
Abstract
Giant cell tumor (GCT) of bone is a rare, benign, osteolytic neoplasm that most commonly occurs in early adulthood and often involves the long bones of the body. Although GCT largely affects the epiphyses of long bones, several reports of GCT involvement of the cranial and facial bones exist in the literature. In addition to reviewing other reported cases of GCT of the lateral skull base in the literature, the authors report here on the clinical presentation, radiographic findings, and management of a patient found to have a GCT of the squamous part of temporal bone invading the middle ear and infratemporal fossae, which was treated by en bloc resection of the lateral skull base.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Giant cell tumors (GCTs) of the bone account for 5% of all primary bone tumors [1], with the commonest sites being the epiphysis of long bones [2]. It usually affects patients between 20 and 40 years of age. For unknown reasons, the incidence of GCTs in females is higher than in males. Its occurrence in the skull, is a rare clinical entity and comprises 1–2% of giant cell tumours of the bone [3], with the majority occurring in the sphenoid bone, followed by the temporal boneThese tumors are usually benign, arises from undifferentiated mesenchymal cells of the bone marrow but are locally aggressive and has the potential to recur locally [4]. Temporal bone GCT causes otalgia, aural fullness, conductive or sensorineural hearing loss, tinnitus, localized swelling in temporal bone or preauricular region, temporomandibular joint dysfunction, and facial paralysis. While considering the benign nature of the tumor and the possibility of malignant change with radiotherapy, surgical removal of the tumor represents the best treatment option, and en bloc resection is recommended to avoid recurrence. Radiotherapy remains the only option for unresectable tumor. The role of adjuvant radiotherapy in eliminating residual tumor tissue remains controversial. Since there have been few cases of GCT of the temporal bone, best practices regarding management and treatment have not been fully developed [5]. This article describes their presentation, diagnostic workup, treatment and follow-up, as well as provides a review of the literature.
Case Summary
A 30-year-old female presented with complaints of swelling and pain in the right temporal region for 1.5 years (Fig. 1). In addition, she experienced hearing loss from right ear, tinnitus and dizziness (on and off) for last 6 months. There was no history of facial weakness or ear discharge. There was no history of trauma or infection. There was no history of headache, nausea, vomiting, or other neurological symptoms. Her family history was unremarkable.

showing the swelling in right temporal region preoperatively
On physical examination, she was healthy in appearance with no apparent abnormalities of the skull or scalp. Examination of the right ear revealed the obliteration of external auditory canal with non visualisation of the tympanic membrane. Audiologic examination demonstrated conductive hearing loss of the right ear with an air–bone gap. The cranial nerve examination showed that the facial nerve was intact. There was restricted mobility of temporomandibular joint and fullness in the preauricular region.
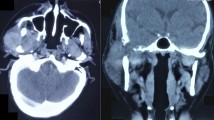
Imaging examinations (Fig. 2) using magnetic resonance imaging (MRI) suggestive of giant cell tumor of temporal bone showing low-signal-intensity area on T2-weighted images and a nonhomogenous high-signal-intensity area on T1-weighted images that measured 49*35*44 mm extending inferiorly upto infratemporal fossa and medially upto the right temporomandibular joint. Laterally the lesion showed extension upto external ear. CT scan of the temporal bone showed a large expansile heterogenous density lesion with solid cystic components noted epicentered in right temporal bone causing its destruction measuring approximately 5*4 cm extending to involve the right mandibular fossa causing erosion and destruction and medially extending into right temporal region. There is extension into the right mastoid bone causing destruction of right mastoid air cells and also extending into right middle ear. The tumor extended to the anterior wall of the right external auditory canal, resulting in obliteration of the canal.

showing a right temporal region mass in radiological imaging
Subsequently, the patient underwent surgery to remove the lesion. Intraoperative assessment demonstrated involvement of the squamous portion of temporal bone, infratemporal fossa, middle cranial fossa duramater and zygoma (Fig. 3). The tumour was removed completely with a clear resected margin. The overlying dura seemed intact. The skull base defect was covered by temporoparietal galeal and temporalis muscle grafting. There were no major intra or post-operative complications.

showing the tumor in right temporal region intraoperatively
Post operative histopathology revealed many multinucleated giant cells along with mononuclear cells in a background of fibrous stroma. Giant cells were distributed uniformly throughout the section (Fig. 4). Nuclei of mononuclear cells were round to oval to spindle and resembled those of giant cells. The final diagnosis of giant cell tumour was made based on the microscopic findings of generally benign cellular cytology, the appearance of large multinucleated giant cells, and lack of sarcomatous features. All the margins were clear except medial margin pathologically. Then the patient underwent radiation therapy and was still in follow up with no complaints. Patient has improvement in hearing, dizziness, tinnitus and pain.

showing multinucleated giant cells and mononuclear histiocytic cells (hematoxylin and eosin staining (40x)
Discussion
Sir Astley Cooper was the first to describe a GCT in 1818. Giant-cell tumors were previously known as myeloid sarcoma, tumor of myeloplexus, osteoblastoclastoma, or osteoclastoma [6]. Since the initial description of this pathological entity, there have been few reports of head and neck involvement. The first GCT affecting the temporal bone was initially described by Doderlein in 1913. These tumors are thought to originate from nonosteogenic neoplastic stromal cells [7] of the bone marrow admixed with multinucleated osteoclastic giant cells. GCT of the cranial and facial bones most commonly affect the temporal [8] and sphenoid bones due to the fact that they arise through endochondral bone formation, as do long bones. Whereas, the remainder of the skull bones develop by intramembranous ossification and are, therefore, unlikely sites for GCT. Because of its rarity in this location, it poses a diagnostic dilemma when it occurs. In the literature, 110 cases of GCT [9] of the skull have been reviewed. Tumor locations were as follows: temporal bone in 37 patients, sphenoid in 20 patients, occipital in 6 patients, frontal in 2 patients, and temporomandibular joint in 2 patients.
The clinical presentation in cases with GCT depends on the site of origin. Clinically, patients with GCT in the temporal bone typically display some degree of pain on the affected side, deafness, swelling in the affected region, and facial weakness, mostly in accordance with the site of tumor occurrence. In comparison, patients with GCT of the sphenoid bone may present with symptoms such as headache, facial hypoesthesia, diplopia, blindness, or visual field defects. Our patient showed fullness in right temporal region and decreased hearing from right ear, which was our guide in the diagnosis through biopsy. Considering the absence of the usual presentation, diagnosis of GCT without radiologic evaluation and biopsy is difficult.
Histopathological examination is essential to confirm the diagnosis of GCT. Grossly, these tumors are gray to yellow–brown, soft, or firm and friable. Small cystic areas and gray-white necrotic foci may be seen. Microscopically, GCT consists of plump spindle-shaped or ovoid cells with admixed multinucleated, cytologically benign giant cells. Variable numbers of benign multinucleated cells are seen amid sheets of benign mononuclear spindle-shaped cells with similar nuclear features. The nuclei are generally hypochromatic with inconspicuous nucleoli, and mitotic figures are uncommon. Rarely, GCT may undergo sarcomatous transformation into a malignant tumour, which may be an osteosarcoma, fibrosarcoma, or undifferentiated pleomorphic sarcoma. Malignancies can either be primary or secondary. Primary malignant GCT of bone is very rare and is evident at the first biopsy. A detailed histological examination is required to confirm diagnosis. Secondary malignant GCT of bone is more common and occurs at site of previously treated GCT. It typically occurs following radiotherapy, but it can follow surgery without adjuvant radiotherapy also. Histopathologically, GCT must be differentiated from giant cell granuloma, aneurysmal bone cyst, non ossifying fibroma, osteogenic sarcoma, benign fibrous histiocytoma, and brown tumour associated with hyperparathyroidism.
Plain radiography shows a radiolucent lesion of the skull that cannot be generally differentiated from other radiolucent lesions. On CT, GCT is seen as a lytic lesion expanding the bony cortex. These tumors are generally contrast enhancing due to their vascular nature. The tumors generally tend to expand and attenuate the bony cortex, rather than erode it. MRI of the tumor demonstrates signal isointensity on T1- weighted imaging and signal hypointensity on both T2- and diffusion-weighted imaging, with the mass showing heterogeneous enhancement after intravenous gadolinium. In this patient, CT and MRI show an expansile mass of the temporal bone extending to the soft tissues of the infratemporal fossa with indistinct margins indicating aggressive behavior.The major radiologic differential diagnoses include aneurysmal bone cyst, chondroblastoma, dermoid cyst, chondrosarcoma, giant cell reparative granuloma, and pigmented villonodular synovitis.
Surgical resection [10] is the primary management choice. Many surgical approaches [11] have been used for GCT in different portions of the temporal bone (Table 1). Although gross total resection of the tumor is ideal, this may not be feasible depending on the extent of structural involvement [12] by the tumor. In this setting, partial resection (i.e., maximal safe resection) followed by adjuvant radiotherapy may be a reasonable alternative. Once this tumor invades into muscle tissue, differentiation of tumor tissue from muscle is difficult. Accordingly, meticulous care was taken to remove the tumor under magnification. Careful follow-up of this case will be continued, although no evidence of recurrence has been seen. A recent systematic literature review of GCTBs involving the skull reported 94 patients who underwent surgery, with 37 of those patients [13] having received adjuvant radiotherapy. In a subanalysis within this review, in 62 patients for whom survival data was available, the 5-year overall survival rate was 84%, with an event-free survival rate (i.e., survival rate without tumor recurrence) of 61.3%. All 16 patients who had gross total resection (with or without adjuvant radiotherapy) were alive and event-free at 5 years. In comparison, patients treated with subtotal resection and adjuvant radiotherapy had an overall survival of 90.3% and event-free survival of 70.1%. Patients treated with subtotal resection without subsequent radiotherapy had an overall survival rate of 50% and an event-free rate of 15.4% at 5 years.
Malone et al [14] reported local control in 19 of 21 patients treated with radiation therapy, with a mean follow-up time of 15.4 years. Most patients received 35 Gy in 15 fractions delivered daily over 3 weeks. 26 Other series have also demonstrated favorable local control rates, with Roeder et al. [15] reporting local control in four of five patients treated with intensity modulated radiation therapy to amedian dose of 64 Gy using conventional fractionation. There are also reports on the use of Gamma Knife (stereotactic) radiosurgery [16] in the treatment of GCTB of the skull base, with marked reduction in tumor size and a meaningful disease-free interval. Recently, several studies have investigated the role of targeted therapy [17] with denosumab, a fully humanized monoclonal receptor activator of nuclear factor kappa-B ligand (RANK-L) antibody, which has shown promise as a potential effective chemotherapeutic treatment option in patients with GCT.
Conclusion
GCTs are rare, particularly those arising in the skull. Thus, GCTs are typically not considered among differential diagnoses of intracranial tumours. The lack of specific imaging findings can contribute to the confusion, and final diagnosis of GCTs can only be achieved with histology evaluation. This is a case of GCT in a very rare location- the squama of the right temporal bone with involvement of the right infratemporal fossa and raise awareness about consideration in the differential diagnosis of skull tumours, particularly those arising from sphenoid or temporal bones. The current study presents a less radical, but effective approach in the treatment of these lesions by means of curettage with no evidence of recurrence till now. This case management strategies are not fully fleshed out, being highly variable based on the location of the tumor and evolving understanding of the tumor's nature. Further analysis of different management strategies and characteristics of GCT of the skull is warranted to better develop treatment strategies for this disease.
References
Chen LK, Su CT, Tsai YF et al (2001) Giant cell tumor of the bone: radiography, CT, MRI, and angiography finding. Chin J Radiol. 26:61–67
Tsai Y-F, Chen L-K, Su C-T, Lee C-C, Wai C-P, Chen S-Y (2000) Giant cell tumor of the skull base: a case report. Chin J Radiol 25:223–227
Zorlu F, Selek U, Soylemezoglu F, Oge K (2006) Malignant giant cell tumor of the skull base originating from clivus and sphenoid bone. J Neurooncol 76:149–152
Venkatesh MD, Vijaya N, Girish N, Galagali JR (2012) Giant cell tumor of temporal bone: A case report. Med J Armed Forces India 68:392–394
Iizuka T et al (2012) Giant cell tumor of the temporal bone with direct invasion into the middle ear and skull base: a case report. Case Rep Otolaryngol 2012:4
Murphey MD, Nomikos GC, Flemming DJ, Gannon FH, Temple HT, Kransdorf MJ (2001) From the archives of AFIP. Imaging of giant cell tumor and giant cell reparative granuloma of bone: radiologic-pathologic correlation. Radiographics 21:1283–1309
Werner M (2006) Giant cell tumour of bone: morphological, biological and histogenetical aspects. Int Orthop 30(06):484–489
Shen Y, Ma C, Wang L, Li J, Wu Y, Sun J (2016) Surgical management of giant cell tumors in temporomandibular joint region involving lateral skull base: a multidisciplinary approach. J Oral Maxillofac Surg 74(11):2295–2311
Freeman JL, Oushy S, Schowinsky J, Sillau S, Youssef AS (2016) Invasive giant cell tumor of the lateral skull base: a systematic review, metaanalysis, and case illustration. World Neurosurg 96:47–57
Roberts DS, Faquin WC, Deschler DG (2010) Giant cell tumors of the temporal bone and infratemporal fossa: a case report and review of the literature. Laryngoscope 120(Suppl 4):S18
Mohamed A, Omi E, Honda K, Suzuki S, Sato T, Fukui N, Ishikawa K (2014) Giant cell tumor of the temporal bone invading into the pterygoid muscle through the temporomandibular joint. J Neurol Surg Rep 75(01):e136–e140. https://doi.org/10.1055/s-0034-1376428
Prasad SC, Piccirillo E, Nuseir A et al (2014) Giant cell tumors of the skull base: case series and current concepts. Audiol Neurootol 19(01):12–21
Zhang Z, Xu J, Yao Y et al (2013) Giant cell tumors of the skull: a series of 18 cases and review of the literature. J Neurooncol 115(03):437–444
Malone S, O’Sullivan B, Catton C, Bell R, Fornasier V, Davis A (1995) Long term follow-up of efficacy and safety of megavoltage radiotherapy in high-risk giant cell tumors of bone. Int J Radiat Oncol Biol Phys 33(03):689–694
Roeder F, Timke C, Zwicker F et al (2010) Intensity modulated radiotherapy (IMRT) in benign giant cell tumors–a single institution case series and a short review of the literature. Radiat Oncol 5:18. https://doi.org/10.1186/1748-717X-5-18
Kim IY, Jung S, Jung TY et al (2012) Gamma knife radiosurgery for giant cell tumor of the petrous bone. Clin Neurol Neurosurg 114(02):185–189
Xu SF, Adams B, Yu XC, Xu M (2013) Denosumab and giant cell tumour of bone-a review and future management considerations. Curr Oncol 20(05):e442–e447
Acknowledgements
None
Funding
None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Consent to participate
The authors certify that they have obtained all appropriate patient’s consent. Consent was taken for maintaining the confidentiality of the identity, images and other clinical information to be reported in the journal. The patient understands that the name and initials will not be published.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gupta, N., Rubina, Rahman, A. et al. Giant Cell Tumor of the Temporal Bone with Direct Invasion into the Middle Ear and Skull Base: A Case Report. Indian J Otolaryngol Head Neck Surg 76, 2890–2894 (2024). https://doi.org/10.1007/s12070-024-04550-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12070-024-04550-w