabolic products, they affect intracellular concentrations of cAMP and Ca2+ concentrations either directly or indirectly depending on the receptor. Activation is responsible in part for immune regulation, allergic/inflammation responses, mobilization of dendric cells, and impaired PGD2-induced sleep to name a few. Although the DP1 amino acid sequence is closely related to other prostaglandin receptors it shares only a 22% percent identity with DP2 which is more closely related to chemoattractant receptors such as the N-formylmethionly-leucyl-phenylalanyl (fMLP) and anaphylatoxin C3a receptors (Wang et al. 2018; Hirai et al. 2003).
DP1 receptor
Introduction
The human prostaglandin D2 receptor DP1 (PTGDR, UniprotKB-Q13258) is translated as a 359 amino acid polypeptide with a calculated molecular weight of 40.3 kDa. There is one additional reported isoform based only on expressed sequence tag (EST) data (Q13258-2) and four coding single nucleotide polymorphic (SNP) variants (R7C, G198E, E301A, and R332Q) (https://genecards.org, Stelzer et al. 2016), none of which involve any known clinical significance or condition. There are no reported X-ray structures, but there are several G-protein coupled receptor database (GPCRdb, https://gpcrdb.org, Pándy-Szekeres et al. 2018) models proposed based on similar proteins (PDB reference 4UHR and 3VG9). Morii and Watanabe (1992) examined the effect of a variety of exo- and endo-glycosidases on the binding of PGD2 to the DP1 receptor on P2 membrane prepared from porcine temporal cortex. They found that the exoglycosidases neuramidase, α-manosidase, β-galactosidase, and β-N-acetylhexoamidase reduced the binding of PGD2 by 6%, 33%, 62%, and 66% respectively, indicating that the integrity of the glycosyl chains was important for ligand binding. Further, Scatchard analysis of the data revealed that modification of the glycosyl chain reduced the affinity of the DP1 receptor for the ligand rather than the maximal binding capacity. Treatment of the P2 membrane with endoglycosidase N-glycohydrolase F which cleaves the N-glycosyl linkage between the carbohydrate and protein reduced PGD2 binding by 66%, confirming the presence of N-linked carbohydrate. Extracellular N-linked glycosylation is predicted for N10 with high probability by NetNGlyc analysis (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004). Extracellular N-linked glycosylations are also predicted for N90, and N297 by sequence analysis, but none of these predictions have yet to be confirmed experimentally (Apweiler et al. 2017). Interestingly, treatment with endo-α-N-acetylgalactosamidase, which cleaves only O-linked carbohydrate from proteins, reduced PGD2 binding by 52%, confirming the presence of O-linked glycosyl chains as well. NetOGlyc analysis (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013) predicts potential O-glycosylation at S238, S254, T240, and S350, all of which are located on the cytoplasmic side of the membrane. Here and throughout this document N-linked and O-linked glycosylation is predicted with these web services. Although the data is strongly suggestive of the presence of both O- and N-glycosylations on the DP1 receptor, one cannot rule out the possibility that at least a portion of the glycosylations may be present on a yet-to-be identified protein associated with the DP1 receptor that served modify the binding affinity for PGD2.
There are numerous potential phosphorylation sites on the human DP1 based on motifs but none are specifically confirmed experimentally. Utilizing the NetPhos 3.1 server (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999), a conservative minimum score of 0.9 and the availability of sites based on topology predictions (UniProtKB), nine potential sites of phosphorylation are predicted: S145 and S300 on extracellular loops, S254 on a cytoplasmic loop, and S347 on the cytoplasmic, C-terminal domain. There are also additional potential G protein-coupled receptor kinases (GRK) phosphorylation sites on the C-terminal domain predicted by the GPS server (http://gps.biocuckoo.cn/, Xue et al. 2011): S347, S350, S352, T353, S357, and S358. There is experimental evidence for GRK-mediated phosphorylation of the N-terminal of the receptor (see below) (Gallant et al. 2007). The aforementioned methodologies for phosphorylation prediction are used throughout the remaining document.
Expression and Characterization
The DP1 receptor is expressed primarily in the gastrointestinal tract, bone marrow and the immune system, and the gall bladder, but is found in many other tissues such as the retinal choroid, ciliary epithelium, longitudinal and circular ciliary muscles, iris, and brain cerebral cortex (Town et al. 1983; Sharif et al. 2000a; http://www.proteinatlas.org, Uhlén et al. (2015)). Although found in many tissues, this receptor is the least abundant of the prostanoid receptors (Boie et al. 1995).
Ligand binding properties for DP1 have been characterized with human recombinant DP1 expressed in human embryonic cell line HEK293 (Wright et al. 1998; Sawyer et al. 2002), mammalian COS-M6 cells (Boie et al. 1995), and a human immortalized myelogenous leukemia cell line K562 (Hirai et al. 2001), as well as human platelets (Table 1). Binding properties for recombinant mouse DP1 expressed in HEK293 cells (Hata et al. 2005; Hata et al. 2003) as well as combined DP in bovine embryonic tracheal cells have also been examined (Sharif et al. 2003). Although prostanoid receptors exhibit preferences with respect to ligand binding, promiscuous cross reactivity has been reported for several receptors (Abramovitz et al. 2000; Narumiya et al. 1999). Both ligand affinities and efficacies have been examined. [H3]-PGD2 equilibrium competition assays provide a good measure of relative ligand binding affinities (Table 1). Binding affinities for recombinant receptor show the highest affinity while binding to human platelets is the lowest of all. Binding of PGE2 and PGF2α is several orders of magnitude poorer than PGD2 for all systems examined, supporting the selectivity of the receptor. Binding of the non-enzymatic dehydration product of PGD2, PGJ2 is 2-10 times weaker than PGD2. Signal efficacy upon binding has been measured as the extent of induced cAMP synthesis as compared to the maximal amount produced by PGD2 at saturation and expressed as the concentration of ligand necessary to produce 50% of the maximal PGD2 levels (EC50). The relative values for the different ligands binding to the same receptor shown in Table 1 compare well with those for the binding assays indicating that affinity is comparable to efficacy.
Examination of the cellular effects resulting from a particular agonist binding to a specific receptor are frequently discerned through judicious use of highly selective agonists and antagonists for a particular receptor. The most frequently used selective agonists for DP1 are BW245C and TS-022, and there are three commonly used selective antagonists, BWA868C, MK-0524, and S-5751. A listing of agonists and antagonists is given in Table 2.
The expression of DP1 from post-translational folding and processing in the Golgi to its expression in the plasma membrane has been examined in detail by Binda et al. (2004). Utilizing recombinant proteins expressed in a HEK293 system, they found that lipocalin-type prostaglandin 2 synthase (L-PGDS) (Binda and Parent 2015) helps facilitate the cell surface expression of DP1. Following glycosylation and maturation in the Golgi, DP1 forms a complex with Hsp90 which then interacts directly with L-PGDS as a requirement for cell surface expression, independent of PGD2 binding. In addition, L-PGDS associates directly with the co-expressed DP1 in the perinuclear region, but not on the plasma membrane, and this association increases the production of PGD2 by L-PGDS which may represent an intracrine signaling system.
Mechanism of Cell Activation
Signal transduction from the DP1 receptor is reported to occur via a Gαs protein signaling system (see Table 3), resulting in an increase in intracellular concentrations of cAMP (Ito et al. 1990; Boie et al. 1995; Sugama et al. 1989 ; Hirata et al. 1994a; Schratl et al. 2007) which in turn activates protein kinase A (PKA), setting off a number of cellular events. Several studies also report a concomitant increase intracellular Ca2+ (Xue et al. 2007; Okuda-Ashitaka et al. 1993), but without an increase in inositol 1,4,5-trisphosphate (IP3) concentration (Boie et al. 1995). The authors suggest that the increase in intracellular calcium could occur through PKA phosphorylation of the L-type Ca2+ channel (LTCC) and the ryanodine receptor (RyR) (Maher et al. 2015; Zaccolo 2009).
Regulation
As is typical for most G-coupled receptors, regulation of the DP1 receptor is accomplished through desensitization and internalization. The regulation of recombinant human DP1 in HEK293 kidney cells with and without recombinant GRKs, arrestins or Ras related proteins (Rab) in the presence or absence of PGD2 has been examined by Gallant et al. (2007). GRKs are known to mediate G-protein receptor desensitization through phosphorylation of the receptor, leading to high affinity binding to arrestins. Bound arrestins are known to inhibit receptor binding to the associated G-protein and at the same time promote receptor uptake into clatherin-coated pits where the receptors are either degraded or recycled back to the membrane, recycling being controlled by Rabs. When stimulated with 1 μM PGD2, Gallant et al. (2007) observed that DP1 expressed alone undergoes approximately 25% internalization, leading to a concomitant reduction of activity. When DP1 is co-expressed with GRK2, PGD2-induced internalization increases by 25% after 2 hours following stimulation but is unaffected by co-expression of either GRK5 or GRK6. In addition, co-expressed GRK2 reduces DP1 signaling as measured by cAMP production by 45% within 15 minutes post-stimulation, whereas co-expressed GRK5 requires 60 minutes to achieve a maximum reduction in cAMP production of 35% and without internalization. Co-expressed GRK 6 has little effect on the signaling. Co-expression with arrestin-3 or arrestin-2 promotes internalization by 53% and 43% respectively. On the other hand, co-expression of Rab4 with DP1 decreases internalization by 64% whereas co-expression with Rab11 has no effect on internalization. These data are consistent with rapid GRK2 phosphorylation of DP1 leading to a slower internalization via arrestins that may be reversed with Rab4, a protein known to be involved in fast endocytic recycling (Wang et al. 1995). Phosphorylation by GRK5 reduces DP1 signaling without subsequent internalization, indicating that the site of phosphorylation may not promote proper arrestin binding but may instead simply reduce G-protein interaction with DP1.
DP2 receptor
Introduction
The human prostaglandin D2 receptor DP2 (PTGDR2, CRTH2, DL1R, GPR44, UniprotKB-Q9Y5Y4) is translated as a 395 amino acid polypeptide with a calculated molecular weight of 43.3 kDa (Nagata and Hirai 2003). There are no additional reported isoforms and three coding SNP variants reported, V204A (Nagata et al. 1999), F179V, P197T (Hsu et al. 2002) and L281F (https://www.ncbi.nlm.nih.gov/clinvar, Landrum et al. 2016), none of which have any reported effect on function. There are two reported X-ray structures (6D26 and 6D27). Extracellular N-linked glycosylations are predicted for N4 and N25 by sequence analysis (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004) and numerous O-linked sites on both the cytoplasmic and extracellular domains (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013), none of which have been confirmed experimentally. However, Nagata et al. (1999) have shown that treatment of DP2 with endo F increases its mobility on sizing gels, indicating a decrease in mass of 20-30 kDa and confirming the presence of N-linked glycosylation.
There are numerous potential phosphorylation sites on the human DP2 based on motifs, but none are specifically confirmed experimentally (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). Eleven potential sites of phosphorylation are predicted: S22 and S27 on the extracellular N-terminal domain, T172 and T174 on extracellular loops, T64 on a cytoplasmic loop, and S318, T344, S345, S346, S352, and S391 on the cytoplasmic, C-terminal domain. There are also additional potential GRK phosphorylation sites in the C-terminal domain predicted for T321, S325, T344, S345, S346, S358, S376, S380, T383, S391, S392, T393, S394, S395 (http://gps.biocuckoo.cn/, Xue et al. 2011).
Expression and Characterization
The DP2 receptor is expressed primarily in the gastrointestinal tract, brain, endocrine system and muscle, but is also found in many other tissues such as immune tissue (eosinophils, basophils and Th2 lymphocytes in particular), gall bladder, ciliary and both male and female reproductive tissue (http://www.proteinatlas.org, Uhlén et al. (2015)).
Ligand binding properties for DP2 have been characterized with human recombinant protein expressed in both HEK293 (Sawyer et al. 2002; Hata et al. 2005) and K562 (Hirai et al. 2001) cell lines, and mouse recombinant protein in a HEK293 cell line (Hata et al. 2003). Ligand affinities as measured by [3H]PGD2 equilibrium competition assays and ligand efficacies as measure by the reduction of [cAMP] in either isoproterenol- or forskilin-stimulated cells are given in Table 1. As observed for the DP1 receptor, the DP2 receptor shows a preference for PGD2, but also cross-reactivity with other ligands such as PGF2α and 9α,11β-PGF2α. The relative cross-reactivities are similar to those observed for the DP1 receptor. The most frequently used selective agonists for DP2 are Indomethacin and L-888,607. There is one commonly used selective antagonist, Ramatroban (Sugimoto et al. 2003) (Table 2).
Mechanism of Cell Activation
Signal transduction from the DP2 receptor occurs via a Gαi protein signaling system (see Table 3) resulting in a decrease in intracellular [cAMP] and an increase in intracellular [Ca2+] (Sawyer et al. 2002; Hirai et al. 2001; Pettipher et al. 2007). Further, DP2 activation in Th2 cells is mediated through a phosphatidylinositol 3-phosphate kinase (PI3K) and Ca2+/calmodulin/calcineurin signaling pathways (Xue et al. 2007).
Regulation
The regulation of DP2 has many similarities to that of DP1 discussed above with some notable exceptions. In addition to their work with DP1, Gallant et al. (2007) also examined the regulation of recombinant DP2 expressed in HEK293 kidney cells. They found that internalization of the DP2 receptor increases 20% in the presence of the PGD2 agonist, 5% less than observed for the DP1 receptor. However, when co-expressed with GRK2, the DP2 receptor’s agonist-induced internalization is increased by 88% compared to an increase of 25% for co-expression of GRK2 with DP1 and the rate desensitization via phosphorylation is considerably slower than observed for DP1. Further, co-expression of GRK5 or GRK6 with DP2 more than doubles the receptor internalization whereas co-expression of either of these GRKs has no effect on DP1 internalization. Co-expression of DP2 with arrestin-2 results in enhancement of agonist-induced internalization by 90%, over twice that observed for DP1, but unlike DP1, co-expression of DP2 with arrestin-3 has no effect on agonist-induced internalization of DP2. As observed for DP1, co-expressed GRKs reduces the DP2 receptor activity (measured by changes in inhibition of forskolin-induced cAMP synthesis) and to a similar degree. Lastly, both DP1 and DP2 recycling is enhanced by Rab proteins, however, for DP2 it is the Rab11 protein that promotes recycling rather than the Rab4 protein that enhances recycling of DP1. These data are consistent with a slow phosphorylation of DP2 by all three GRKs on a time scale comparable to internalization that is in turn reversed by Rab11 activation.
Schröder et al. (2009) examined the effect of DP2’s C-terminal tail on receptor internalization through comparison of the activity of recombinant DP2 with the native 86 amino acid C-terminal tail to that that of a recombinant truncated version with an 8 amino acid tail. They found that the truncated version exhibits enhanced signaling compared to wild type DP2, indicating that the wild type C-terminus inhibits interaction with Gαi to reduce signaling activity. Further, they found that the C-terminal is the site for β-arrestin-2 binding, a prerequisite for internalization, leading to a reduction in signaling. Interestingly, they report that DP2 is not phosphorylated upon agonist binding. This directly contrasts with the Gallant et al. (2007) report where GRKs were found to significantly increase internalization, presumably through phosphorylation of the receptor. Further, this report also indicates that inhibition of PKA with H-89 decreases internalization, again suggesting a phosphorylation event. Further study is clearly required to rationalize these contrasting results.
Regulation on the transcription level has also been examined. MacLean Scott et al. (2018) revealed that the transcription factor GATA3 enhances expression of DP2 during Th2 cell differentiation and within innate lymphoid cells. They also report that over-expression of the NFAT1 transcription factor reduces GATA3 promotor activity. They suggest that a dynamic relationship between GATA3 and NFAT1 competition for the promotor site may play a role in DP-mediated allergic inflammation.
Synergy and Opposing Actions of the DP1 and DP2 Receptors
PGD2 is generated by mast, Th2, and dendritic cells and is found in high concentrations at sites of inflammation. The DP1 and DP2 receptors act jointly in the development and maintenance of the allergic response.
PGD2 stimulation of DP2 receptors is directly involved in activation of Th2 lymphocytes, eosinophils and basophils, with little effect on neutrophils (Emery et al. 1989), as well as chemotaxis of these cells to the site of inflammation (Xue et al. 2007; Schratl et al. 2007). Further, efficient synthesis of the microvasculature permeability enhancer cystenyl LTs (LTC4 LTD4 and LTE4) by eosinophils requires simultaneous stimulation of the DP1 and DP2 receptors (Mesquita-Santos et al. 2011). However, these two receptors have opposing effects on eosinophil activation. Binding of PGD2 to the DP2 receptor activates eosinophils by stimulating the production of the leukocyte integrin CD11b, an event required for adhesion, migration and accumulation at the site of inflammation (Maiguel et al. 2011). Binding of PGD2 to the DP1 receptor, however, negatively regulates the production of CD11b, serving to modulate the activation of eosinophils (Monneret et al. 2001).
Activation of both DP1 and DP2 receptors by PGD2 results in the inhibition of apoptosis of particular immune cells, thus prolonging the inflammatory burden in regions of inflammation. Xue et al. (2009) have shown that binding of PGD2 to DP2 receptors on Th2 cells suppresses annexin V binding, mitochondrial cytochrome C release, and caspase activities associated with cell apoptosis. Peinhaupt et al. (2018) have shown that stimulation of DP1 inhibits the onset of intrinsic apoptosis of eosinophils apparently though the activation of anti-apoptotic proteins such as the Bcl2 family of proteins. Further, they found that DP1 signaling induced the serum response element (SRE) that regulates genes responsible for cytoskeleton production and survival.
Prostaglandin E2 Receptors
Introduction
There are four known distinct subtypes of PGE2 receptors: 1) the EP1 receptor (PGE2 receptor EP1 subtype, Gene: PTGER1), 2) the EP2 receptor (PGE2 receptor EP2 subtype, Gene: PTGER2, 3) the EP3 receptor (PGE2 receptor EP3 subtype, PGE2-R, Gene: PTGER3), and 4) the EP4 receptor (PGE2 receptor EP4 subtype, Gene: PTGER3). Sequence homology between the subtypes is in the 28-33% range (Sugimoto and Narumiya 2007). Expression levels are both tissue and receptor subtype dependent.
EP1 receptor
Introduction
The human prostaglandin E2 receptor EP1 (PTGER1, UniprotKB-P34995) is translated as a 402 amino acid polypeptide with a calculated molecular weight of 41.8 kDa. There are no additional reported isoforms and three coding SNP variants (A71T, T223M and H256R) (https://genecards.org, Stelzer et al. 2016), none of which involve any known clinical significance or condition. There are no reported X-ray structures. Extracellular N-linked glycosylations are predicted for N8 and N25 by sequence analysis (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004) and numerous O-linked sites on both the cytoplasmic and extracellular domains (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013), none of which have been confirmed experimentally.
There are numerous potential phosphorylation sites on the human PTGER1 based on motifs but none are specifically confirmed experimentally (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). Potential sites of phosphorylation are predicted for S69, S150, S238, S249, S260, S262, S265, S282, S285 on cytoplasmic loops, and S390, S393, S394, and S397 on the cytoplasmic, C-terminal domain. There are also potential GRK phosphorylation sites predicted for T382, S384, S389, and S400 (http://gps.biocuckoo.cn/, Xue et al. 2011).
Expression and Characterization
The EP1 receptor is primarily expressed in the kidney and spleen, but is also found in the gastrointestinal tract, lung, smooth muscle, and the central nervous system (http://www.proteinatlas.org, Uhlén et al. (2015)).
Ligand binding properties for recombinant human EP1 (Abramovitz et al. 2000; Sharif and Davis 2002; Ungrin et al. 2001) and recombinant mouse EP1 (Kiriyama et al. 1997) have been characterized in HEK 293 and Chinese hamster ovary (CHO) cell lines respectively (Table 4). Ligand efficacies for human EP1 were determined by the increase in intracellular [Ca2+] accompanying agonist binding (Table 5). The data show that the EP1 receptor exhibits the weakest PGE2 binding of all EP receptors, but by most reports has efficacy similar to other EP receptors. Interestingly, although the binding of alternate ligands PGE1 and PGF2α are 10 and 40-fold weaker than PGE2 respectively, the differences in efficacies are much smaller. The most frequently used selective agonists for EP1 are ONO-DI-004 and 17-phenyl PGE2 (Sugimoto and Narumiya 2007; Dey et al. 2006). There is one commonly used selective antagonist, ONO-8713 (Sugimoto and Narumiya 2007) (Table 2).
EP1 has been found to form heterodimers or higher order oligomers with the β2-androgenic receptor (β2AR) airway smooth muscle (ASM) cells and when both are transfected into COS-7 cells. Although EP1 stimulation alone does not appear to have any significant effect on ASM cells itself, in the presence of β2AR the heterodimeric complex forms and stimulation of EP1 serves to alter the β2AR structure leading to a reduction in binding of β2AR to its Gαs protein, resulting in a reduction β3AR-mediated ASM relaxation (McGraw et al. 2006).
Mechanism of Cell Activation
Signal transduction from the EP1 receptor has been controversial with respect to the particular G-proteins involved and the mechanisms that lead to the observed increase in intracellular calcium (Sugimoto and Narumiya 2007; Tsuboi et al. 2002). It is now generally accepted that EP1 interacts with Gαq (Table 3) which in turn activates phosphoinositol-phospholipase C (PI-PLC) causing the release of IP3 intracellularly, and a subsequent increase in intracellular [Ca2+] (Tang et al. 2005; Markovič et al. 2017). Ji et al. (2010) have not only confirmed the interaction of Gαq/11 with EP1 for recombinant human EP1 in a HEK cell line, but have also shown that EP1 can couple to Gαi/o and activate the PI3K/protein kinase B (PKB or AKT)/mTOR kinase signaling pathway in the same experimental system.
Regulation
The regulation of the EP1 receptor is facilitated by several different mechanisms. Nasrallah et al. (2015) examined the expression of EP1 in MCT cells and found this receptor is constitutively expressed and the expression is enhanced following a 24 hr incubation with either PGE2 or transforming growth factor beta (TGFβ). They found that PGE2 alone and TGFβ alone increase expression 2.5- and 3.8-fold respectively and combined they increased EP1 expression 7-fold. Further, utilizing MCT cells transfected with EP1 siRNA the effect of PGE2 on EP1 expression is reduced by 50%, suggesting that the effect of PGE2 is on the transcriptional level.
Expression of EP1 can also be altered under conditions of low oxygen. Under hypoxic conditions of 5% and 2% O2, Lee et al. (2007) found that PGE2 release and EP1 receptor expression is strongly elevated when compared to 21% O2 conditions. It is not known if the increase in EP1 expression results from a localized increase in PGE2 levels as described above or is due to another mechanism.
The expression of EP1 is regulated by co-expressed proteins as well. Sood et al. (2014) have found that overexpression of COX-2 increases the membrane expression of EP1 and does so non-transcriptionally. This mechanism provides a feedback loop to resolve inflammation, as COX-2 expression is down-regulated by EP1 stimulation of COX-2 degradation.
EP2 receptor
Introduction
The human prostaglandin E2 receptor EP2 (PTGER2, UniprotKB-P43116) is translated as a 358 amino acid polypeptide with a calculated molecular weight of 39.8 kDa. There are no additional reported isoforms and two coding SNP variants (C83G and Y285C) (https://www.ncbi.nlm.nih.gov/clinvar/, Landrum et al. 2016), both of which appear to be benign. There are no reported X-ray structures. There are extracellular N-linked glycosylations predicted for N3 and N6 on the N-terminal and N96 and N287 on extracellular loops (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004). There are also O-linked sites predicted for S237 on a cytoplasmic loop and S5 extracellular N-terminal domain (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013). None of these specific glycosylations are confirmed experimentally, however, western blot analysis of HaCaT and COS-7 cells reveal three EP2 reactive bands at 30 kDa, 43-45 kDa, and 51 kDa (Konger et al. 2002), consistent with multiple glycoforms.
There are numerous potential phosphorylation sites on the human EP2 based on motifs but none are specifically confirmed experimentally. Phosphorylation is predicted for S8 and S10 on the extracellular N-terminal domain, S291 on an extracellular loop, S62, S63, S149, S151, S229, S232, S240, and S255 on cytoplasmic loops, and T344 and S353 on the cytoplasmic, C-terminal domain (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). There are also potential GRK phosphorylation sites predicted for the C-terminal domain at S328, S335, T338, T342, S345, S347, and S350 (http://gps.biocuckoo.cn/, Xue et al. 2011).
Expression and Characterization
The EP2 receptor is widely expressed, albeit in lower abundance when compared to other receptors (http://www.proteinatlas.org, Uhlén et al. (2015)). Ligand binding properties for recombinant human EP2 (Abramovitz et al. 2000) and recombinant mouse EP2 (Kiriyama et al. 1997; Nishigaki et al. 1995) have been characterized in HEK 293 and Chinese hamster ovary (CHO) cell lines respectively (Table 4). Ligand efficacies for human and murine EP2 were determined by the increase in intracellular cAMP (Table 5). The data show that the EP2 receptor exhibits moderate PGE2 binding, similar to EP3 (see below). The efficacy is like other EP receptors in CHO cells, but exhibit much lower efficiency in COS-7 cells. The most frequently used selective agonist for EP2 is ONO-AE1-259. The most highly selective antagonist for EP2 is PF-04418948 (af Forselles et al. 2011) with greater than 10,000-fold selectivity for EP2 receptors than other prostanoid receptors. There are also several moderately selective antagonists, TG6-129, TG4-155 and TG6-10 available (Ganesh 2014).
Mechanism of Cell Activation
Signal transduction for the EP2 receptor has been shown to occur through both G-protein-dependent and G-protein-independent pathways. The G protein-dependent pathway is similar to that of DP1 (Table 3) in that it is coupled to Gαs (Hirata and Narumiya 2011), leading to activation of adenylate cyclase and the production of cAMP (Honda et al. 1993; Regan et al. 1994a) which in turn activates PKA (Chun et al. 2009; Regan 2003) and exchange proteins that are activated by cAMP (Epacs) (Sands and Palmer 2008). EP2 can also switch from signaling through Gαs to signaling through Gαq/11 in human myometrium, leading to an increase in [Ca2+] and pro-inflammatory pathways, promoting a pro-labor condition (Kandola et al. 2014). The G-protein-independent pathway involves the formation of an EP2-β-arrestin1-Src tyrosine protein-kinase complex that leads to the activation of the epidermal growth factor receptor (EGFR) which can then lead to the activation of H-Ras (protein p21), PKB and the MAP kinases ERK1/2 (Chun et al. 2009).
Regulation
The regulation of the EP2 receptor is facilitated by a number of different mechanisms. The effect of various agonists on the expression of prostanoid receptors have been presented. Perchick and Jabbour (2003) examined the effect of upstream prostanoid production on the expression of EP type receptors. Here they overexpressed transfected human cyclooxygenase 2 (COX-2) into the Ishikawa human endometrial cell line to increase the production of PGH2, the direct precursor for PGE2. They found that not only was the production of PGE2 elevated, but that EP2 and EP3 receptor expression was increased with no effect on EP1 and EP4 expression.
Agonist expression can also direct signaling pathways. Agonist-induced conformational changes in the EP2 receptor allows for phosphorylation of specific sites on the C-terminus that alter the receptor response in a phosphorylation site dependent manner (Tobin 2008). Phosphorylation may be accomplished by G-protein receptor kinases (GRK) (Ferguson 2007), protein kinase C (PKC), PKA, or PKB to name a few and the sequence specificity of the kinase determines the site or sites of modification, leading to a specific outcome (Tobin 2008). One such outcome is the binding of β-arrestin which results in the displacement of Gαs, thus halting the cAMP production pathway. Although β-arrestin binding is also known to lead to desensitization through internalization of GPCRs via a clatherin-mediated pathway, expression of recombinant EP2 in 293-EBNA cells (Invitrogen) and COS-1 cells produces little internalization (Desai et al. 2000; Penn et al. 2001). However, when co-expressed with a modified arrestin (ARR2 (R169E)), an arrestin that does not require phosphorylation to bind to GPCRs, EP2 exhibits a four-fold increase in internalization compared to cells co-expressing wild type recombinant arrestin (Penn et al. 2001). These results indicate that the failure of wild type arrestin to cause internalization of EP2 is due to the absence of specific phosphorylation and suggests that the required kinase was not active under the experimental conditions.
Agonist-induced desensitization is a common feature of GPCRs. EP2 is not readily desensitized through phosphorylation as noted above, but over time the formation of PGE2 metabolic products which have considerably longer half-lives than PGE2 and lower efficacies, accumulate to effectively compete with PGE2 for the receptor and in doing so reduce the response to PGE2 (Nishigaki et al. 1996).
Regulation of EP2 on the transcriptional level has also been observed. It is well established that treatment of macrophages with bacterial lipopolysaccharide (LPS) stimulates macrophages and leads to their releasing large amounts of PGE2. Ikegami et al. (2001) reported that LPS treatment of cultured mouse macrophages results in a transient five-fold up-regulation of EP2 mRNA at 3 hours after stimulation, which returns to previous levels by 5 hours. They showed further that no protein synthesis is required for the up-regulation and suggest that up-regulation of EP2 mRNA is a direct effect of LPS stimulation. They also observed that changing media promotes up-regulation of EP2 mRNA, but to a lesser degree than observed for LPS and suggest that induction factors in the media may be responsible. Kashmiry et al. (2018) reported that up-regulation of EP2 receptor mRNA in human monocytes and THP-1 cells following LPS stimulation is controlled by the PGE2 stimulation of the EP4 receptor.
Steroid hormone regulation of the transcription of a large number of genes is well established. Lim and Dey (1997) examined EP2 mRNA expression in the mouse preimplantation uterus as a function of experimentally elevated progesterone (P4) and 17β-estradiol (E2). They found that P4 up-regulates EP2 mRNA and the presence of E2 enhances this effect, while E2 alone down-regulates EP2 mRNA and suggest that the synergy between P4 and E2 on EP2 expression is essential in preparing the uterus for implantation.
EP3 receptor
Introduction
The human prostaglandin E2 receptor EP3 (PTGER3, UniprotKB-P43115) is translated as a 390 amino acid polypeptide with a calculated molecular weight of 43.3 kDa. There are 14 reported isoforms and 3 coding SNP variants (N366S, T319M, M169L) (https://genecards.org, Stelzer et al. 2016) of the canonical EP3A sequence, none of which have any reported effect on function. Each of the isoforms is a splice variant of the C-terminal tail and all are identical for the first 359 residues but differ in G-protein interactions and signaling. There are two reported X-ray structures (PDB entry 6AK3 and 6M9T). Extracellular N-linked glycosylation are predicted for N18 and N36 on the N-terminal and with lower confidence N217 and N308 on extracellular loops (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004). There are O-linked sites predicted with high confidence for S20, T22, and S30 on the extracellular N-terminal domain and S369, S371, S373, and S380 on the intracellular C-terminal tail (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013). None of these specific glycosylations are confirmed experimentally, however, site-specific mutagenesis studies of rat EP3 (UniProtKB-P34980) confirm N-glycosylation of N16 and N194 (Böer et al. 2000) which correspond to N18 and N217 in human EP3. Further, the glycosylation for the rat analog is required for correct sorting to the plasma membrane, but not for correct folding of EP3.
There are numerous potential phosphorylation sites on the human EP3 based on motifs but none are specifically confirmed experimentally (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). For all known isoforms, residues 1-359 are identical. High probability phosphorylation sites are predicted for all isoforms at S43 on the extracellular N-terminal domain, and S82, S258 and S270 on cytoplasmic loops. There are also multiple potential GRK phosphorylation sites on the C-terminal domain for all but isoforms EP3-III and EP3-IV (http://gps.biocuckoo.cn/, Xue et al. 2011).
Expression and Characterization
The EP3 receptor is widely expressed in low abundance, but is highly expressed in smooth muscle, kidney, endometrium, and in adipose tissue (http://www.proteinatlas.org, Uhlén et al. (2015)). Ligand binding properties for recombinant human EP3 isoforms have been characterized in HEK293 (Abramovitz et al. 2000), COS-1 (Kunapuli et al. 1994), COS-7 (Kotani et al. 1995; Regan et al. 1994b) and COS-M6 (Adam et al. 1994) cell lines (Table 4). Ligand efficacies for human recombinant EP3 isoforms were determined in CHO cells by the increase in intracellular [Ca2+] (Sekido et al. 2016) and guinea pig vas deferens smooth muscle by changes in twitch contraction values (Savage et al. 1993) (Table 5).
The data show that the EP3 receptor on the average exhibits PGE2 binding that is greater than most EP receptors, but slightly less than observed for EP4. The average efficacy is similar to other EP receptors. The most selective agonists for EP3 in general are SC-46275, ONO-AE-248, MB28767 and sulprostone (Norel et al. 2004; Savage et al. 1993; Kotani et al. 2000; Jin et al. 1997; Abramovitz et al. 2000) (Table 2). There are two EP3 specific antagonists available, DG-041 and ONO-AE3-240 (Abramovitz et al. 2000) (Table 2).
Mechanism of Cell Activation
Signal transduction by the EP3 receptor is dependent on the isoform present (Table 3). As noted above, there are at least 14 different isoforms, each of which differs only in the composition and length of the C-terminal tail from residue 360 onward (Kotani et al. 1995; Kotani et al. 1997). These differences determine which G-proteins bind and hence determine the signal pathway actuated.
EP3-I (UniprotKB isoform EP3A, identifier P4535115-1) the canonical isoform is coupled to two secondary messenger systems, involving inhibition of cAMP and stimulation of phosphoinositide turnover that is consistent with coupling to Gαi and Gαq proteins respectively (Kotani et al. 1995).
EP3-II (UniprotKB isoform EP3C, identifier P4535115-2) is functionally equivalent to bovine EP3D which is known to be coupled to Gαi, Gαs, and Gαq (Kotani et al. 1995; Kotani et al. 1997) that initiate cAMP repression, an increase cAMP production, and an increase in both inositol 1,4,5-trisphosphate (IP3) and intracellular [Ca2+] respectively.
EP3-III (UniprotKB isoform EP3B, identifier P4535115-3) is coupled to the inhibition of cAMP production (Regan et al. 1994b) with an increase in intracellular [Ca2+] (Schmid et al. 1995), does not stimulate IP3 production (Kotani et al. 1995), and its activity is inhibited by the presence of pertussis toxin, a known inhibitor of Gαi, all of which clearly indicate that it couples to Gαi (Jin et al. 1997).
EP3-IV (UniprotKB isoform EP3D, identifier P4535115-4) is coupled to both stimulation of cAMP production and reduction in cAMP accumulation with no reports of IP3 generation (Kotani et al. 1995). The activity of this receptor is also inhibited by the presence of pertussin toxin, indicating that the reduction of cAMP is coupled through Gαi (Jin et al. 1997). The production of cAMP is consistent with coupling to Gαs.
EP3-V (UniprotKB isoform EP3E, identifier P4535115-5) is coupled to the inhibition of cAMP production. This fact and the loss of activity in the presence of pertussis toxin indicates a coupling to Gαi (Kotani et al. 2000). Associated changes in intracellular [Ca2+] have not been reported.
EP3-VI (UniprotKB isoform EP3F, identifier P4535115-6) like EP3-IV is also coupled to both stimulation of cAMP production and reduction in cAMP accumulation with no reports of IP3 generation (Kotani et al. 2000). Observed inhibition of cAMP reduction with pertussis toxin and stimulation of cAMP production with the EP3-specific agonist M&B28767 indicate coupling to Gαi and Gαs respectively.
There are eight additional EP3 isoforms reported in the literature of which only EP3.e and EP3.f have been partially characterized as reducing [cAMP] upon PGE2 activation (Schmid et al. 1995). Interestingly sequences for these two are not listed in UniProtKB, however, P435115-11 and P435115-10 have identical sequences to EP3.e and EP3.f respectively with the exception of an additional exon sequence VANAVSSCSNDGQKGQPISLSNEIIQTEA (360-388) to the N-terminal side of the defining C-terminal sequence.
Regulation
Regulation by receptor internalization is isoform dependent. In HEK293 cells expressing human recombinant EP3-I, EP3-II , EP3-III EP3-IV, EP3-V, EP3.e, and EP3.f, all receptors are found to be located primarily on the cell surface under non-stimulating conditions (Bilson et al. 2004). Isoforms EP3-III, EP3-IV, EP3-V, EP3.f are also observed intracellularly. When stimulated with PGE2, isoform EP3-I translocate robustly to intracellular punctate vesicles along with β-arrestin. EP3.f behaves similarily to EP3-I but is internalized to a lesser extent. Isoforms EP3-II, EP3-V, EP3-VI internalize upon stimulation to a lesser extent than EP3-I and this is not accompanied by β-arrestin migration into the vesicles. Isoforms EP3-III and EP3-IV do not internalize in response to PGE2 binding. This is expected, as the C-terminal tails of these isoforms contain no Ser of Thr residues, and thus the phosphorylation required for β-arrestin facilitated internalization cannot occur.
Receptor response to agonist activation of EP3 isoforms I-IV has been examined in detail in terms of inhibition of cAMP production (Jin et al. 1997). Isoforms EP3-I and EP3-II exhibit typical agonist concentration-dependent behavior whereas EP3-III exhibits full constitutive activity and EP3-IV exhibits partial constitutive activity. In contrast, agonist response for both the EP3-III and EP3-IV receptors in terms of increased intracellular [Ca2+] is agonist dependent (Regan et al. 1994b; Schmid et al. 1995; Sekido et al. 2016).
Regulation of EP3 on the transcriptional level has been reported without reference to specific isoforms. Human leukemic T cells of the HSB.2 T cell line express EP2, EP3 and EP4 receptor subtypes. Exposure of these cells to the T cell mitogen concanavalin A (Con A) increases interleukin 6 (IL-6) secretion in response to PGE2 while at the same time downregulates the expression of EP3 mRNA (Zeng et al. 1998). In contrast, forced overexpression of the ERG transcription factor in immortalized PCA cell line DU145 results in the upregulation of EP3 while simultaneously increasing IL-6 secretion (Merz et al. 2016).
EP4 receptor
Introduction
The human prostaglandin E4 receptor EP4 (PTGER4, UniprotKB-P35408) is translated as a 488 amino acid polypeptide with a calculated molecular weight of 53.1 kDa. There are no reported isoforms and one reported coding SNP variant (V294I) which is not reported to have any effect on function (https://www.ncbi.nlm.nih.gov/clinvar/, Landrum et al. 2016). There are two reported X-ray structures (PDB 5YHL and 5YWY). Extracellular N-linked glycosylation is predicted with high confidence for N7 and two with lower confidence, N177 on an extracellular loop and N482 on the intracellular C-terminal domain (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004). There are also numerous O-linked sites predicted for both the N- and C-terminal domains as well as on one extracellular loop (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013). None of these specific glycosylations are confirmed experimentally. There are five known phosphorylation sites on the intracellular C-terminal domain, S370, S374, S377, S379, and S382 found in various combinations that are involved in to β-arrestin binding and internalization (Neuschäfer-Rube et al. 2004). There are additional potential serine phosphorylation sites predicted for S11, S13, S19 on the extracellular N-terminal, S222, S252, S259 on a cytoplasmic loop, and S371, S437, S440, S442, S443, S448, S460, and S480 on the C-terminal domain (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). Additional potential GRK phosphorylation sites are predicted for S366, S442, S443, S450, and S484 with high probability (http://gps.biocuckoo.cn/, Xue et al. 2011). None of these phosphorylations have been confirmed experimentally.
Expression and Characterization
The EP4 receptor is widely expressed with the highest amounts in smooth muscle, and the immune system (http://www.proteinatlas.org, Uhlén et al. (2015)). Ligand binding properties for recombinant human EP4 have been characterized in HEK293 cell lines (Abramovitz et al. 2000; Sharif et al. 2003) (Table 4). Ligand efficacies for human recombinant EP4 were determined in CHO cells by the increase in intracellular cAMP obtained by enzyme immunoassay (Sharif et al. 2003) and response to stimulated short circuit current (SSC) and slope conductance (SG) in tissue from human duodenal biopsies (Larsen et al. 2005) (Table 5). The data show that the EP4 receptor exhibits the tightest PGE2 binding of all EP receptors, however, the average efficacy is significantly less than observed for other EP receptors. The most selective agonists for EP4 are ONO-AE1-329 and tetrazolo PGE1, (Sugimoto and Narumiya 2007; Jones et al. 2009) and the most selective antagonists are ONO-AE3-208 and CJ-042794 (Sugimoto and Narumiya 2007; Jones et al. 2009) (Table 2).
Mechanism of Cell Activation
Similar to EP2, signal transduction for the EP4 receptor has been shown to occur through both G-protein-dependent (Table 3) and G-protein-independent pathways. G protein-dependent coupling to Gαs (Nishigaki et al. 1996), leads to activation of adenylate cyclase and the production of cAMP which in turn activates PKA (Regan 2003) that phosphorylates downstream proteins, including the cAMP response element binding protein (CREBP) (Takayama et al. 2002). EP4 has also been shown to switch from signaling through Gαs to signaling through a cAMP independent and PI3K- dependent mechanism, that leads to phosphorylation of extracellular signal-related kinases (ERKs), which in turn leads to the expression of early growth response factor-1 (EGR-1) that is not observed in EP2 signaling (Fujino et al. 2003; Fujino and Regan 2006). PI3K-dependent signaling may also result in phosphorylation of PKB which in turn regulates cell survival through protection from apoptotic stimuli (George et al. 2007). Similarly, EP4 stimulation also activates the T-cell-factor (Tcf)/lymphoid enhancer factor (Lef), known to be activated by the Gαq-coupled FPB prostanoid receptor through a PI3K-dependent signaling pathway (Fujino et al. 2002). EP4 signaling can also occur through coupling with Gαi/o, resulting reduction of intracellular cAMP via inhibition of adenylate cyclase. Leduc et al. (2009) examined the differential coupling of EP4 to various G-proteins and found that selectivity was dependent on the identity of the agonist binding to the receptor. They also found that PGE2 was the most selective for Gαs signaling whereas PGF2 and PGE1-OH were more selective for Gαi/o signaling and β-arrestin binding.
Regulation
As observed for EP2, Nishigaki et al. (1996) found that EP4 is desensitized through agonist-induced GRK-mediated phosphorylation. However, the two receptors differ in sensitivity to agonist where EP4 is more rapidly desensitized than EP2 and is much less responsive to PGE2 metabolic products than EP2, suggesting that EP4 is involved in a short-term and rapid response while EP2 is involved in a more long-lasting response. Similar to EP2, desensitization of EP4 leads to replacement of Gαs with a β-arrestin isoform that leads to internalization into punctate vesicles. However, the phosphorylation state of EP4 is not as critical to the internalization as it is for EP2 (Penn et al. 2001). In fact, Desai et al. (2000) have shown that mutant EP4 receptors with Ala substituted for each and every Ser and Thr in the C-terminal domain internalized to the same extent as wild type EP4 (Desai et al. 2000) indicating that phosphorylation is unnecessary for internalization. However the same study showed that EP4 mutants with C-terminal tails truncated before residue 383 had a reduced internalization and those truncated at the proximal end of the C-terminus (residue 350) did not internalize at all, indicating that the presence of the C-terminal tail is necessary for internalization.
Regulation of EP4 on the transcriptional level has also been observed in mouse macrophages. As noted above for EP2, LPS treatment of cultured macrophages results in changes in EP receptor mRNA expression. However, the mechanisms and results for EP4 are quite different. In contrast to the LPS-induced upregulation of EP2, Ikegami et al. (2001) report that EP4 is down-regulated within 3 hours of treatment and the EP4 mRNA expression levels drop to less than 10% of the control levels. The mechanism apparently involves the LPS-induced over-production of PGE2 which in turn mediates EP4 stimulated cAMP production that results in a reduction of gene expression. The likely regulatory mechanism under inflammatory conditions would involve an interaction of cAMP-induced CREBP and NFκB transcription factors with the EP4 gene.
In human monocytes the transcriptional regulation is quite different (Kashmiry et al. 2018). Here, LPS treatment alone results in a nearly three-fold increase in EP4 mRNA rather than the decrease observed in the mouse study and in the presence of added PGE2 the increase is significantly lower at 25% above control.
In human glioblastomas EP4 transcription is regulated in part by the specificity protein 1 (Sp-1) transcription factor (Kambe et al. 2008). Phosphorylation of Sp-1 reduces its ability to bind to DNA which in turn leads to the suppression of EP4 transcription. Phosphorylation of Sp-1 is known to be facilitated by several kinases including ERK. The fact that ERKs are known to be activated by EP4 signaling (Fujino et al. 2003) suggests that this mechanism may represent a negative feedback loop.
IP receptor
Introduction
The human prostacyclin (PGI2) receptor hIP (PTGIR, UniprotKB-P43119) is translated as a 386 amino acid polypeptide with a calculated molecular weight of 41.0 kDa. No X-ray structures have been reported. There are four other reported isoforms of which three represent structures utilizing fewer than the total exons found in the canonical sequence and one with an additional exon not found in the canonical form. All are predicted to have a least one transmembrane helix and the biological functions are currently unknown. There are 18 reported SNP variants, eight of which exhibit various degrees of biological dysfunction and represent less than 2% (n = 1,761) of the hIP receptors examined (Stitham et al. 2002; Stitham et al. 2011). One N-linked extracellular glycosylation is predicted with high confidence for N7 and with lower confidence for N78 and N203 (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004) and one high confidence cytosolic O-linked predicted for S337 (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013). Only glycosylation at N7 and N78 have been experimentally confirmed (Zhang et al. 2001). Although specific glycosylation of other residues has not been confirmed experimentally, glycosylation at multiple unknown sites have been reported (Miggin et al. 2002; Donnellan and Kinsella 2009) and glycosylation of the C-terminal region, possibly S337, may be involved in membrane localization (Smyth et al. 1998). One prenylation site is predicted by PrePS ((http://mendel.imp.ac.at/sat/PrePS/index.html, Maurer-Stroh et al. 2007) for C383, and confirmed experimentally (Miggin et al. 2002). Palmitoylation was originally reported for C308 and C311, however, the publication has since been retracted (Miggin et al. 2003). Predictions for palmitoylation with CSS-Palm (http://csspalm.biocuckoo.org/, Zhou et al. 2006) reveal high confidence predictions at C5, C308, C309, and C-311 with the highest for C5. However, C5-C165 and C92-C170 disulfides have been confirmed experimentally, potentially ruling out a C5 palmitoylation (Giguère et al. 2004). Further, it has been shown that formation of these disulfides is required for expression and may also be involved in dimerization and oligomerization of the receptor. There are potential serine phosphorylation sites predicted for S14 on the extracellular N-terminal, S268 and S269 on an extracellular loop, T230 on a cytoplasmic loop, and S328, S337, and S374 on the C-terminal domain (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). Only phosphorylation at S328 has been confirmed experimentally (Smyth et al. 1998) and shown to be accomplished by PKC (Smyth et al. 1998; Smyth et al. 1996). There are 11 potential high probability GRK phosphorylation sites predicted but as yet not experimentally confirmed (http://gps.biocuckoo.cn/, Xue et al. 2011).
Expression and Characterization
The human IP receptor (hIP) is widely expressed with the highest amounts in lung, muscle, female tissues, and the immune system (http://www.proteinatlas.org, Uhlén et al. (2015)). Ligand binding properties for hIP have been characterized in platelet membranes by displacement of [3H]iloprost, an IP-specific agonist (Jones et al. 2009; Siegl et al. 1979; Tsai et al. 1989; Crider et al. 2001) (Tables 2 and 6) instead of PGI2 due to the short half-life of the latter. Ligand efficacies for hIP have been determined in platelet membranes (Fitscha et al. 1987; Stürzebecher and Losert 1987; Kobzar et al. 2001), vascular smooth muscle (Angulo et al. 2002; Hadházy et al. 1986; Baxter et al. 1995, lymphocytes (Kilfeather et al. 1984), and recombinant human IP expressed in COS-1 (Crider et al. 2001) and MEG-01 (Tunaru et al. 2016) cells (Table 6). Although other PG receptors are present in the human tissue samples, the EC50 values clearly indicate that any cross-reactivity of PGI2 with the other receptors would result in negligible response. The most selective agonists for IP are cicaprost, carbacyclin, and iloprost, the former two being slightly more potent than the latter (Jones et al. 2009) (Table 2). Interestingly, the eicosanoid 19(S)-hydroxy-eicosatetraenoic acid (19(S)-HETE) also serves as a moderately potent hIP agonist and stimulates the production of cAMP (Tunaru et al. 2016). The most selective antagonists are RO-1138452 and RO-3244794 (Jones et al. 2009) (Table 2).
hIP receptors have the ability to form homodimers, homooligomers and heterodimers with other GPCRs. Homodimerization and oligomerization occurs through disulfide linkages, specifically C5-C165 and C92-C170, and monomers, dimers and oligomers are expressed on the cell surface in well-defined ratios (Giguère et al. 2004). Furthermore, the polymerization process occurs intracellularly and is independent of agonist stimulation. Whether polymerization is a requirement for initial cell surface expression is currently unknown. Heterodimers of hIP and the human thromboxane receptor hTPα have been reported by Wilson et al. (2004). Expression of both receptors in HEK293 and human aortic smooth muscle cells (hAMSC) were demonstrated and when activated with a TPα agonist, rather than the expected increase in IP3 and intracellular [Ca2+] normally associated with TPα stimulation, an intracellular cAMP increase is observed as would be expected for the IP receptor. Clearly the G-protein specificity of the heterodimer is defined by the IP receptor subunit.
Mechanism of Cell Activation
Similar to EP2 and EP4 receptors, signal transduction for the hIP receptor occurs through both Gαs and Gαq protein pathways (Table 3). Iloprost initiates cAMP production (EC50 = 0.1 ± 0.03 nM) and is favored over IP3 production (EC50 = 43.1 ± 10 nM) in HEK293 cells expressing recombinant hIP, resulting in the activation of PKA and PKC and their associated pathways respectively (Smyth et al. 1996). Further, basal receptor phosphorylation rapidly occurs and does so only when agonist concentrations exceeds 1 nM, well above that required for cAMP production, indicating that the PKC and not the PKA pathway is responsible. Phosphorylation assays in the presence of PKA- and PKC-specific inhibitors confirm these observations. Although not specifically proven, these results strongly suggest that phosphorylation alters the G-protein specificities and hence the preferred pathway (Smyth et al. 1998).
hIP signal transduction in preadipocytes is somewhat different (Vassaux et al. 1992). Both systems produce cAMP at low concentrations of IP agonist, carbaprostacyclin (cPGI2) in the case preadipocytes. However, in preadipocytes, the cPGI2 also produces a transient increase in intracellular [Ca2+] that does not depend on cAMP production, extracellular [Ca2+] or calcium channel blockers, and is independent of IP3 production. The pathway for the increase in [Ca2+] is not currently known, but the latter observations suggest that the mechanism for this increase differs from the IP3-independent pathway suggested for DP1.
Regulation
Zhang et al. (2001) show that the regulation of expressed hIP is modulated by the glycosylation state of the hIP receptor. They found that the extent of glycosylation at both N7 and N78 and in particular the glycosylation of N78 is paramount to the efficient binding of receptor agonist. Lack of glycosylation at N7 itself has little effect (≤ 10%) on iloprost binding. However, lack of glycosylation at N78 increases the iloprost Kd seven-fold and lack of glycosylation at both N7 and N78 increases the Kd to undetectable levels, effectively inhibiting binding. Glycosylation also has an effect on signaling efficiency. Similar to the pattern observed for the binding efficiency, lack of glycosylation at N7 reduces cAMP production efficiency by increasing the EC50 value nearly 10-fold, lack of glycosylation at N78 increases EC50 over a 1000 fold, and lack of glycosylation at both sites eliminates cAMP signaling altogether. Interestingly, although the wild type receptor exhibits activation of phospholipase C (PLC) and IP3 production, deglycosylation at either N7 or N78 eliminates this signal pathway.
The isoprenylation state of C383 also has a direct effect on hIP signaling (Miggin et al. 2002). Although the isoprenylation state does not alter iloprost binding, isoprenylation is required for efficient binding of both Gαs and Gαq proteins and for this reason is required for competent signaling through both the cAMP and IP3 pathways.
Regulation of hIP also occurs through independent pathways that are agonist stimulated and typically involve phosphorylation of the C-terminal as well. As noted above, elevated concentrations of the hIP agonist iloprost stimulate the activation of PKC, resulting in the phosphorylation of the hIP receptor and the production of IP3 (Smyth et al. 1996). Later work reveals that the PKC release results in the phosphorylation of S328 and that this event reduces the production of cAMP to effectively basal levels while increasing the production of IP3 2-3 fold (Smyth et al. 1998). These results strongly suggest that this phosphorylation alters the G-protein affinity from Gαs to another G-protein, presumably Gαq. It should be noted that phosphorylation at S328 does not account for all agonist-induced receptor phosphorylation and the function of these phosphorylation sites is currently unknown.
G-protein preferences also depend on interactions with other regions of the receptor. Chimeric constructs of IP and TP receptors reveal that the 116-134 intracellular loop of the IP (intracellular loop 2) receptor is necessary for Gαs-mediated cAMP production and that deletion of the YLYAQ sequence from this region not only drastically reduces cAMP production, but also enhances the intracellular [Ca2+] through a Gαq signaling pathway (Chakraborty et al. 2013). Further, the intracellular loop 209-235 (intracellular loop 3) is involved in both Gαs and Gαq binding.
Sequestration of hIP through internalization is another known pathway for regulation (Giovanazzi et al. 1997). Although internalization of other PG receptors is mediated by GRK-mediated phosphorylation and β-arrestin binding, hIP receptor internalization has been shown to be independent of GRK-2, -3, -5, -6 and arrestin-2 (Smyth et al. 2000). Further, iloprost-induced internalization requires agonist concentrations (EC50 = 27.6 ± 5.7 nM) similar to that required for PKC-induced phosphorylation of S328 but is independent of this phosphorylation event. Sequestration of hIP proceeds through a dynamin-dependent clathrin-coated vesicular endocytotic pathway and a dynamin-independent pathway (Smyth et al. 2000). One mechanism explaining the dynamin-dependent hIP internalization pathway involves the Rab5a protein, a Ras-like GTPase (O'Keeffe et al. 2008). Upon binding GTP, the now active Rab5a binds to agonist (circaprost)-bound hIP and in concert with dynamin, internalizes the Rab5a-receptor complex into punctate endocytotic vesicles until 30-40% of hIP is internalized. The internalization is clearly directed by Rab5a, as over co-expression of Rab5a with hIP significantly increases internalization. Rab5a is released upon hydrolysis of GTP and the hIP receptor remains internalized for several hours post-stimulation. By 4 hours after stimulation, 50% of the sequestered receptor returns to the surface. Upon prolonged stimulation (4-8 h), 50% of the internalized hIP fails to return to the surface and thus likely undergoes degradation through trafficking to lysosomes. Interestingly, the C-terminal tail, directly involved in other PG receptor internalizations, is not involved in facilitating Rab5a-mediated endocytosis itself, but instead appeared to play a role in hIP sorting following the Rab5a-mediated endocytosis. Additional mechanisms have been presented showing the involvement of the related Rab11a and Rab4a proteins (Wikström et al. 2008). hIP has been shown to localize to both Rab11a- and Rab4a-positive vesicles in response to cicaprost binding. As observed for Rab5a, Rab11a also interacts directly with hIP, however, the C-terminal tail of hIP is required for interaction with Rab11a, unlike the Rab5a interaction. On the other hand, Rab4a does not directly interact with hIP, nor does it direct hIP to Rab4a positive vesicles.
Although the precise pathway for sequestration is currently unknown, it has been shown that glycosylation of the extracellular N7 and not the extracellular N78 is required for internalization (Zhang et al. 2001). Internalization is also affected by the prenylation state of C383. Farsenyl or geranylgeranyl prenylation of C383 is also required for efficient internalization of the IP receptor, possibly by allowing the insertion of the prenyl group into the membrane thus forming a fourth cytoplasmic loop that aids internalization (Miggin et al. 2002).
As noted above, hIP undergoes internalization in response to agonist stimulation and a portion of the internalized receptors are later recycled back to the membrane while some are transported to lysosomes for degradation. As reported for other proteins, including other GPCR receptors (Hanyaloglu and von Zastrow 2008), targeting to lysozomes requires prior polyubiquitination of the protein. Polyubiquitinated hIP has been observed in HEK cells and is required for transport to lysozomes, but it is not known if polyubiquitination occurs prior to internalization or post-internalization (Donnellan and Kinsella 2009) [99].
Regulation at the transcriptional level has also been reported. Turner and Kinsella (2010) report that estrogen significantly increases the transcription of hIP in primary aortic smooth muscle cells and that the increase is modulated through the binding of the ERα nuclear estrogen receptor to the hIP promotor region on the DNA itself. The ERβ nuclear receptor is apparently not involved. The authors postulate that this interaction in part explains the vasodilatory effect of estrogen as well as its anti-atherogenic effects (see also Tostes et al. 2003). Eivers and Kinsella (2016) report that androgens also confer cardioprotective effects through stimulation of hIP transcription in human erythroleukemia (HEL) cells and human endothelial cells (EA.hy926). Utilizing dihydrotestosterone (DHT) as stimulus, hIP mRNA levels were significantly increased, 3.9-fold in the case of HEL cells. Further, in combination with low serum cholesterol, DHT enhances the binding of the sterol regulatory element binding protein (SREBP1) to the hIP promoter thus increasing transcription. The mechanism by which low cholesterol also helps facilitate this process is thoroughly discussed by Heemers et al. (2006).
TP receptor
Introduction
The human thromboxane A2 (TXA2) receptor is expressed as two isoforms referred to as hTRXα and hTRXβ. hTRXα (TBXA2R, UniprotKB-P21731) is translated as a 343 amino acid polypeptide with a calculated molecular weight of 37.5 kDa whereas hTRXβ (UniprotKB-P21731-2) is translated as a 407 amino acid polypeptide with a calculated molecular weight of 44.2 kDa (Hirata et al. 1996). The two isoforms are identical for the first 328 amino acids and differ in their C-terminal tails due to differential splicing of exon 3, leading to vastly different intracellular signaling. There are numerous coding SNP variants reported (R60L, P28S, C68S, V80E, E94V, A160T, V176E, V217I, D238G, V241G, S283C, and D304N) of which several result in receptor dysfunction. R60L results in a receptor with a defective response to its natural agonist TXA2 (Hirata et al. 1994a). R60L, V241G, and D304 N result in susceptibility to platelet-type bleeding disorder 13 (https://www.ncbi.nlm.nih.gov/clinvar, Landrum et al. 2016). S283C and D238G lead to impaired platelet aggregation (https://www.ncbi.nlm.nih.gov/clinvar, Landrum et al. 2016). Both receptors are seven helix transmembrane proteins. There are no reported X-ray structures and one reported structural model for the alpha isomer (PDB reference 1LBN). Two N-linked extracellular glycosylations are predicted with high confidence for N4 and N16 (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004) and confirmed definitively for hTRXα and potentially for hTRXβ by Walsh et al. (1998). Confirmation for hTRXα was obtained through examination of N to Q site directed mutagenesis of recombinant receptor. Treatment of HEK293 cells expressing individual isomers with tunicamycin, a potent inhibitor of N-glycosylation, reduces the expression of both isomers indicating that glycosylation is not only present on both isomers but is also required for proper folding in the ER. Further, treatment of the same cell lines individually with endo-H, an enzyme that cleaves off the glycol portion of N-linked sugars, reduces the ligand binding efficiencies by approximately 50%, confirming the presence of N-linked glycosylation on both isoforms and indicating that the glycosylation is involved in ligand binding. There are also numerous high confidence cytosolic O-linked sites predicted (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013). Specific sites for these glycosylations have not been experimentally confirmed. There is potential phosphorylation predicted for both isomers, T286 on the second extracellular loop, S139 and S239 on cytoplasmic loops, and S324 on the C-terminal domain. Both isomers have additional and different phosphorylation sites predicted for the C-terminal tail (hTRXα: S329 and S331; hTRXβ: S330, S355, S360 and S404) (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). Although in vivo and in vitro phosphorylation of both receptors has been confirmed (Kinsella et al. 1994; Habib et al. 1999), not all potential sites have been confirmed experimentally. Deletion and substitution mutants have confirmed hTRXα phosphorylation at T337 (Kelley-Hickie and Kinsella 2004) and S329 (Walsh et al. 2000a) and hTRXβ phosphorylation at S145, S239, S357 (Kelley-Hickie and Kinsella 2006), and T399 (Kelley-Hickie and Kinsella 2004). Numerous additional GRK phosphorylations are also predicted but not experimentally confirmed (http://gps.biocuckoo.cn/, Xue et al. 2011).
Expression and Characterization
The human TP receptor (hTP) is widely expressed with the highest amounts in the endocrine system, female tissues, and the immune system (http://www.proteinatlas.org, Uhlén et al. (2015)). Most tissues express both isoforms with hTRXα predominating over hTRXβ. Levels of hTRXα mRNA expression are relatively constant throughout most tissue types while mRNA levels of hTRXβ vary significantly (Miggin and Kinsella 1998).
Many ligand binding and efficacy studies do not specify which particular isoform was under study, albeit hTRXα is the likely candidate. Further, tissue studies of TP receptors would be sampling both isoforms simultaneously. For such studies hTP is used to designate either or both human isoforms. Fortunately, the ligand binding properties of the two isoforms were found to be identical (Hirata et al. 1996) and thus differentiation between isoforms is unnecessary for ligand binding studies (Table 7). Efficacy studies are a bit more complex, as these receptors can couple to an array of G-proteins and in a isoform-dependent manner for which the cellular response may differ. Ligand binding properties for hTP have been characterized with recombinant receptor in HEK293 (Abramovitz et al. 2000; Capra et al. 2013) and COS-m6 (Hirata et al. 1996) cell lines as well as in human platelet membranes (Modesti et al. 1989; Miki et al. 1992; Armstrong et al. 1993; Dorn 1989) by displacement of synthetic radiolabeled agonists (Table 7). Ligand binding to recombinant murine TP expressed in CHO cells (Sawyer et al. 2002; Kiriyama et al. 1997) has also been examined. Ligand efficacies for hTP have been determined using recombinant receptor in COS-m6 (Hirata et al. 1996), CHO (Hirata et al. 1996, 1994b), and HEK293 cell lines (Capra et al. 2013) as well as in human hand vein (Arner et al. 1991), lung (McKenniff et al. 1988) tissues and platelets (Dorn 1989, 1991; Mayeux et al. 1988; Tymkewycz et al. 1991; Ushikubi et al. 1989b), all with synthetic agonists (Table 7). TXA2 cannot be used as an agonist itself, as it is an unstable AA metabolite with a half-life of about 30 seconds (Ricciotti and FitzGerald 2011). PGH2 can also bind and activate TP receptors (Gluais et al. 2005), but its short in vitro half-life of 5 min (Yu et al. 2011) severely limits its usefulness in such studies. Conversion of PGH2 to multiple prostaglandins products in vivo shortens the half-life even further. The most commonly used selective TP agonists are U-46619, I-BOP, and STA2 (Abramovitz et al. 2000; Hirata et al. 1996) and the most commonly used selective TP antgonists are SQ29548 and S-145 (Wright et al. 1998; Ushikubi et al. 1989b). Ah-23848, Ramatroban, and Vaproprost have also been used (Jones et al. 2009) (Table 2).
Mechanism of Cell Activation
The TP receptor is rather promiscuous in its G-protein coupling and thus the ultimate effect of activating the TP receptor is dependent on the availability of particular G-proteins which in turn is cell type and tissue dependent (Table 3). Early work characterized hTP receptor in platelets as coupling with Gαq (Shenker et al. 1991), known to stimulate IP3 and diacylglycerol (DAG) production via PLC-β and a subsequent increase in intracellular [Ca2+] and PKC. Later work with recombinant proteins expressed in HEK293 cells reveals that two specific members of the Gαq family, Gα11 and Gα16, couple to hTP (Kinsella et al. 1997; Walsh et al. 2000b). Although both hTP isoforms were shown to couple through the Gαq family, only hTRXβ also coupled to Gαi, which leads to a decrease in cAMP production by inhibiting adenylate cyclase, whilst hTRXα appears to also couple with Gαs, as stimulation with the I-BOP agonist leads to an increase in cAMP production (Hirata et al. 1996). The interaction between hTP and the specific G-protein appears to be regulated by different domains of the receptor where residue R60 of hTP is involved in the binding of Gαq and Gαs, but not Gαi (Hirata et al. 1996; Hirata et al. 1994b). Both hTRXα and hTRXβ couple to Gα12, and quite likely to Gα13 as well (Offermanns et al. 1994; Becker et al. 1999), and upon stimulation, intracellular [Ca2+] increases in an IP3/DAG independent manner (Walsh et al. 2000b). Apparently, Gα12 couples both hTP isoforms to verapamil-sensitive, L-type Ca2+ channels. The hTRXα receptor couples to Gα13, leading to an increase in Na+/H+ exchange. Agonist binding to the alpha isoform is enhanced when coupled to Gα13 (Becker et al. 1999). Activation of Gα12 or Gα13 also leads to activation of guanine nucleotide exchange factor (GEF) for Rho, p115RhoGEF, that initiates Rho-mediated signaling (Kozasa et al. 1998). Both hTP isoforms also bind to the high molecular weight (70 kDa) Gh protein (Vezza et al. 1999). Upon stimulation, the hTRXα-Gh complex enhances IP3/DAG production via activation of PLC-d1 and ultimately an increase in intracellular [Ca2+] and active PKC (Feng et al. 1996). The hTRXβ-Gαh complex does not activate IP3/DAG production and its biological function has yet to be determined. Interestingly, Gαh also functions as a tissue transglutaminase that is inhibited by GTP binding, suggesting that the β-isoform may be involved in regulation of the transglutaminase function.
Regulation
Regulation of expressed hTP is modulated by the glycosylation state of the receptor by multiple mechanisms. Walsh et al. (1998) reported that reduction of N-glycosylation of hTRXα through the use of N to Q mutants reveals that loss of N4, N16 or both glycosylations results in a reduction of agonist binding by 53%, 42% and 92% respectively. The reductions are caused by a reduction in maximal binding (Bmax) and not by changes in affinity. The reduction in Bmax is not a function of membrane expression, as the expression of mutants lacking either N4 or N16 glycosylation are the same as wild type. In addition, the membrane expression of the double mutant is reduced by 45% compared to wild type. As would be expected, N-glycosylation also affects the second messenger signaling. Intracellular [Ca2+] mobilization by hTRXα for which glycosylation is removed from N4, N16 or both showed reductions in [Ca2+] of 22%, 11%, and 58% respectively, significantly less than the reduction in ligand binding. Intracellular cAMP production is likewise reduced in the absence of glycosylation at N4, N16 or both where reductions of 12%, 25%, and 58% respectively were observed. Similar results would be expected for hTRXβ, with the exception of cAMP production, as the structures are identical with the exception of the cytosolic C-terminal tail. This expectation is supported by the observation by Walsh et al. (1998) that tunicamycin, a potent inhibitor of N-linked glycosylation, treatment of HEK293 cell lines individually expressing recombinant hTRXα and hTRXβ reduces Ca2+ mobilization by 84% and 88% respectively. The larger reduction in Ca2+ mobilization for hTRXα in this experiment compared to the double mutant is likely due to retention of residual signal transducing activity by the double mutant.
Regulation through desensitization occurs through both homologous (receptor specific) and heterologous (highly specific for other receptors) modalities, each isoform taking a different pathway (Parent et al. 1999; Murray et al. 1990). Habib et al. (1997) examined the agonist-dependent phosphorylation of both hTPα and hTPβ transfected into HEK293 cells. Using the specific stable TP-specific agonist U46619 they found that both isoforms are phosphorylated with similar time and dose dependency and phosphorylation reduces both the production of IP3 and the increase in intracellular [Ca2+]. Current thought is that the phosphorylation prevents proper docking of G-proteins to the receptor, thus preventing or reducing signal transmission. Further, their data suggest that the phosphorylation leading to desensitization is mediated primarily by kinases other than PKA or PKC. However, they did report that PKC and PKA marginally influence receptor phosphorylation, but not at residues that influence desensitization and suggest that phosphorylation by these kinases may be involved in a heterologous desensitization. In a related set of experiments, Zhou et al. (2001) have shown that treatment of HEK293 cells expressing recombinant hTPα with the TP-specific agonist I-BOP induces both receptor phosphorylation and Ca2+ release in a time- and dose-dependent manner. Following pretreatment (10 min) with I-BOP, a second challenge with I-BOP abolishes Ca2+ induction, clearly demonstrating homologous desensitization. Further, co-transfection of hTPα with GRK5 and GRK6 augments the I-BOP phosphorylation and inhibits the I-BOP-induced Ca2+ release. Without pre-stimulation with I-BOP, the GRKS do not facilitate receptor phosphorylation, showing that GRKs must be recruited to the receptor by agonist-induced phosphorylation before GRK-facilitated phosphorylation can occur. Wang et al. (1998) suggest a potential source for the initial phosphorylation. They report that a cGMP-dependent G kinase phosphorylates the TP receptor that in turn inhibits IP3 release and increases intracellular [Ca2+]; specific α or β isoforms were not investigated. They also show that PKA is not involved. Inactivation of hTPβ and not hTPα is also facilitated by internalization. Parent et al. (1999) report that following stimulation with U46619 of hTPβ transfected in HEK293 cells, the receptor internalizes to a to a plateau of about 40% in 2-3 hours whereas transfected hTPα do not internalize after 3 hours of stimulation. Further internalization requires the presence of GRK (GRK2 and potentially either GRK 3, GRK5, or GRK6), arrestin-2 or -3 and dynamin, suggesting the involvement of clatherin-coated pits that lead to punctate vesicle formation. Overexpression of arrestins significantly promotes internalization whereas GRK co-expression only increases internalization slightly, suggesting that arrestins are the limiting factor for internalization. Utilizing hTPβ transfected into HEK293 cells, Kelly-Hickie and Kinsella (2006) show that PKC does indeed phosphorylate hTPβ and this leads to 20% partial and transient agonist-induced impairment of activity. In addition, they observed that GRK-mediated, agonist-induced phosphorylation leads to a sustained desensitization. Utilizing specific serine to alanine mutations in transfected hTPβ receptors they found that S145 on a cytoplasmic loop is the target of PKC phosphorylation and that S239 and S357 on the C-terminal tail are the targets for GRK phosphorylation. In a complementary study Kelley-Hickie et al. (2007) demonstrated that like hTPβ, phosphorylation of S135 by PKC partially and transiently impairs hTPα signaling. Unlike hTPβ, GRK phosphorylation of the C-terminal tail is not involved in sustained desensitization. Instead they found that both PKC and cGMP-dependent protein kinase (PKG) phosphorylated hTPα at T337 and S331 respectively, leading to profound desensitization of hTPα, each kinase of which is stimulated through the TP signaling pathway.
Heterologous desensitization occurs through both the desensitization of the G-protein rather than the receptor itself and through the action of other receptors. Examples of the former have been reported by several groups. Both Gα12 an Gα13 are known signal transducers for hTP. They have been shown to be phosphorylated by PKC in response to increasing cAMP levels created by other receptor signaling and that this phosphorylation results in the loss of hTP signaling (Manganello et al. 1999; Offermanns et al. 1996). The phosphorylation reduces the affinity of the α subunit for the βγ subunits and thus prevents signaling of both receptor isoforms through the affected G-protein (Kozasa and Gilman 1996). Murray et al. (1990) have reported that stimulation of the hIP receptor with receptor specific agonists results in a PKC-independent desensitization of only one of the two isoforms hTP, the one with strong binding to the antagonist GR3291 shown by others to be the hTPα receptor (Habib et al. 1999). These results were confirmed by Walsh et at (Walsh et al. 2000a) and they further show that the desensitization of hTPα occurs through direct PKA phosphorylation of S329 which is located on the C-terminal domain of the hTPα receptor.
FP receptor
Introduction
The human prostaglandin F2α receptor (hFP, PTGFR, UniprotKB-P43088) is expressed as a 395 amino acid transcript with a calculated molecular weight of 40.0 kDa. There are six reported splice isoform variants (Liang et al. 2008a) obtained from mRNA data that differ only after residue 266, each of which are calculated to have only 6 transmembrane helices rather than the seven found in the canonical isoform (Cserzo et al. 2002). No X-ray structures are available. Although none of these variants have been found naturally occurring at the protein level to date, all have been transfected into HEK293 cells and are translated into functional protein (Liang et al. 2008a; Vielhauer et al. 2004) (see below). There are two reported coding SNP variants (Q8L and L218S) (https://www.ncbi.nlm.nih.gov/clinvar/, Landrum et al. 2016) neither of which has been reported to affect function. There are two N-linked glycoforms predicted for N4 and N19 but are not experimentally confirmed (http://www.cbs.dtu.dk/services/NetNGlyc/, Blom et al. 2004). However, N-glycosylation of the FP receptor in rats has been shown to be necessary for biological activity (Kitanaka et al. 1994). O-glycosylation is predicted for S234 (http://www.cbs.dtu.dk/services/NetOGlyc/, Steentoft et al. 2013) and has not been observed experimentally. There are potential phosphorylation sites predicted for S29, S94, and S279 on extracellular loops, S62 on a cytoplasmic loop, and S337 and S341 on the C-terminal intracellular domain (http://www.cbs.dtu.dk/services/NetPhos/, Blom et al. 1999). Over a dozen potential GRK phosphorylation sites are predicted for the C-terminal domain as well (http://gps.biocuckoo.cn/, Xue et al. 2011). None of these phosphorylation sites have been confirmed experimentally.
Expression and Characterization
The human FP receptor (hFP) is widely expressed with the highest amounts in smooth muscle, female tissues, urinary bladder, and the gall bladder (http://www.proteinatlas.org, Uhlén et al. (2015)). Only the cannonical isoform (hFP) has thus far been shown to produce an intracellular signaling in response to PGF2α or the analog and specific agonist fluprostenol (see below). Agonist-induced signaling for recombinant variant 1 (isoform 2) and 4 (hFPs, isoform 5) have been tested and results show that the latter does not produce an intracellular signal in response to PGF2α (Liang et al. 2008a) and the former does not even bind the agonist (Vielhauer et al. 2004). These findings make sense, as it has been shown that the seventh transmembrane domain, missing in all variants, is required for agonist binding (Neuschäfer-Rube et al. 2003). Ligand binding properties for FP have been characterized for human recombinant receptors, for recombinant mouse receptor (mFP), as well as bovine receptor (bFP) in corpus luteum tissue homogenates by displacement of radiolabeled agonists (Table 8). Ligand efficacies for recombinant hFP, mFP and rat receptor (rFP) as well as in rat hepatocytes and astrocytes have been determined with a variety of agonists. FP receptors are the least selective of the prostanoid receptors; reported EC50 values are given in Table 8. The most selective agonists are Fluprostenol, Cloprostenol, and Latanoprost (Abramovitz et al. 2000; Anderson et al. 1999; Sharif et al. 2000b) whereas the most highly selective antagonists are OBE022, THG113 and AS604872 (Pohl et al. 2018; Peri et al. 2002; Jones et al. 2009) (Table 2).
Mechanism of Cell Activation
The hFP receptor couples primarily through Gaq leading to an IP3/Ca2+ signaling pathway (Table 3) (Breyer et al. 2001; Abramovitz et al. 1994). Potential coupling to Gαs has also been reported as an explanation for the increase in cAMP that follows agonist-induced desensitization of FP in bovine iris sphincter (Tachado et al. 1993). Coupling with other G-proteins has not been reported. However, the rabbit kidney recombinant FP receptor known to couple Gαi has a 98.3% sequence homology to hFP, suggesting a potential for Gαi coupling. Recombinant ovine FP (FPA) expressed in HEK293 cells has been reported to couple through Gα12/13, resulting in stimulation of the small G-protein Rho (Pierce et al. 1999). However, the lower overall sequence homology between ovine FPA and hFP (83.9%) make it less likely that hFP might also couple with Gα12/13, especially since the C-terminal domains that interact with the G-proteins have a lower sequence homology (78.7%).
As noted above, there are six truncated variants of FP, none of which have been shown to exhibit biological signaling activity when expressed alone. However, an hFP dimer with variant 4 (hFP-altFP4) expressed in HEL293 cells has been shown to be activated by the prostamide PGF2α analog bimatoprost whereas hFP alone shows less than 20% the response and altFP4 shows no response at all to this amide analog (Liang et al. 2008a). Further, both the hFP monomer and the heterodimer exhibit identical signaling responses to PGF2α. Clearly, the addition of the altFP species extends the agonist repertoire to amide analogs, lending a possible biological function for these receptor isoforms. Although not yet shown, heterodimer formation with the other altFP isoforms may also modify the agonist specificity of the FP receptor.
Regulation
Regulation of hFP has been shown to be modulated in several different ways. Tachado et al. (1993) confirmed the coupling of bovine FP to Gαq leading to the activation of PLC. They also observed that PGF2α stimulation of FP leads to short-term desensitization that results in an increase in cAMP and attenuation of PLC activity. Both events involve PKC, known to activate adenylate cyclase. The authors suggest a possible explanation involving FP switching from Gαq signaling to Gαs signaling, the latter known to activate adenyl cyclase, thus increasing basal cAMP. The suggestion is qualified by the potential for a yet-to-be discovered FP receptor that normally signals through Gαs. It is interesting to note that the potential phosphorylation sites on the C-terminal domain are high probability sites for PKC phosphorylation and that phosphorylation of other receptors in this region is known to modify G-protein binding specificity. Agonist-induced hFP desensitization has also been observed for recombinant hFP expressed in HEK293 and NIH3T3 cells (Kunapuli et al. 1997), but the connection to either cAMP or PKC has not been explored. FP regulation on the transcriptional level has also been reported. Liang et al. (2008b) examined regulation of hFP expression in human uterine myocytes. Here they found that hFP mRNA expression is negatively regulated by progesterone and cAMP and upregulated by the inflammatory cytokines IL-1β and TNFα that in turn activate the transcription factor NFkB and PKC, the latter also shown to participate in the upregulation of hFP. The results explain the decrease in myometrial expression of hFP during pregnancy and the increase associated with labor.
Conclusion
In this review the expression, characterization, regulation, and mechanism of action of prostanoid receptors are summarized with a focus on human receptors. Prostanoid receptors control numerous biological functions not only through the diversity in prostaglandins themselves, but through the diversity of receptor isoforms and the heterogeneity of G-protein coupling. Commonality is found in regulation where agonist-induced receptor desensitization is accomplished through phosphorylation by PKA, PKB, PKC or a variety of different GRKs. Although much is known about prostanoid receptor signaling, there are still many missing pieces. It is our hope that this review will help underscore these areas and stimulate research to find these missing pieces.
Availability of data and material
Not applicable
References
Abramovitz M, Adam M, Boie Y, Carrière M, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM (2000) The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta 1483:285–293. https://doi.org/10.1016/S1388-1981(99)00164-X
Abramovitz M, Boie Y, Nguyen T, Rushmore TH, Bayne MA, Metters KM, Slipetz DM, Grygorczyk R (1994) Cloning and expression of a cDNA for the human prostanoid FP receptor. J Biol Chem 269:2632–2636
Adam M, Boie Y, Rushmore TH, Müller G, Bastien L, McKee KT, Metters KM, Abramovitz M (1994) Cloning and expression of three isoforms of the human EP3 prostanoid receptor. FEBS Lett 338:170–174. https://doi.org/10.1016/0014-5793(94)80358-7
Af Forselles KJ, Root J, Clarke T, Davey D, Aughton K, Dack K, Pullen N (2011) In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2 receptor antagonist. Br J Pharmacol 164:1847–1856. https://doi.org/10.1111/j.1476-5381.2011.01495.x
Anderson LE, Schultz MK, Wiltbank MC (1999) Prostaglandin moieties that determine receptor binding specificity in the bovine corpus luteum. J. Reprod. Fertil. 116:133–141. https://doi.org/10.1530/jrf.0.1160133
Angulo J, Cuevas P, La Fuente JM, Pomerol JM, Ruiz-Castañé E, Puigvert A, Gabancho S, Fernández A, Ney P, Sáenz De Tejada I (2002) Regulation of human penile smooth muscle tone by prostanoid receptors. Br J Pharmacol 136:23–30. https://doi.org/10.1038/sj.bjp.0704675
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh L-SL (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45:D158–D169. https://doi.org/10.1093/nar/gkh131
Arimura A, Yasui K, Kishino J, Asanuma F, Hasegawa H, Kakudo S, Ohtani M, Arita H (2001) Prevention of allergic inflammation by a novel prostaglandin receptor antagonist, S-5751. J Pharmacol Exp Ther 298:411–419
Armstrong RA, Humphrey PP, Lumley P (1993) Characteristics of the binding of [3H]-GR32191 to the thromboxane (TP-) receptor of human platelets. Br J Pharmacol 110:539–547. https://doi.org/10.1111/j.1476-5381.1993.tb13844.x
Arner M, Högestätt ED, Uski TK (1991) Characterization of contraction-mediating prostanoid receptors in human hand veins: effects of the thromboxane receptor antagonists BM13,505 and AH23848. Acta Physiol Scand 141:79–86. https://doi.org/10.1111/j.1748-1716.1991.tb09047.x
Baxter GS, Clayton JK, Coleman RA, Marshall K, Sangha R, Senior J (1995) Characterization of the prostanoid receptors mediating constriction and relaxation of human isolated uterine artery. Br J Pharmacol 116:1692–1696. https://doi.org/10.1111/j.1476-5381.1995.tb16393.x
Becker KP, Garnovskaya M, Gettys T, Halushka PV (1999) Coupling of thromboxane A2 receptor isoforms to Galpha13: effects on ligand binding and signalling. Biochim Biophys Acta 1450:288–296. https://doi.org/10.1016/S0167-4889(99)00068-3
Bilson HA, Mitchell DL, Ashby B (2004) Human prostaglandin EP3 receptor isoforms show different agonist-induced internalization patterns. FEBS Lett 572:271–275. https://doi.org/10.1016/j.febslet.2004.06.089
Binda C, Génier S, Cartier A, Larrivée JF, Stankova J, Young JC, Parent JL (2004) A G protein-coupled receptor and the intracellular synthase of its agonist functionally cooperate. J Cell Biol 204:377–393. https://doi.org/10.1083/jcb.201304015
Binda C, Parent JL (2015) Characterization of the interaction between the prostaglandin D2 DP1 receptor and the intracellular L-prostaglandin D synthase. Methods Mol Biol 1234:53–67. https://doi.org/10.1007/978-1-4939-1755-6_6
Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294:1351–1362. https://doi.org/10.1006/jmbi.1999.3310
Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4:1633–1649. https://doi.org/10.1002/pmic.200300771
Böer U, Neuschäfer-Rube F, Möller U, Püschel GP (2000) Requirement of N-glycosylation of the prostaglandin E2 receptor EP3beta for correct sorting to the plasma membrane but not for correct folding. Biochem J 350:839–847. https://doi.org/10.1042/bj3500839
Boie Y, Sawyer N, Slipetz DM, Metters KM, Abramovitz M (1995) Molecular cloning and characterization of the human prostanoid DP receptor. J Biol Chem 270:18910–18916. https://doi.org/10.1074/jbc.270.32.18910
Breyer RM, Bagdassarian CK, Myers SA, Breyer MD (2001) Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41:661–690. https://doi.org/10.1146/annurev.pharmtox.41.1.661
Capra V, Busnelli M, Perenna A, Ambrosio M, Accomazzo MR, Galés C, Chini B, Rovati GE (2013) Full and partial agonists of thromboxane prostanoid receptor unveil fine tuning of receptor superactive conformation and G protein activation. PLoS One 8:e60475. https://doi.org/10.1371/journal.pone.0060475
Chakraborty R, Pydi SP, Gleim S, Bhullar RP, Hwa J, Dakshinamurti S, Chelikani P (2013) New insights into structural determinants for prostanoid thromboxane A2 receptor- and prostacyclin receptor-G protein coupling. Mol Cell Biol 33:184–193. https://doi.org/10.1128/MCB.00725-12
Chang SW, Reddy V, Pereira T, Dean BJ, Xia YQ, Seto C, Franklin RB, Karanam BV (2007) The pharmacokinetics and disposition of MK-0524, a Prosglandin D2 Receptor 1 antagonist, in rats, dogs and monkeys. Xenobiotica 37:514–533. https://doi.org/10.1080/00498250601175565
Chun KS, Lao HC, Trempus CS, Okada M, Langenbach R (2009) The prostaglandin receptor EP2 activates multiple signaling pathways and beta-arrestin1 complex formation during mouse skin papilloma development. Carcinogenesis 30:1620–1627. https://doi.org/10.1093/carcin/bgp168
Crider JY, Griffin BW, Sharif NA (2000) Endogenous EP4 prostaglandin receptors coupled positively to adenylyl cyclase in Chinese hamster ovary cells: pharmacological characterization. Prostaglandins Leukot. Essent. Fatty Acids 62:21–26. https://doi.org/10.1054/plef.1999.0120
Crider JY, Xu SX, Sharif NA (2001) Pharmacology of functional endogenous IP prostanoid receptors in NCB-20 cells: comparison with binding data from human platelets. Prostaglandins Leukot Essent Fatty Acids 65:253–258. https://doi.org/10.1054/plef.2001.0322
Cserzo M, Eisenhaber F, Eisenhaber B, Simon I (2002) On filtering false positive transmembrane protein predictions. Protein Eng 15:745–752. https://doi.org/10.1093/protein/15.9.745
Desai S, April H, Nwaneshiudu C, Ashby B (2000) Comparison of agonist-induced internalization of the human EP2 and EP4 prostaglandin receptors: role of the carboxyl terminus in EP4 receptor sequestration. Mol Pharmacol 58:1279–1286. https://doi.org/10.1124/mol.58.6.1279
Dey I, Lejeune M, Chadee K (2006) Prostaglandin E2 receptor distribution and function in the gastrointestinal tract. Br J Pharmacol 149:611–623. https://doi.org/10.1038/sj.bjp.0706923
Donnellan PD, Kinsella BT (2009) Immature and mature species of the human Prostacyclin Receptor are ubiquitinated and targeted to the 26S proteasomal or lysosomal degradation pathways, respectively. J Mol Signal 4:7. https://doi.org/10.1186/1750-2187-4-7
Dorn GW 2nd (1989) Distinct platelet thromboxane A2/prostaglandin H2 receptor subtypes. A radioligand binding study of human platelets. J Clin Invest 84:1883–1891. https://doi.org/10.1172/JCI114375
Dorn GW 2nd (1991) Tissue- and species-specific differences in ligand binding to thromboxane A2 receptors. Am J Physiol 261:R145–R153. https://doi.org/10.1152/ajpregu.1991.261.1.R145
Eivers SB, Kinsella BT (2016) Regulated expression of the prostacyclin receptor (IP) gene by androgens within the vasculature: Combined role for androgens and serum cholesterol. Biochim Biophys Acta 1859:1333–1351. https://doi.org/10.1016/j.bbagrm.2016.06.011
Emery DL, Djokic TD, Graf PD, Nadel JA (1989) Prostaglandin D2 causes accumulation of eosinophils in the lumen of the dog trachea. J Appl Physiol 67:959–962. https://doi.org/10.1152/jappl.1989.67.3.959
Feng JF, Rhee SG, Im MJ (1996) Evidence that phospholipase delta1 is the effector in the Gh (transglutaminase II)-mediated signaling. J Bio Chem. 271:16451–16454. https://doi.org/10.1074/jbc.271.28.16451
Ferguson SS (2007) Phosphorylation-independent attenuation of GPCR signaling. Trends Pharmacol Sci 28:73–179. https://doi.org/10.1016/j.tips.2007.02.008
Fitscha P, Tiso B, Krais T, Sinzinger H (1987) Effect of iloprost on in vivo and in vitro platelet function in patients with peripheral vascular disease (PVD)B. In: Samuelsson, R. Paloetti, P.W. Ramwell (eds). Adv Prostaglandin Thromboxane Leukot Res, Raven Press, New York 17A:450–454
Fujino H, Regan JW (2006) EP(4) prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G protein. Mol Pharmacol 69:5–10. https://doi.org/10.1124/mol.105.017749
Fujino H, West KA, Regan JW (2002) Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J Biol Chem 277:2614–2619. https://doi.org/10.1074/jbc.M109440200
Fujino H, Xu W, Regan JW (2003) Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem 278:12151–12156. https://doi.org/10.1074/jbc.M212665200
Gallant MA, Slipetz D, Hamelin E, Rochdi MD, Talbot S, de Brum-Fernandes AJ, Parent JL (2007) Differential regulation of the signaling and trafficking of the two prostaglandin D2 receptors, prostanoid DP receptor and CRTH2. Eur J Pharmacol 557:115–123. https://doi.org/10.1016/j.ejphar.2006.11.058
Ganesh T (2014) Prostanoid receptor EP2 as a therapeutic target. J Med Chem 57:4454–4465. https://doi.org/10.1021/jm401431x
George RJ, Sturmoski MA, Anant S, Houchen CW (2007) EP4 mediates PGE2 dependent cell survival through the PI3 kinase/AKT pathway. Prostaglandins Other Lipid Mediat 83:112–120. https://doi.org/10.1016/j.prostaglandins.2006.10.005
Gervais FG, Morello JP, Beaulieu C, Sawyer N, Denis D, Greig G, Malebranche AD, O'Neill GP (2005) Identification of a potent and selective synthetic agonist at the CRTH2 receptor. Mol Pharmacol 67:1834–1839. https://doi.org/10.1124/mol.104.009068
Giguère V, Gallant MA, de Brum-Fernandes AJ, Parent JL (2004) Role of extracellular cysteine residues in dimerization/oligomerization of the human prostacyclin receptor. Eur J Pharmacol 494:11–22. https://doi.org/10.1016/j.ejphar.2004.04.041
Giovanazzi S, Accomazzo MR, Letari O, Oliva D, Nicosia S (1997) Internalization and down-regulation of the prostacyclin receptor in human platelets. Biochem J 325:71–77. https://doi.org/10.1042/bj3250071
Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Feletou M (2005) Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol 146:834–845. https://doi.org/10.1038/sj.bjp.0706390
Griffin BW, Magnino PE, Pang IH, Sharif NA (1998) Pharmacological characterization of an FP prostaglandin receptor on rat vascular smooth muscle cells (A7r5) coupled to phosphoinositide turnover and intracellular calcium mobilization. J Pharmacol Exp Ther 286:411–418
Habib A, FitzGerald GA, Maclouf J (1999) Phosphorylation of the thromboxane receptor alpha, the predominant isoform expressed in human platelets. J Biol Chem 274:2645–2651. https://doi.org/10.1074/jbc.274.5.2645
Habib A, Vezza R, Créminon C, Maclouf J, FitzGerald GA (1997) Rapid, agonist-dependent phosphorylation in vivo of human thromboxane receptor isoforms. Minimal involvement of protein kinase C. J Biol Chem 272:7191–7200. https://doi.org/10.1074/jbc.272.11.7191
Hadházy P, Nagy L, Dömötör L, Magyar K (1986) Effects of indomethacin, prostaglandins E2 and I2 on the tone of human isolated femoral arteries. Eur J Clin Invest 16:248–251. https://doi.org/10.1111/j.1365-2362.1986.tb01337.x
Hanyaloglu AC, von Zastrow M (2008) Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmaco Toxicol. 48:537–568. https://doi.org/10.1146/annurev.pharmtox.48.113006.094830
Hata AN, Lybrand TP, Breyer RM (2005) Identification of determinants of ligand binding affinity and selectivity in the prostaglandin D2 receptor CRTH2. J Biol Chem 280:32442–32451. https://doi.org/10.1074/jbc.M502563200
Hata AN, Zent R, Breyer MD, Breyer RM (2003) Expression and molecular pharmacology of the mouse CRTH2 receptor. J Pharmacol Exp Ther 306:463–470. https://doi.org/10.1124/jpet.103.050955
Hébert RL, Carmosino M, Saito O, Yang G, Jackson CA, Qi Z, Breyer RM, Natarajan C, Hata AN, Zhang Y, Guan Y, Breyer MD (2005) Characterization of a rabbit kidney prostaglandin F(2{alpha}) receptor exhibiting G(i)-restricted signaling that inhibits water absorption in the collecting duct. J Biol Chem 280:35028–35037. https://doi.org/10.1074/jbc.M505852200
Heemers HV, Verhoeven G, Swinnen JV (2006) Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol Endocrinol 20:2265–2277. https://doi.org/10.1210/me.2005-0479
Hirai H, Tanaka K, Takano S, Ichimasa M, Nakamura M, Nagata K (2002) Cutting edge: agonistic effect of indomethacin on a prostaglandin D2 receptor, CRTH2. J Immunol 168:981–985. https://doi.org/10.4049/jimmunol.168.3.981
Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K (2001) Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med 193:255–261. https://doi.org/10.1084/jem.193.2.255
Hirai H, Abe H, Tanaka K, Takatsu K, Sugamura K, Nakamura M, Nagata K (2003) Gene structure and functional properties of mouse CRTH2, a prostaglandin D2 receptor. Biochem Biophys Res Commun 307:797–802. https://doi.org/10.1016/S0006-291X(03)01266-X
Hirata M, Kakizuka A, Aizawa M, Ushikubi F, Narumiya S (1994a) Molecular characterization of a mouse prostaglandin D receptor and functional expression of the cloned gene. Proc Natl Acad Sci U S A 91:11192–11196. https://doi.org/10.1073/pnas.91.23.11192
Hirata T, Kakizuka A, Ushikubi F, Fuse I, Okuma M, Narumiya S (1994b) Arg60 to Leu mutation of the human thromboxane A2 receptor in a dominantly inherited bleeding disorder. J Clin Invest 94:1662–1667. https://doi.org/10.1172/JCI117510
Hirata T, Narumiya S (2011) Prostanoid receptors. Chem Rev 111:6209–6230. https://doi.org/10.1021/cr200010h
Hirata T, Ushikubi F, Kakizuka A, Okuma M, Narumiya S (1996) Two thromboxane A2 receptor isoforms in human platelets. Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J Clin Invest 97:949–956. https://doi.org/10.1172/JCI118518
Honda A, Sugimoto Y, Namba T, Watabe A, Irie A, Negishi M, Narumiya S, Ichikawa A (1993) Cloning and expression of a cDNA for mouse prostaglandin E receptor EP2 subtype. J Biol Chem 268:7759–7762
Hsu SC, Chen LC, Kuo ML, Huang JL, Huang SK (2002) Novel SNPs in a candidate gene, CRTH2, for allergic diseases. Genes Immun 3:114–116. https://doi.org/10.1038/sj.gene.6363826
Ikegami R, Sugimoto Y, Segi E, Katsuyama M, Karahashi H, Amano F, Maruyama T, Yamane H, Tsuchiya S, Ichikawa A (2001) The expression of prostaglandin E receptors EP2 and EP4 and their different regulation by lipopolysaccharide in C3H/HeN peritoneal macrophages. J Immunol 166:4689–4696. https://doi.org/10.4049/jimmunol.166.7.4689
Ito S, Okuda E, Sugama K, Negishi M, Hayaishi O (1990) Evaluation of ZK110841 and AH6809, an agonist and an antagonist of prostaglandin DP-receptors on human platelets, with a PGD2-responsive cell line from bovine embryonic trachea. Br J Pharmacol 99:13–14. https://doi.org/10.1111/j.1476-5381.1990.tb14645.x
Ji R, Chou CL, Xu W, Chen XB, Woodward DF, Regan JW (2010) EP1 prostanoid receptor coupling to G i/o up-regulates the expression of hypoxia-inducible factor-1 alpha through activation of a phosphoinositide-3 kinase signaling pathway. Mol Pharmacol 77:1025–1036. https://doi.org/10.1124/mol.110.063933
Jin J, Mao GF, Ashby B (1997) Constitutive activity of human prostaglandin E receptor EP3 isoforms. Br J Pharmacol 121:317–323. https://doi.org/10.1038/sj.bjp.0701121
Jones RL, Giembycz MA, Woodward DF (2009) Prostanoid receptor antagonists: development strategies and therapeutic applications. Br J. Pharmacol 158:104–145. https://doi.org/10.1111/j.1476-5381.2009.00317.x
Kambe A, Iguchi G, Moon Y, Kamitani H, Watanabe T, Eling TE (2008) Regulation of EP4 expression via the Sp-1 transcription factor: inhibition of expression by anti-cancer agents. Biochim Biophys Acta 1783:1211–1219. https://doi.org/10.1016/j.bbamcr.2008.01.032
Kandola MK, Sykes L, Lee YS, Johnson MR, Hanyaloglu AC, Bennett PR (2014) EP2 receptor activates dual G protein signaling pathways that mediate contrasting proinflammatory and relaxatory responses in term pregnant human myometrium. Endocrinology 155:605–617. https://doi.org/10.1210/en.2013-1761
Kashmiry A, Tate R, Rotondo G, Davidson J, Rotondo D (2018) The prostaglandin EP4 receptor is a master regulator of the expression of PGE2 receptors following inflammatory activation in human monocytic cells. Biochim Biophys Acta Mol Cell Biol Lipids 1863:1297–1304. https://doi.org/10.1016/j.bbalip.2018.07.003
Kelley-Hickie LP, Kinsella BT (2004) EP1- and FP-mediated cross-desensitization of the alpha (alpha) and beta (beta) isoforms of the human thromboxane A2 receptor. Br J Pharmacol 142:203–221. https://doi.org/10.1038/sj.bjp.0705695
Kelley-Hickie LP, Kinsella BT (2006) Homologous desensitization of signalling by the beta (beta) isoform of the human thromboxane A2 receptor. Biochim Biophys Acta 1761:1114–1131. https://doi.org/10.1016/j.bbalip.2006.07.012
Kelley-Hickie LP, O'Keeffe MB, Reid HM, Kinsella BT (2007) Homologous desensitization of signalling by the alpha (alpha) isoform of the human thromboxane A2 receptor: a specific role for nitric oxide signalling. Biochim Biophys Acta 1773:970–989. https://doi.org/10.1016/j.bbamcr.2007.03.012
Kilfeather SA, Massarella A, Gorgolewska G, Ansell E, Turner P (1984) Beta-adrenoceptor and epoprostenol (prostacyclin) responsiveness of lymphocytes in migraine patients. Postgrad Med J 60:391–393. https://doi.org/10.1136/pgmj.60.704.391
Kinsella BT, O'Mahony DJ, FitzGerald GA (1994) Phosphorylation and regulated expression of the human thromboxane A2 receptor. J Biol Chem 269:29914–29919
Kinsella BT, O'Mahony DJ, Fitzgerald GA (1997) The human thromboxane A2 receptor alpha isoform (TP alpha) functionally couples to the G proteins Gq and G11 in vivo and is activated by the isoprostane 8-epi prostaglandin F2 alpha. J Pharmacol Exp Ther 281:957–964
Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S (1997) Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol 122:217–224. https://doi.org/10.1038/sj.bjp.0701367
Kitanaka J, Hamano T, Gotoh M, Hashimoto H, Baba A (1994) Tunicamycin inhibits prostaglandin F2 alpha receptor-mediated phosphoinositide hydrolysis in cultured rat astrocytes. Neurochem Res 19:1545–1550
Kobzar G, Mardla V, Järving I, Samel N (2001) Comparison of anti-aggregatory effects of PGI2, PGI3 and iloprost on human and rabbit platelets. Cell Physiol Biochem 11:279–284. https://doi.org/10.1159/000047814
Konger RL, Scott GA, Landt Y, Ladenson JH, Pentland AP (2002) Loss of the EP2 prostaglandin E2 receptor in immortalized human keratinocytes results in increased invasiveness and decreased paxillin expression. Am J Pathol 161:2065–2078. https://doi.org/10.1016/S0002-9440(10)64485-9
Kotani M, Tanaka I, Ogawa Y, Suganami T, Matsumoto T, Muro S, Yamamoto Y, Sugawara A, Yoshimasa Y, Sagawa N, Narumiya S, Nakao K (2000) Multiple signal transduction pathways through two prostaglandin E receptor EP3 subtype isoforms expressed in human uterus. J Clin Endocrinol Metab 85:4315–4322. https://doi.org/10.1210/jcem.85.11.6989
Kotani M, Tanaka I, Ogawa Y, Usui T, Mori K, Ichikawa A, Narumiya S, Yoshimi T, Nakao K (1995) Molecular cloning and expression of multiple isoforms of human prostaglandin E receptor EP3 subtype generated by alternative messenger RNA splicing: multiple second messenger systems and tissue-specific distributions. Mol Pharmacol 48:869–879
Kotani M, Tanaka I, Ogawa Y, Usui T, Tamura N, Mori K, Narumiya S, Yoshimi T, Nakao K (1997) Structural organization of the human prostaglandin EP3 receptor subtype gene (PTGER3). Genomics 40:425–434. https://doi.org/10.1006/geno.1996.4585
Kozasa T, Gilman AG (1996) Protein kinase C phosphorylates G12 alpha and inhibits its interaction with G beta gamma. J Biol Chem 271:12562–12567. https://doi.org/10.1074/jbc.271.21.12562
Kozasa T, Jiang X, Hart MJ, Sternweis PM, Singer WD, Gilman AG, Bollag G, Sternweis PC (1998) p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13. Science 280:2109–2111. https://doi.org/10.1126/science.280.5372.2109
Kunapuli P, Lawson JA, Rokach J, FitzGerald GA (1997) Functional characterization of the ocular prostaglandin f2alpha (PGF2alpha) receptor. Activation by the isoprostane, 12-iso-PGF2alpha. J Biol Chem 272:27147–27154. https://doi.org/10.1074/jbc.272.43.27147
Kunapuli P, Lawson JA, Rokach JA, Meinkoth JL, FitzGerald GA (1998) Prostaglandin F2alpha (PGF2alpha) and the isoprostane, 8, 12-iso-isoprostane F2alpha-III, induce cardiomyocyte hypertrophy. Differential activation of downstream signaling pathways. J Biol Chem 273:22442–22452. https://doi.org/10.1074/jbc.273.35.22442
Kunapuli SP, Fen Mao G, Bastepe M, Liu-Chen LY, Li S, Cheung PP, JK DR, Ashby B (1994) Cloning and expression of a prostaglandin E receptor EP3 subtype from human erythroleukaemia cells. Biochem J 298:263–267. https://doi.org/10.1042/bj2980263
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, Jang W, Katz K, Ovetsky M, Riley G, Sethi A, Tully R, Villamarin-Salomon R, Rubinstein W, Maglott DR (2016) ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44(D1):D862–D868. https://doi.org/10.1093/nar/gkv122
Larsen R, Hansen MB, Bindslev N (2005) Duodenal secretion in humans mediated by the EP4 receptor subtype. Acta Physiol Scand 185:133–140. https://doi.org/10.1111/j.1365-201X.2005.01471.x
Leduc M, Breton B, Galés C, Le Gouill C, Bouvier M, Chemtob S, Heveker H (2009) Functional selectivity of natural and synthetic prostaglandin EP4 receptor ligands. J Pharmacol Exp Ther 331:297–307. https://doi.org/10.1124/jpet.109.156398
Lee CM, Genetos DC, You Z, Yellowley CE (2007) Hypoxia regulates PGE(2) release and EP1 receptor expression in osteoblastic cells. J Cell Physiol 212:182–188. https://doi.org/10.1002/jcp.21017
Liang Y, Woodward DF, Guzman VM, Li C, Scott DF, Wang JW, Wheeler LA, Garst ME, Landsverk K, Sachs G, Krauss AH, Cornell C, Martos J, Pettit S, Fliri H (2008a) Identification and pharmacological characterization of the prostaglandin FP receptor and FP receptor variant complexes. Br J Pharmacol 154:1079–1093. https://doi.org/10.1038/bjp.2008.142
Liang Z, Sooranna SR, Engineer N, Tattersall M, Khanjani S, Bennett PR, Myatt L, Johnson MR (2008b) Prostaglandin F2-alpha receptor regulation in human uterine myocytes. Mol Hum Reprod 14:215–223. https://doi.org/10.1093/molehr/gan008
Lim H, Dey SK (1997) Prostaglandin E2 receptor subtype EP2 gene expression in the mouse uterus coincides with differentiation of the luminal epithelium for implantation. Endocrinology 138:4599–4606. https://doi.org/10.1210/endo.138.11.5528
MacLean Scott E, Solomon LA, Davidson C, Storie J, Palikhe NS, Cameron L (2018) Activation of Th2 cells downregulates CRTh2 through an NFAT1 mediated mechanism. PLoS One 13:e0199156. https://doi.org/10.1371/journal.pone.0199156
Maher SA, Birrell MA, Adcock JJ, Wortley MA, Dubuis ED, Bonvini SJ, Grace MS, Belvisi MG (2015) Prostaglandin D2 and the role of the DP1, DP2 and TP receptors in the control of airway reflex events. Eur Respir J 45:1108–1118. https://doi.org/10.1183/09031936.00061614
Maiguel D, Faridi MH, Wei C, Kuwano Y, Balla KM, Hernandez D, Barth CJ, Lugo G, Donnelly M, Nayer A, Moita LF, Schürer S, Traver D, Ruiz P, Vazquez-Padron RI, Ley K, Reiser J, Gupta V (2011) Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci Signal 4:ra57. https://doi.org/10.1126/scisignal.2001811
Manganello JM, Djellas Y, Borg C, Antonakis K (1999) G.C. Le Breton GC, Cyclic AMP-dependent phosphorylation of thromboxane A(2) receptor-associated Galpha(13). J Biol Chem 274:28003-28010. 0.1074/jbc.274.39.28003
Markovič T, Jakopin Ž, Dolenc MS, Mlinarič-Raščan I (2017) Structural features of subtype-selective EP receptor modulators. Drug Discov Today 22:57–71. https://doi.org/10.1016/j.drudis.2016.08.003
Maurer-Stroh S, Koranda M, Benetka W, Schneider G, Sirota F, Eisenhaber F (2007) Towards complete sets of farnesylated and geranylgeranylated proteins. PLoS Comput Biol 3:e66. https://doi.org/10.1371/journal.pcbi.0030066
Mayeux PR, Morton HE, Gillard J, Lord A, Morinelli TA, Boehm A, Mais DE, Halushka PV (1988) The affinities of prostaglandin H2 and thromboxane A2 for their receptor are similar in washed human platelets. Biochem Biophys Res Commun 157:733–739. https://doi.org/10.1016/S0006-291X(88)80311-5
McGraw DW, Mihlbachler KA, Schwarb MR, Rahman FF, Small KM, Almoosa KF, Liggett SB (2006) Airway smooth muscle prostaglandin-EP1 receptors directly modulate beta2-adrenergic receptors within a unique heterodimeric complex. J Clin Invest 116:1400–1409. https://doi.org/10.1172/JCI25840
McKenniff M, Rodger IW, Norman P, Gardiner PJ (1988) Characterisation of receptors mediating the contractile effects of prostanoids in guinea-pig and human airways. Eur J Pharmacol 153:149–159. https://doi.org/10.1016/0014-2999(88)90601-2
Meisdalen K, Dajani OF, Christoffersen T, Sandnes D (2007) Prostaglandins Enhance EGF-induced DNA synthesis in Hepatocytes by Stimulation of EP3 and FP Receptors. J Pharm Exp Ther 322:1044–1050. https://doi.org/10.1124/jpet.107.121277
Merz C, von Mässenhausen A, Queisser A, Vogel W, Andrén O, Kirfel J, Duensing S, Perner S, Nowak M (2016) IL-6 Overexpression in ERG-Positive Prostate Cancer Is Mediated by Prostaglandin Receptor EP2. Am J Pathol 186:974–984. https://doi.org/10.1016/j.ajpath.2015.12.009
Mesquita-Santos FP, Bakker-Abreu I, Luna-Gomes T, Bozza PT, Diaz BL, Bandeira-Melo C (2011) Co-operative signalling through DP(1) and DP(2) prostanoid receptors is required to enhance leukotriene C(4) synthesis induced by prostaglandin D(2) in eosinophils. Br J Pharmacol 162:1674–1685. https://doi.org/10.1111/j.1476-5381.2010.01086.x
Miggin SM, Kinsella BT (1998) Expression and tissue distribution of the mRNAs encoding the human thromboxane A2 receptor (TP) alpha and beta isoforms. Biochim Biophys Acta 1425:543–559. https://doi.org/10.1016/S0304-4165(98)00109-3
Miggin SM, Lawler OA, Kinsella BT (2002) Investigation of a functional requirement for isoprenylation by the human prostacyclin receptor. Eur J Biochem 269:1714–1725. https://doi.org/10.1046/j.1432-1327.2002.02817.x
Miggin SM, Lawler OA, Kinsella BT (2003) Palmitoylation of the human prostacyclin receptor. Functional implications of palmitoylation and isoprenylation. J Biol Chem 278:6947–6958. https://doi.org/10.1074/jbc.M210637200
Miki I, Kishibayashi N, Nonaka H, Ohshima E, Takami H, Obase H, Ishii A (1992) Effects of KW-3635, a novel dibenzoxepin derivative of a selective thromboxane A2 antagonist, on human, guinea pig and rat platelets. Jpn J Pharmacol 59:357–364. https://doi.org/10.1254/jjp.59.357
Modesti PA, Colella A, Abbate R, Gensini G, Neri Serneri G (1989) Competitive inhibition of platelet thromboxane A2 receptor binding by picotamide. Eur J Pharmacol 169:85–93. https://doi.org/10.1016/0014-2999(89)90820-0
Monneret G, Gravel S, Diamond M, Rokach J (2001) Powell WS, Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood 98:1942–1948. https://doi.org/10.1182/blood.V98.6.1942
Morii H, Watanabe Y (1992) A possible role of carbohydrate moieties in prostaglandin D2 and prostaglandin E2 receptor proteins from the porcine temporal cortex. Arch Biochem Biophys 292:121–127. https://doi.org/10.1016/0003-9861(92)90059-6
Murray R, Shipp E, FitzGerald GA (1990) Prostaglandin endoperoxide/thromboxane A2 receptor desensitization. Cross-talk with adenylate cyclase in human platelets. J Biol Chem 265:21670–21675
Nagata K, Hirai H (2003) The second PGD(2) receptor CRTH2: structure, properties, and functions in leukocytes. Prostaglandins Leukot Essent Fatty Acids 69:169–177. https://doi.org/10.1016/S0952-3278(03)00078-4
Nagata K, Tanaka K, Ogawa K, Kemmotsu K, Imai T, Yoshie O, Abe H, Tada K, Nakamura M, Sugamura K, Takano S (1999) Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol 162:1278–1286
Narumiya S, Sugimoto Y, Ushikubi F (1999) Prostanoid receptors: structures, properties, and functions. Physiol Rev 79:1193–1226. https://doi.org/10.1152/physrev.1999.79.4.1193
Nasrallah R, Hassouneh R, Zimpelmann J, Karam AJ, Thibodeau JF, Burger D, Burns KD, Kennedy CR, Hébert RL (2015) Prostaglandin E2 increases proximal tubule fluid reabsorption, and modulates cultured proximal tubule cell responses via EP1 and EP4 receptors. Lab Invest 95:1044–1055. https://doi.org/10.1038/labinvest.2015.79
Neuschäfer-Rube F, Engemaier E, Koch S, Böer U, Püschel GP (2003) Identification by site-directed mutagenesis of amino acids contributing to ligand-binding specificity or signal transduction properties of the human FP prostanoid receptor. Biochem J 371:443–449. https://doi.org/10.1042/bj20021429
Neuschäfer-Rube F, Hermosilla R, Rehwald M, Rönnstrand L, Schülein R, Wernstedt C, Püschel GP (2004) Identification of a Ser/Thr cluster in the C-terminal domain of the human prostaglandin receptor EP4 that is essential for agonist-induced beta-arrestin1 recruitment but differs from the apparent principal phosphorylation site. Biochem J 379:573–585. https://doi.org/10.1042/bj20031820
Nishigaki N, Negishi M, Honda A, Sugimoto Y, Namba T, Narumiya S, Ichikawa A (1995) Identification of prostaglandin E receptor 'EP2' cloned from mastocytoma cells EP4 subtype. FEBS Lett 364:339–341. https://doi.org/10.1016/0014-5793(95)00421-5
Nishigaki N, Negishi M, Ichikawa A (1996) Two Gs-coupled prostaglandin E receptor subtypes, EP2 and EP4, differ in desensitization and sensitivity to the metabolic inactivation of the agonist. Mol Pharmacol 50:1031–1037
Norel X, de Montpreville V, Brink C (2004) Vasoconstriction induced by activation of EP1 and EP3 receptors in human lung: effects of ONO-AE-248, ONO-DI-004, ONO-8711 or ONO-8713. Prostaglandins Other Lipid Mediat 74:101–112. https://doi.org/10.1016/j.prostaglandins.2004.07.003
Offermanns S, Hu YH, Simon MI (1996) Galpha12 and galpha13 are phosphorylated during platelet activation. J Biol Chem 271:26044–26048. https://doi.org/10.1074/jbc.271.42.26044
Offermanns S, Laugwitz KL, Spicher K, Schultz G (1994) G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc Natl Acad Sci U S A 91:504–508. https://doi.org/10.1073/pnas.91.2.504
Ohmichi M, Koike K, Kimura A, Masuhara K, Ikegami H, Ikebuchi Y, Kanzaki T, Touhara K, Sakaue M, Kobayashi Y, Akabane M, Miyake A, Murata Y (1997) Role of mitogen-activated protein kinase pathway in prostaglandin F2alpha-induced rat puerperal uterine contraction. Endocrinology 138:3103–3111. https://doi.org/10.1210/endo.138.8.5305
O'Keeffe MB, Reid HM, Kinsella BT (2008) Agonist-dependent internalization and trafficking of the human prostacyclin receptor: a direct role for Rab5a GTPase. Biochim Biophys Acta 1783:1914–1928. https://doi.org/10.1016/j.bbamcr.2008.04.010
Okuda-Ashitaka E, Sakamoto K, Giles H, Ito S, Hayaishi O (1993) Cyclic-AMP-dependent Ca2+ influx elicited by prostaglandin D2 in freshly isolated nonchromaffin cells from bovine adrenal medulla. Biochim Biophys Acta 1176:148–154. https://doi.org/10.1016/0167-4889(93)90190-Z
Pándy-Szekeres G, Munk C, Tsonkov TM, Mordalski S, Harpsøe K, Hauser AS, Bojarski AJ, Gloriam DE (2018) GPCRdb in 2018: adding GPCR structure models and ligands. Nucleic Acids Res 46:D440–D446. https://doi.org/10.1093/nar/gkx1109
Parent JL, Labrecque P, Orsini MJ, Benovic JL (1999) Internalization of the TXA2 receptor alpha and beta isoforms. Role of the differentially spliced cooh terminus in agonist-promoted receptor internalization. J Biol Chem 274:8941–8948. https://doi.org/10.1074/jbc.274.13.8941
Peinhaupt M, Roula D, Theiler A, Sedej M, Schicho R, Marsche G, Sturm EM, Sabroe I, Rothenberg ME, Heinemann A (2018) DP1 receptor signaling prevents the onset of intrinsic apoptosis in eosinophils and functions as a transcriptional modulator. J Leukoc Biol 104:159–171. https://doi.org/10.1002/JLB.3MA1017-404R
Penn RB, Pascual RM, Kim YM, Mundell SJ, Krymskaya VP, Panettieri RA Jr, Benovic JL (2001) Arrestin specificity for G protein-coupled receptors in human airway smooth muscle. J Biol Chem 276:32648–32656. https://doi.org/10.1074/jbc.M104143200
Perchick GB, Jabbour HN (2003) Cyclooxygenase-2 overexpression inhibits cathepsin D-mediated cleavage of plasminogen to the potent antiangiogenic factor angiostatin. Endocrinology 144:5322–5328. https://doi.org/10.1210/en.2003-0986
Peri KG, Quiniou C, Hou X, Abran D, Varma DR, Lubell WD, Chemtob S (2002) THG113: a novel selective FP antagonist that delays preterm labor. Semin Perinatol 26:389–397. https://doi.org/10.1053/sper.2002.37307
Pettipher R, Hansel TT, Armer R (2007) Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat Rev Drug Discov 6:313–325. https://doi.org/10.1038/nrd2266
Pierce KL, Fujino H, Srinivasan D, Regan JW (1999) Activation of FP prostanoid receptor isoforms leads to Rho-mediated changes in cell morphology and in the cell cytoskeleton. J Biol Chem 274:35944–35949. https://doi.org/10.1074/jbc.274.50.35944
Pohl O, Chollet A, Kim SH, Riaposova L, Spézia F, Gervais F, Guillaume P, Lluel P, Méen M, Lemaux F, Terzidou V, Bennett PR, Gotteland JP (2018) OBE022, an Oral and Selective Prostaglandin F2α Receptor Antagonist as an Effective and Safe Modality for the Treatment of Preterm Labor. J Pharmacol Exp Ther 366:349–364. https://doi.org/10.1124/jpet.118.247668
Rangachari PK, Betti PA, Prior ET, Roberts LJ 2nd (1995) Effects of a selective DP receptor agonist (BW 245C) and antagonist (BW A868C) on the canine colonic epithelium: an argument for a different DP receptor? J Pharmacol Exp Ther 275:611–617
Regan JW (2003) EP2 and EP4 prostanoid receptor signaling. Life Sci 74:143–153. https://doi.org/10.1016/j.lfs.2003.09.031
Regan JW, Bailey TJ, Donello JE, Pierce KL, Pepperl DJ, Zhang D, Kedzie KM, Fairbairn CE, Bogardus AM, Woodward DF, Gil DW (1994b) Molecular cloning and expression of human EP3 receptors: evidence of three variants with differing carboxyl termini. Br J Pharmacol 112:377–385. https://doi.org/10.1111/j.1476-5381.1994.tb13082.x
Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, Fairbairn CE, Kedzie KM, Woodward DF, Gil DW (1994a) Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol 46:213–220
Ricciotti E, FitzGerald GA (2011) Prostaglandins and Inflammation. Arterioscler Thromb Vasc Biol 31:986–1000. https://doi.org/10.1161/ATVBAHA.110.207449
Sands WA, Palmer TM (2008) Regulating gene transcription in response to cyclic AMP elevation. Cell Signal 20:460–466. https://doi.org/10.1016/j.cellsig.2007.10.005
Savage MA, Moummi C, Karabatsos PJ, Lanthorn TH (1993) SC-46275: a potent and highly selective agonist at the EP3 receptor. Prostaglandins Leukot Essent Fatty Acids 49:939–943. https://doi.org/10.1016/0952-3278(93)90179-Z
Sawyer N, Cauchon E, Chateauneuf A, Cruz RP, Nicholson DW, Metters KM, O'Neill GP, Gervais FG (2002) Molecular pharmacology of the human prostaglandin D2 receptor, CRTH2. Br J Pharmacol 137:1163–1172. https://doi.org/10.1038/sj.bjp.0704973
Schmid A, Thierauch KH, Schleuning WD, Dinter H (1995) Splice variants of the human EP3 receptor for prostaglandin E2. Eur J Biochem 228:23–30. https://doi.org/10.1111/j.1432-1033.1995.0023o.x
Schratl P, Royer JF, Kostenis E, Ulven T, Sturm EM, Waldhoer M, Hoefler G, Schuligoi R, Lippe IT, Peskar BA, Heinemann A (2007) The role of the prostaglandin D2 receptor, DP, in eosinophil trafficking. J Immunol 179:4792–4799. https://doi.org/10.4049/jimmunol.179.7.4792
Schröder R, Merten N, Mathiesen JM, Martini L, Kruljac-Letunic A, Krop F, Blaukat A, Fang Y, Tran E, Ulven T, Drewke C, Whistler J, Pardo L, Gomeza J, Kostenis E (2009) The C-terminal tail of CRTH2 is a key molecular determinant that constrains Gα and downstream signaling cascade activation. J Biol Chem 284(2):1324–1336. https://doi.org/10.1074/jbc.M806867200
Sekido N, Kida J, Mashimo H, Wakamatsu D, Okada H, Matsuya H (2016) Promising Effects of a Novel EP2 and EP3 Receptor Dual Agonist, ONO-8055, on Neurogenic Underactive Bladder in a Rat Lumbar Canal Stenosis Model. J Urol 196:609–616. https://doi.org/10.1016/j.juro.2016.02.064
Sharif NA, Crider JY, Xu SX, Williams GW (2000b) Affinities, selectivities, potencies, and intrinsic activities of natural and synthetic prostanoids using endogenous receptors: focus on DP class prostanoids. J Pharmacol Exp Ther 293:321–328
Sharif NA, Davis TL (2002) Cloned human EP1 prostanoid receptor pharmacology characterized using radioligand binding techniques. J Pharm Pharmacol 54:539–547. https://doi.org/10.1211/0022357021778655
Sharif NA, Kelly CR, Crider JY, Williams GW, Xu SX (2003) Ocular hypotensive FP prostaglandin (PG) analogs: PG receptor subtype binding affinities and selectivities, and agonist potencies at FP and other PG receptors in cultured cells. J Ocul Pharmacol Ther 19:501–515. https://doi.org/10.1089/108076803322660422
Sharif NA, Williams GW, Davis TL (2000a) Pharmacology and autoradiography of human DP prostanoid receptors using [(3)H]-BWA868C, a DP receptor-selective antagonist radioligand. Br J Pharmacol 131:1025–1038. https://doi.org/10.1038/sj.bjp.0703686
Shenker A, Goldsmith P, Unson CG, Spiegel AM (1991) The G protein coupled to the thromboxane A2 receptor in human platelets is a member of the novel Gq family. Trans Assoc Am Physicians 104:11–20
Siegl AM, Smith JB, Silver MJ, Nicolaou KC, Ahern D (1979) Selective binding site for [3H]prostacyclin on platelets. J Clin Invest 63:215–220. https://doi.org/10.1172/JCI109292
Smyth EM, Austin SC, Reilly MP, FitzGerald GA (2000) Internalization and sequestration of the human prostacyclin receptor. J Biol Chem 275:32037–32045. https://doi.org/10.1074/jbc.M003873200
Smyth EM, Li WH, FitzGerald GA (1998) Phosphorylation of the prostacyclin receptor during homologous desensitization. A critical role for protein kinase C. J Biol Chem 273:23258–23266. https://doi.org/10.1074/jbc.273.36.23258
Smyth EM, Nestor PA, FitzGerald GA (1996) Agonist-dependent phosphorylation of an epitope-tagged human prostacyclin receptor. J Biol Chem 271:33698–33704. https://doi.org/10.1074/jbc.271.52.33698
Sood R, Flint-Ashtamker G, Borenstein D, Barki-Harrington L (2014) Upregulation of prostaglandin receptor EP1 expression involves its association with cyclooxygenase-2. PLoS One 9:e91018. https://doi.org/10.1371/journal.pone.0091018
Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos-Silva L, Gupta R, Bennett EP, Mandel U, Brunak S, Wandall HH, Levery SB (2013) Clausen H, Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J 32:1478–1488. https://doi.org/10.1038/emboj.2013.79
Stelzer G, Rosen R, Plaschkes I, Zimmerman S, Twik M, Fishilevich S, Iny Stein T, Nudel R, Lieder I, Mazor Y, Kaplan S, Dahary D, Warshawsky D, Guan-Golan Y, Kohn A, Rappaport N, Safran M, and Lancet D (2016) The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analysis. Current Protocols in Bioinformatics 54:1.30.1-1.30.33. https://doi.org/10.1002/cpbi.5
Stitham J, Arehart E, Elderon L, Gleim SR, Douville K, Kasza Z, Fetalvero K, MacKenzie T, Robb J, Martin KA, Hwa J (2011) Comprehensive biochemical analysis of rare prostacyclin receptor variants: study of association of signaling with coronary artery obstruction. J Biol Chem 286:7060–7069. https://doi.org/10.1074/jbc.M110.124933
Stitham J, Stojanovic A, Hwa J (2002) Impaired receptor binding and activation associated with a human prostacyclin receptor polymorphism. J Biol Chem 277:15439–15444. https://doi.org/10.1074/jbc.M201187200
Stürzebecher CS, Losert W (1987) Effects of Iloprost on Platelet Activation In Vitro. In: Gryglewski RJ, Stock G (eds) Prostacyclin and Its Stable Analogue Iloprost. Springer, Berlin, pp 39–45
Sugama K, Tanaka T, Yokohama H, Negishi M, Hayashi H, Ito S, Hayaishi O (1989) Stimulation of cAMP formation by prostaglandin D2 in primary cultures of bovine adrenal medullary cells: identification of the responsive population as fibroblasts. Biochim Biophys Acta 1011:75–80. https://doi.org/10.1016/0167-4889(89)90081-5
Suganami A, Fujino H, Okura I, Yanagisawa N, Sugiyama H, Regan JW, Tamura Y, Murayama T (2016) Human DP and EP2 prostanoid receptors take on distinct forms depending on the diverse binding of different ligands. FEBS 283:3853–4011. https://doi.org/10.1111/febs.13899
Sugimoto H, Shichijo M, Iino T, Manabe Y, Watanabe A, Shimazaki M, Gantner F, Bacon KB (2003) An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY u3405), inhibits prostaglandin D2-induced eosinophil migration in vitro. J Pharmacol Exp Ther 305:347–352. https://doi.org/10.1124/jpet.102.046748
Sugimoto Y, Narumiya S (2007) Prostaglandin E receptors. J Biol Chem. 282:11613–11617. https://doi.org/10.1074/jbc.R600038200
Tachado SD, Zhang Y, Abdel-Latif AA (1993) Protein kinase C is involved in cyclic adenosine monophosphate formation due to PGF2 alpha desensitization in bovine iris sphincter. Invest Ophthalmol Vis Sci 34:2023–2032
Takayama K, García-Cardena G, Sukhova GK, Comander J, Gimbrone MA Jr, Libby P (2002) Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J Biol Chem 277:44147–44154. https://doi.org/10.1074/jbc.M204810200
Tang CH, Yang RS, Fu WM (2005) Prostaglandin E2 stimulates fibronectin expression through EP1 receptor, phospholipase C, protein kinase Calpha, and c-Src pathway in primary cultured rat osteoblasts. J Biol Chem 280:22907–22916. https://doi.org/10.1074/jbc.M500130200
Tobin AB (2008) G-protein-coupled receptor phosphorylation: where, when and by whom. Br. J Pharmacol 153:S167–S176. https://doi.org/10.1038/sj.bjp.0707662
Tostes RC, Nigro D, Fortes ZB, Carvalho MH (2003) Effects of estrogen on the vascular system. Braz J Med Biol Res 36:1143–1158. https://doi.org/10.1590/S0100-879X2003000900002
Town MH, Casals-Stenzel J, Schillinger E (1983) Pharmacological and cardiovascular properties of a hydantoin derivative, BW 245 C, with high affinity and selectivity for PGD2 receptors. Prostaglandins 25:13–28. https://doi.org/10.1016/0090-6980(83)90131-4
Tsai AL, Hsu MJ, Vijjeswarapu H, Wu KK (1989) Solubilization of prostacyclin membrane receptors from human platelets. J Biol Chem 264:61–67
Tsuboi K, Sugimoto Y, Ichikawa A (2002) Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat 68-69:535–556. https://doi.org/10.1016/S0090-6980(02)00054-0
Tunaru S, Chennupati R, Nüsing RM, Offermanns S (2016) Arachidonic Acid Metabolite 19(S)-HETE Induces Vasorelaxation and Platelet Inhibition by Activating Prostacyclin (IP) Receptor. PLoS One Sep 23;11(9):e0163633. https://doi.org/10.1371/journal.pone.0163633
Turner EC, Kinsella BT (2010) Estrogen increases expression of the human prostacyclin receptor within the vasculature through an ERalpha-dependent mechanism. J Mol Biol 396:473–486. https://doi.org/10.1016/j.jmb.2010.01.010
Tymkewycz PM, Jones RL, Wilson NH, Marr CG (1991) Heterogeneity of thromboxane A2 (TP-) receptors: evidence from antagonist but not agonist potency measurements. Br J Pharmacol 102:607–614. https://doi.org/10.1111/j.1476-5381.1991.tb12220.x
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F (2015) Tissue-based map of the human proteome. Science 347:1260419. https://doi.org/10.1126/science.1260419
Ungrin MD, Carrière MC, Denis D, Lamontagne S, Sawyer N, Stocco R, Tremblay N, Metters KM, Abramovitz M (2001) Key structural features of prostaglandin E(2) and prostanoid analogs involved in binding and activation of the human EP(1) prostanoid receptor. Mol Pharmacol 59:1446–1456. https://doi.org/10.1124/mol.59.6.1446
Ushikubi F, Nakajima M, Hirata M, Okuma M, Fujiwara M, Narumiya S (1989a) Purification of the thromboxane A2/prostaglandin H2 receptor from human blood platelets. J Biol Chem 264:16496–16501
Ushikubi F, Nakajima M, Yamamoto M, Ohtsu K, Kimura Y, Okuma M, Uchino H, Fujiwara M, Narumiya S (1989b) [3H]S-145 and [125I]I-S-145-OH: new radioligands for platelet thromboxane A2 receptor with low nonspecific binding and high binding affinity for various receptor preparations. Eicosanoids 2:21–27
Ushikubi F, Nakamura K, Narumiya S (1994) Functional reconstitution of platelet thromboxane A2 receptors with Gq and Gi2 in phospholipid vesicles. Mol Pharmacol 46:808–816
Vassaux G, Gaillard D, Ailhaud G, Négrel R (1992) Prostacyclin is a specific effector of adipose cell differentiation. Its dual role as a cAMP- and Ca(2+)-elevating agent. J Biol Chem 267:11092–11097
Vezza R, Habib A, FitzGerald GA (1999) Differential signaling by the thromboxane receptor isoforms via the novel GTP-binding protein, Gh. J Biol Chem 274:12774–12779. https://doi.org/10.1074/jbc.274.18.12774
Vielhauer GA, Fujino H, Regan JW (2004) Cloning and localization of hFP(S): a six-transmembrane mRNA splice variant of the human FP prostanoid receptor. Arch Biochem Biophys 421:175–185. https://doi.org/10.1016/j.abb.2003.10.021
Walsh M, Foley JF, Kinsella BT (2000b) Investigation of the role of the carboxyl-terminal tails of the alpha and beta isoforms of the human thromboxane A(2) receptor (TP) in mediating receptor:effector coupling. Biochim Biophys Acta 1496:164–182. https://doi.org/10.1016/S0167-4889(00)00031-8
Walsh MT, Foley JF, Kinsella BT (1998) Characterization of the role of N-linked glycosylation on the cell signaling and expression of the human thromboxane A2 receptor alpha and beta isoforms. J Pharmacol Exp Ther 286:1026–1036
Walsh MT, Foley JF, Kinsella BT (2000a) The alpha, but not the beta, isoform of the human thromboxane A2 receptor is a target for prostacyclin-mediated desensitization. J Bio Chem 275:20412–20423. https://doi.org/10.1074/jbc.M907881199
Wang GR, Zhu Y, Halushka PV, Lincoln TM, Mendelsohn ME (1998) Mechanism of platelet inhibition by nitric oxide: in vivo phosphorylation of thromboxane receptor by cyclic GMP-dependent protein kinase. Proc Natl Acad Sci U S A 95:4888–4893. https://doi.org/10.1073/pnas.95.9.4888
Wang L, Yao D, Deepak RNVK, Liu H, Xiao Q, Fan H, Gong W, Wei Z, Zhang C (2018) Structures of the Human PGD2 Receptor CRTH2 Reveal Novel Mechanisms for Ligand Recognition. Mol Cell 72:48–59. https://doi.org/10.1016/j.molcel.2018.08.009
Wang XL, Akhtar RA, Abdel-Latif AA (1995) Purification and properties of D-myo-inositol 1,4,5-trisphosphate 3-kinase from bovine iris sphincter smooth muscle: effects of protein phosphorylation in vitro and in intact muscle. Biochem J 308:1009–1016. https://doi.org/10.1042/bj3081009
Wheeldon A, Vardey CJ (1993) Characterization of the inhibitory prostanoid receptors on human neutrophils. Br J Pharmacol 108:1051–1054. https://doi.org/10.1111/j.1476-5381.1993.tb13504.x
Wikström K, Reid HM, Hill M, English KA, O'Keeffe MB, Kimbembe CC, Kinsella BT (2008) Recycling of the human prostacyclin receptor is regulated through a direct interaction with Rab11a GTPase. Cell Signal. 20:2332–2346. https://doi.org/10.1016/j.cellsig.2008.09.003
Wilson SJ, Roche AM, Kostetskaia E, Smyth EM (2004) Dimerization of the human receptors for prostacyclin and thromboxane facilitates thromboxane receptor-mediated cAMP generation. J Biol Chem 279:53036–53047. https://doi.org/10.1074/jbc.M405002200
Woodward DF, Krauss AH, Chen J, Gil DW, Kedzie KM, Protzman CE, Shi L, Chen R, Krauss HA, Bogardus A, Dinh HT, Wheeler LA, Andrews SW, Burk RM, Gac T, Roof MB, Garst ME, Kaplan LJ, Sachs G, Pierce KL, Regan JW, Ross RA, Chan MF (2000) Replacement of the carboxylic acid group of prostaglandin f(2alpha) with a hydroxyl or methoxy substituent provides biologically unique compounds. Br J Pharmacol 130:1933–1943. https://doi.org/10.1038/sj.bjp.0703462
Woodward DF, Krauss AH, Chen J, Liang Y, Li C, Protzman CE, Bogardus A, Chen R, Kedzie KM, Krauss HA, Gil DW, Kharlamb A, Wheeler LA, Babusis D, Welty D, Tang-Liu DD, Cherukury M, Andrews SW, Burk RM, Garst ME (2003) Pharmacological characterization of a novel antiglaucoma agent, Bimatoprost (AGN 192024). J Pharmacol Exp Ther 305:772–785. https://doi.org/10.1124/jpet.102.047837
Wright DH, Metters KM, Abramovitz M, Ford-Hutchinson AW (1998) Characterization of the recombinant human prostanoid DP receptor and identification of L-644,698, a novel selective DP agonist. Br J Pharmacol 123:1317–1324. https://doi.org/10.1038/sj.bjp.0701708
Xue I, Gyles SL, Barrow A, Pettipher R (2007) Inhibition of PI3K and calcineurin suppresses chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2)-dependent responses of Th2 lymphocytes to prostaglandin D(2). Biochem Pharmacol 73:843–853. https://doi.org/10.1016/j.bcp.2006.11.021
Xue L, Barrow A, Pettipher R (2009) Novel function of CRTH2 in preventing apoptosis of human Th2 cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol 182:7580–7586. https://doi.org/10.4049/jimmunol.0804090
Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L, Ren J (2011) GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng Des Sel 24:255–260. https://doi.org/10.1093/protein/gzq094
Yu R, Xiao L, Zhao G, Christman JW, van Breemen RB (2011) Competitive enzymatic interactions determine the relative amounts of prostaglandins E2 and D2. J Pharmacol Exp Ther 339:716–725. https://doi.org/10.1124/jpet.111.185405
Zaccolo M (2009) cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br J Pharmacol 158:50–60. https://doi.org/10.1111/j.1476-5381.2009.00185.x
Zeng L, An S, Goetzl EJ (1998) EP4/EP2 receptor-specific prostaglandin E2 regulation of interleukin-6 generation by human HSB.2 early T cells. J Pharmacol Exp Ther 286:1420–1426
Zhang Z, Austin SC, Smyth EM (2001) Glycosylation of the human prostacyclin receptor: role in ligand binding and signal transduction. Mol Pharmacol 60:480–487
Zhou F, Xue Y, Yao X, Xu Y (2006) CSS-Palm: palmitoylation site prediction with a clustering and scoring strategy (CSS). Bioinformatics 22:894–896. https://doi.org/10.1093/bioinformatics/btl013
Zhou H, Yan F, Tai HH (2001) Phosphorylation and desensitization of the human thromboxane receptor-alpha by G protein-coupled receptor kinases. J Pharmacol Exp Ther 298:1243–1251
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest/Competing interests
The author declares that he has no conflicts of interest or competing interests.
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Code availability
Not applicable
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Biringer, R.G. A Review of Prostanoid Receptors: Expression, Characterization, Regulation, and Mechanism of Action. J. Cell Commun. Signal. 15, 155–184 (2021). https://doi.org/10.1007/s12079-020-00585-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-020-00585-0