
Abstract
Functionalisation of nitrogen–nitrogen bonds of antisite defective boron nitride nanotubes (BNNTs), including exchange antisite defect which is produced by the rotation of BN bond, and substitutional antisite defect which is formed by substitution of an N with B, is investigated through their interaction with a \(\mathrm{B}^{-}_{6}\) cluster. The smaller defect formation energies for the substitutional antisite defects indicate that the substitution of an N atom with B atom is easier than rotation of a BN bond. The formation of antisite defects at the edge or near the edges is more favourable than that in the middle of the tubes. When complexation between double ring \(\mathrm{B}^{-}_{6}\) and nitrogen–nitrogen bonds of antisite defective BNNTs occurs, two-fold coordination, double ring configuration of boron cluster and N–N bond cleavage are seen. In the most stable complex, the \(\mathrm{B}^{-}_{6}\) pulls apart the B–N bond and becomes an integral part of the tube by expanding the hexagonal BN ring, while in the other BNNT-B6 clusters, double ring \(\mathrm{B}^{-}_{6}\) acts as a bridge at the top of the decagon. Functionalisation of N–N bonds at the edge or near the edges is more favourable than that in the middle of tubes.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In the past decades, considerable attention has been devoted to boron nitride nanotubes (BNNTs) due to their unique physical and chemical properties for various applications [1,2,3,4,5]. BNNTs, in which the boron (B) and nitrogen (N) atoms are alternatively positioned to form hexagonal boron nitride (BN) network, are isoelectric analogues of carbon nanotubes (CNTs) [6,7,8]. However, in contrast to CNTs, BNNTs have homogeneous electronic behaviours: BNNTs of different chiralities are semiconductors with almost the same optical band gaps, which are theoretically found to be 6.2 eV and experimentally determined to be 5.8 eV [2, 8]. Besides, due to the polar B–N bonds, these materials have larger surface reactivity compared to their carbon analogues [9, 10]. The effect of doping of B and N atoms on the thermoelectric properties of the \(\hbox {C}_{60}\) cage sandwiched between two metallic electrodes has been investigated by Yaghobi and Larijani [11]. Furthermore, BN-based materials are predicted to possess high sensitivity and low limits of detection, which make them suitable in applications like sensing and detecting harmful gases, such as CO, NO, \(\hbox {NO}_{2}\) and \(\hbox {NH}_{3}\) [12,13,14,15].
There are also some theoretical evidences that indicate greater adsorption energies of molecular hydrogen over BN nanostructures than their carbon analogues [16, 17].
On the other hand, it is known that similar to CNTs, BNNTs are not defect-free [18]. In fact, due to the many limitations in experimental synthesis or purification, a variety of defects are unavoidably formed in BNNTs, including substitutional impurities, vacancies, antisite defects and Stone–Wales (SW) defects [19,20,21,22,23,24]. The SW defects are topological defects involving a change in connectivity of a B–N pair due to the rotation of the midpoint of the pair.
The antisite defects could be produced by exchanging the positions of adjacent B and N atoms, or by the substitution of B / N atoms with N / B atoms [24]. The latter leads to the formation of B- or N-rich defective BNNTs in comparison with their parents. It should be noted that both of them generate energetically unfavourable homonuclear B–B or N–N bonds. Experimental as well as theoretical studies have indicated that these defective BNNTs exhibit high reactivity at the defective sites [4, 25, 26]. In fact, because of the strain of the B–B and N–N bonds, defective sites can act as Lewis acid (B-sites) and Lewis base (N-sites) centres [27]. This reactivity feature leads to chemical modifications which not only allow tuning several properties of BNNTs but also create new functionalised materials which have been an active research area [1]. Such modifications include substitutional doping and covalent and non-covalent functionalisation by a wide range of chemicals at the surface. For example, Lin et al [28] and Nag et al [29] have recently found that the amine molecules form Lewis acid–base complexes with the electron-deficient B atoms on the h-BN surface, based on a mechanism similar to that involved in the functionalisation of BNNTs [30,31,32,33,34,35]. Cano Ordaz et al [36] investigated the functionalisation of homonuclear N bonds of a BN fullerene with a B cluster to the adsorption of the antibiotic dapsone molecule, suggesting that this functionalised fullerene is a good candidate for use as a nanovehicle for drug delivery.
Anota et al [37] performed a density functional theory (DFT) study on the adsorption, activation and possible dissociation of a glucose molecule on the magnetic \([\hbox {BN fullerene}{-}\hbox {B}_{6}]^{-}\) system. They proposed that BN fullerene functionalised with a magnetic \(\hbox {B}_{6}\) cluster can be used as magnetic nanovehicles for drug delivery. Moreover, the low values of the work function indicate that they are good for technological applications such as design of magnetic devices based on organic molecules. In another work, they analysed the adsorption of the carbon monoxide (CO) molecules onto the magnetic \([\hbox {BN} ~\hbox {fullerene}{-}\hbox {B}_{6}]^{-}\) and \([\hbox {BN}~ \hbox {fullerene}{-}\hbox {C}_{6}]^{-}\) nanocomposites [38]. Based on global quantum descriptors such as polarity, the average chemical reactivity and work function, they show high retention capacity of CO molecules for these nanocomposites.
The thermal stability and chemical properties of B clusters have widely increased their use in nanomedicine devices [39,40,41,42,43]. \(\hbox {B}_{6}\) particle is able to stabilise an extra-added electron, leading to the symmetric octahedral \(\hbox {B}_{6}^{-}\) ion which is highly stable among B clusters and shows magnetic and semiconductor behaviours. These properties make the \(\hbox {B}_{6}^{-}\) cluster a promising candidate for searching novel nanodevices [44]. The absorption of the stable \(\hbox {B}_{6}^{-}\) and \(\hbox {C}_{6}^{-}\) clusters on octagraphene nanosheets induces magnetic behaviour on the functionalised sheets. The quantum descriptors obtained for these systems reveal that they are feasible candidates for designing molecular circuits, magnetic devices and nanovehicles for drug delivery [45].
For these reasons, we explore, in the present investigation, the interaction between B cluster \(\hbox {B}_{6}^{-}\) and homonuclear N–N bonds (defective sites) at the surfaces of antisite defective BN nanotubes. This study first addresses the formation of two types of antisite defects, which can be produced by \(180{^{\circ }}\) rotation of a BN bond (exchange antisite defect) or by the substitution of an N atom with a B atom (substitutional antisite defect), the former leads to defective BNNTs isoelectric with their parents while the latter leads to the N-rich defective BNNTs in comparison with their parents. Then, defective BNNTs are functionalised with the \(\hbox {B}_{6}^{-}\) anion. Such systems are chosen to address the following questions: Is the formation of antisite defects in BNNTs favourable? Which kind of antisite defects is energetically more preferable, exchange or substitutional antisite defect? Which defect site is the most favourable to be functionalised with B cluster? How do the antisite defects affect the electronic properties of perfect BNNTs? And how does the B cluster affect the electronic properties of antisite defective BNNTs?
2 Computational method
Finite models of BNNTs containing 46, 60 and 72 B atoms are used to represent defect-free (4,4), (5,5) and (6,6) BNNTs, respectively, where tips are saturated with hydrogens to avoid dangling bonds. As possible active sites of BNNTs for chemical modifications, defective or functionalised sites are at the edges or near the edges. Besides, in the middle of the tubes, periodic models are not appropriate; periodic boundary conditions cannot be applied to mimic edge modifications due to the interruption of translational symmetry. Thus, the molecular models seem a reasonable approach. It is shown that the inclusion of dispersion corrections and diffuse sp-functions for electronegative atoms is compulsory for studying the chemical reactivity and interaction energy of nanotubes. The M06-2X functional belongs to a new generation of hybrid metageneralised gradient approximation exchange correlation functional which includes an accurate treatment of the dispersion energy. Therefore, the M06-2X functional [46] in combination with the 6-311\(+\)G(d,p) basis set is used for geometry optimisations and single-point energy calculations [47,48,49,50]. Frequency calculations are carried out for all the systems at the same level of theory, and the actual obtained frequencies confirm that all of them are structures with minimum energies. All DFT calculations are performed using GAMESS suite of programs [51, 52].
3 Results and discussion
The prefect (4,4), (5,5) and (6,6) armchair BNNTs, which are fully optimised at the M06-2X / 6-311\(+\)G(d, p) level, are chosen as the parent molecules for defective BNNTs. The B–N bond lengths in the middle of the optimised prefect BNNTs are found to be 1.449–1.455 Å, which correspond to the previously reported values (1.401 and 1.458 Å) [53]. As shown in figure 1, 6 non-equivalent B atoms and 12 non-equivalent BN bonds can be recognised in these BNNTs. Then the antisite defects are produced at different positions (at the edges, near the edges or in the middle of the tubes), based on two ways: antisite defective BNNTs are presented as (\(\hbox {N}_{i}\))BNNT (\(i = 1{-}6\)), in which the antisite defect is produced by the substitution of an N atom with the \(\hbox {B}_{i}\) atom, and as (\(\hbox {NB}_{i}\))BNNT (\(i = 1{-}12\)), in which the antisite defect is formed by exchanging the position of the \(\hbox {B}_{i}\) atom and its adjacent N atom. Fully optimised at the M06-2X / 6–311\(+\)G(d,p) level, the geometries of substitutional and exchange antisite defective BNNTs are shown in figures 2 and 3, respectively. As observed, the substitutional antisite defects lead to the formation of N-rich BNNTs. At this point, it is necessary to mention that the primary purpose of this research is to study the possibility of antisite defects in the BN nanotubes and the trend of some structural and electronic parameters in the considered models. Therefore, to reduce the computational cost, we consider only the armchair BNNTs in this study.

Optimised structures of (4,4) BNNT. Six non-equivalent B atoms and 12 non-equivalent BN bonds are shown in bold and italic forms.

The optimised geometries of the six configurations of substitutional antisite defective BNNTs (N1–N6–BNNTs) in which the antisite defects were produced by the substitution of B atoms with N atoms.

The optimised geometries of 12 configurations of exchange antisite defective BNNTs, (NB1)–(NB12)–BNNTs, in which the antisite defects were produced by exchange positions of adjacent B and N atoms.
The defect formation energies with zero-point energy (ZPE) corrections, \(E_{\mathrm{D}}\), required to form exchange and substitutional antisite defects are obtained using eqs (1) and (2), respectively:
where \(E_{\mathrm{defective}}\) and \(E_{\mathrm{perfect}}\) stand for the total energies of an antisite defective BNNT and its corresponding perfect BNNT, respectively, \(E_{\mathrm{N}}\) and \(E_{\mathrm{B}}\) represent the energies of N and B atoms. The total energy (\(E_{\mathrm{T}}\)), defect formation energy (\(E_{\mathrm{D}}\)) and HOMO–LUMO (highest occupied molecular orbital–lowest unoccupied molecular orbital) energy gap (\(E_{\mathrm{g}}\)) of the antisite defective BNNTs along with those of their parents are listed in table 1.
As seen in table 1, defect formation energies for the antisite defective BNNTs are found to be positive, indicating that the formation of exchange and substitutional antisite defects in the BNNTs is endothermic. However, the substitutional antisite defective BNNTs, (\(\hbox {N}_{i}\))BNNTs, have smaller defect formation energies than the corresponding exchange antisite defective BNNTs, (\(\hbox {NB}_{i}\))BNNTs, which indicate that the substitutional antisite defects are energetically easier to be formed in the armchair BNNTs than with those in exchange antisite defects. This can be due to the formation of fewer unfavourable homonuclear N–N and B–B bonds in the former. It can also be seen in figure 4 that the formation of antisite defects at the edge or near the edges is more favourable than that in the middle of the tubes. The most stable isomers among the exchange antisite defective BNNTs and among the substitutional antisite defective BNNTs are mainly found to be (\(\hbox {NB}_{1}\))BNNTs and (\(\hbox {N}_{1}\))BNNTs, respectively. Isomers of (\(\hbox {N}_{i}\))BNNTs and (\(\hbox {NB}_{i}\))BNNTs with \(i \ge 3\), where anitsite defects are produced in the middle of the tube, have almost the same values of defect formation energies. Therefore, the orientation of the defects on the BNNT surface has an energetically minor effect on the formation of antisite defective BNNTs. The defect formation energies for the most stable antisite defective isomers of BNNTs are calculated to be 2.73, 2.90 and 2.99 eV in \(\hbox {N}_{1}\)(4,4), \(\hbox {N}_{1}\)(5,5) and \(\hbox {N}_{1}\)(6,6), and 3.46, 3.62 and 4.08 eV in \(\hbox {NB}_{1}\)(4,4), \(\hbox {NB}_{1}\)(5,5) and \(\hbox {NB}_{1}\)(6,6), respectively, which means that the formation of antisite defects in the narrower BNNTs is more favourable which can be due to the higher curvature and more reactivity of these BNNTs.

Defect formation energies and HOMO–LUMO gaps of substitutional and exchange antisite defective BNNTs.
Due to the formation of more homonuclear B–B and N–N bonds, defect formation energies for the antisite defective BNNTs are found to be higher than those for SW defective BNNTs previously obtained by Li et al [54]. In fact, the conversion of B–N bonds to homonuclear B–B or N–N bonds leads to an instability in the structures of the tubes, known as the bond frustration effect [55]. Inspection of the structures of the antisite defective BNNTs shows that the change in bond lengths only occurs for B–N bonds near the defective sites, which means that the bond frustration effect is mainly due to a local strain. The lengths of the homonuclear N–N bonds in the exchange antisite defective BNNTs are found to be 1.464–1.499 Å, which are shorter than those obtained in the substitutional antisite defective BNNTs (1.493–1.548 Å). Moreover, homonuclear B–B bonds in the exchange antisite defective BNNTs are found to have lengths of 1.620–1.692 Å. This kind of bond, as seen in figure 2, is not found in the substitutional antisite defective BNNTs. These values are similar to those previously computed in SW defective BNNTs [54, 55].
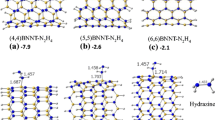
Six homonuclear N–N bonds with different positions at the surface of isomers of antisite defective BNNTs are considered for chemical functionalisation with the charged B cluster \(\hbox {B}_{6}^{-}\). Sites 1, 3 and 5 refer to the N–N bonds (defective sites) that lie perpendicular to the tube axis, and sites 2, 4 and 6 are diagonal N–N bonds. F1 refers to the functionalisation of N–N bonds that lie at the edges of tubes, while functionalised N–N bonds in F2 and F3 are near the edges, and finally N–N bonds that are functionalised in F4–F6 positions are close to the middle of the tubes (see figure 5). To produce these complexes, the charged B cluster \(\hbox {B}_{6}^{-}\) is initially placed directly above the N–N bond of the defective BNNTs, and then geometries are fully optimised. As the \(\hbox {B}_{6}^{-}\) cluster is composed of two recursive tetrahedrons with high symmetry, it has practically the same possibility to be adsorbed through each end. The lowest energy states for these configurations are found with the anionic charge (\(Q = -1\)) and multiplicity of \(M = 2S_{\mathrm{T}} + 1 = 2\), where \(S_{\mathrm{T}}\) is the total spin. To avoid the states of energetic degeneracy, the energy differences (\(\Delta E\)) between the multiplicities close to the most energetic of F1–F6 are calculated and found to be in the range between 1.25 and 2.66 eV, where the energies are computed using the M06-2X / 6-311\(+\)G(d,p) for \(M = 4\). The optimised geometries of the \(\hbox {BNNT}{-}\hbox {B}_{6}\) complexes, F1–F6, are presented in figure 5.

Functionalisation of the homonuclear N–N bonds of antisite defective BNNTs by the \(\hbox {B}_{6}\) boron cluster and with respect to different antisite defects on the BNNT surface.
Interaction energies (\(E_{\mathrm{Int}}\)) with ZPE corrections are calculated based on the following equation, referring to the energy difference between the complex and the constituents, and summarised in table 2:
where E is the electronic energy. The values of \(E_{\mathrm{Int}}\) are found to be negative, which shows the exothermic character of the functionalisation process. This means that the interactions are attractive and favourable.
The interaction of B cluster with the N–N bonds at the defective sites leads to the cleavage of these bonds, which is indicated by the (non-bonding) 3.13–3.75 Å distance. As seen from figure 5, two-fold coordination (binding in two points) occurs between double ring \(\hbox {B}_{6}^{-}\) and defective BNNT surface. In the most stable complexes of (4,4) defective \(\hbox {BNNT}{-}\hbox {B}_{6}^{-}\), i.e. F1, the \(\hbox {B}_{6}\) cluster pulls apart the N–N bond of the tube at site 1 and becomes an integral part of the tube by expanding the original hexagonal ring at the tube surface (see figure 5). This N–N bond cleavage and expansion of the surface stabilise the complex F1 by more than 10 kcal / mol. Drastic structural changes are noticed for the B cluster functionalised on the antisite defective BNNT surfaces. B–B bond lengths of the adsorbed double ring \(\hbox {B}_{6}\) cluster are found to be within 1.571–1.838 Å.
In the other \(\hbox {BNNT}{-}\hbox {B}_{6}\) clusters (F2–F6), the \(\hbox {B}_{6}\) cluster acts as a bridge at the top of the BNNT, rather than being a part of the tube as in F1. In these complexes, the bond lengths of the adsorbed double ring \(\hbox {B}_{6}\) cluster are found to be within 1.555–1.835 Å and all BN bonds of the formed decagon are calculated to be in the range of 1.449–1.512 Å.
It should be noted that the functionalisation of the N–N defective sites at the edges or near the edges (F1–F3) is found to be more favourable than that of the defective sites in the middle of the tube. Also as seen in table 2, functionalisation of diagonal N–N bonds is more favourable than the N–N bonds that lie perpendicular to the tube axis. For example, \(E_{\mathrm{Int}}\) values obtained for the diagonal N–N bonds positioned close to the middle of the tubes are about \(-7.85\) and \(-8.90\) eV, in F6, and F4, respectively, more negative than the \(E_{\mathrm{Int}}\) value of \(-7.07\) eV obtained for the perpendicular N–N bond in F5. However, functionalisation of diagonal and perpendicular N–N bonds near the edges leads to the essentially isoenergetic isomers of F2 and F3, lying within 0.01 eV of one another.
Previous theoretical studies pointed out that the electronic structures of nanotubes can be modified in the presence of some defects or addends [56]. Therefore, one purpose of the study of the defects and exohedral functionalisation of nanotubes is to modify the electronic structures of these systems. The energy differences between the HOMO and the LUMO for the pristine, defective and functionalised models of the tubes are calculated and listed in tables 1 and 2. From the results of \(E_{\mathrm{g}}\), it can be concluded that the antisite defects noticeably influence the electronic properties of the semiconductor BNNTs. In other words, antisite defects would result in the reduction of \(E_{\mathrm{g}}\) from 5.972, 5.989 and 6.087 eV in the (4,4), (5,5) and (6,6)BNNTs to 5.333–5.565, 5.165–5.322 and 5.125–5.268 eV in the exchange antisite defective (4,4), (5,5) and (6,6), respectively, BNNTs, and to 3.993–5.506, 3.925–5.467 and 4.034–5.412 eV in the substitutional antisite defective (4,4), (5,5) and (6,6)BNNTs, respectively [54]. On the other hand, functionalisation of N–N bonds at the edges (F1), near the edges (F2 and F3) and the middle of the tubes (F4–F6) leads to a decrease of \(E_{\mathrm{g}}\) values to 3.12 eV (by about 27.99% change), 3.51 and 3.52 eV (by about 13.33 and 13.67% changes), and 3.06–3.27 eV (by about 20.93–25.54% changes), respectively. It is noted that the main purpose of this study at this point is the evolution of the HOMO–LUMO gaps in the defective and functionalised models, and this is just an approximated comparison, not to find precise HOMO–LUMO gaps for a given composition.
The efficiency of a molecule as an electron acceptor depends not only on its electron affinity to effectively absorb electrons from adjacent donor molecules but also on its resistance against electron back transfer to donor molecules. In this respect, a global index called electrophilicity has been defined by Parr et al [57] in terms of the quantitative chemical concepts in DFT. As most of the reactions can be analysed through the electrophilicity of various species involved in the process, a proper understanding of these properties becomes essential. Anafcheh et al [58] and Anafcheh and Ghafouri [59] used electrophilicity index to investigate the propensity of the \(\hbox {C}_{60}\) and \(\hbox {C}_{70}\) fullerene derivatives to acquire an electron as the acceptor in bulk heterojunction solar cells and \((\hbox {SiH})_{48}\hbox {X}_{12}\) heterofullerenes in which X atoms are the groups III and V dopants. Such a quantity can be assigned as a basis for evaluating the electrophilic competence of a system by applying \(\mu \) as a reliable factor for measuring the propensity of the system to acquire an additional electron from the adjacent electron-rich species and simultaneously using \(\eta \) for describing the resistance of the system to exchange electron with the environment. Then, the electrophilicity index is given by [60]
in which electronic chemical potential (\(\mu \)) and hardness (\(\eta \)) can be given by
It is worthwhile to note that these relations are just approximations for \(\mu \) and \(\eta \). A way of defining \(\mu \) and \(\eta \) is through Janak’s approximation [33] where the first ionisation potential (IP) and electron affinity (EA) are calculated under the Koopmans’ theorem, based on the frozen orbital approximations, and the finite difference approach, so that they are expressed in terms of the HOMO energy (\(\upvarepsilon _{\mathrm{HOMO}}\)) and the LUMO energy (\(\varepsilon _{{\mathrm{LUMO}})}\!: \hbox {IP} \approx - \varepsilon _{\mathrm{HOMO}}\), \(\hbox {EA}\approx - \varepsilon _{\mathrm{LUMO}}\).
Therefore, \(\omega \) values are calculated for the pristine, antisite defective (4,4), (5,5) and (6,6) BNNTs and functionalised models based on the above formula. Electrophilicity for the pristine (4,4), (5,5) and (6,6) BNNTs are found to be 0.92, 0.90 and 0.88 eV, respectively.
Our results indicate that both exchange and substitutional defects lead to an increase in the electrophilicity values (1.13–1.59 eV). Moreover, the electrophilicity values of the functionalised antisite defective BNNTs (\(\hbox {BNNT}{-}\hbox {B}_{6}\) clusters F2–F6) are found to be greater than those of their parents (2.35–4.02 eV). It is worthwhile to note that the electrophilicity values of the complexes of \(\hbox {BNNT}{-}\hbox {B}_{6}\) in which \(\hbox {B}_{6}\) cluster binds to a N–N bond at the edge or near the edges (F1–F3) are smaller than those of the complexes in which the \(\hbox {B}_{6}\) cluster binds to defective sites in the middle of the tubes (F4–F6).
4 Conclusion
We have performed a computational study to investigate the functionalisation of homoelemental N–N bonds of antisite defective BNNTs through their interaction with the boron \(\hbox {B}_{6}\) cluster. The formation of two types of antisite defects, exchange antisite defect formed by \(180{^{\circ }}\) rotation of a BN bond and the substitutional antisite defect produced by the substitution of the natural N atom with a B atom, are investigated. According to the obtained results, we emphasise the following points: (1) the smaller defect formation energies of the substitutional antisite defective BNNTs indicate that the substitution of an N atom with a B atom in the BNNTs is energetically easier than with the \(180{^{\circ }}\) rotation of a BN bond, (2) the formation of antisite defects at the edge or near the edges is energetically more favourable than the defects in the middle of the tubes, (3) the formation of antisite defects in the narrower BNNTs is more energetically favourable, which can be due to the higher curvature in these BNNTs, (4) drastic structural changes, double ring configuration, two-fold coordination and the N–N bond cleavage were observed in the complexes of BNNT–\(\hbox {B}_{6}\), (5) in the most stable complex of BNNT–\(\hbox {B}_{6}\), (F1), the \(\hbox {B}_{6}\) cluster pulls apart the B–N bond of the tube and becomes an integral part of the tube by expanding the original hexagonal BN ring at the tube surface while in the other BNNT-\(\hbox {B}_{6}\) complexes (F2–F6), double ring \(\hbox {B}_{6}\) acts as a bridge at the top of the decagon and (6) it is noted that the N–N bonds at the edge or near the edges are more reactive than the N–N bonds in the middle of the tube and functionalisation of diagonal N–N bonds at the middle of the tubes is more favourable than the N–N bonds that lie perpendicular to the tube axis.
References
C W Chang, W Q Han and A Zettle, Appl. Phys. Lett. 86, 173102 (2005)
X Blase, A Rubio, S Louie and M Cohen, Eur. Phys. Lett. 28, 335 (1994)
D Golberg, Y Bando, M Eremets, K Takemura, K Kurashima, K Tamiya and H Yusa, Chem. Phys. Lett. 279, 191 (1997)
W An, X Wu, J L Yang and X C Zeng, J. Phys. Chem. C 111, 14105 (2007)
L Lai, W Song, J Lu, Z Gao, S Nagase, M Ni, W N Mei, J Liu, D Yu and H Ye, J. Phys. Chem. B 110, 14092 (2006)
N Hamada, S L Sawada and A Oshiyama, Phys. Rev. Lett. 68, 1579 (1992)
D Golberg, Y Bando, Y Huang, T Terao, M Mitome, C Tang and C Zhi, ACS Nano 4, 2979 (2010)
M Terauchi, M Tanaka, M Takehisa and Y Saito, J. Electron. Microsc. 47, 319 (1998)
X Blase, A Rubio, S G Louie and M L Cohen, Europhys. Lett. 28, 335 (1994)
A Rubio, J L Corkill and M L Cohen, Phys. Rev. B 49, 5081 (1994)
M Yaghobi and F A Larijani, Pramana – J. Phys. 84, 155 (2015)
R J Baierle, T M Schmidt and A Fazzio, Solid State Commun. 142, 49 (2007)
S S Varghese, S Swaminathan, S K Kumar and V Mittal, Comput. Condens. Matter 9, 40 (2016)
K Xu, C Fu, Z Gao, F Wei, Y Ying, C Xu and G Fu, Instrum. Sci. Technol. 46, 115 (2018)
W Yang, L Gan, H Li and T Zhai, Inorg. Chem. Front. 3, 433 (2016)
T Oku, Energies 8, 319 (2015)
Q Sun, Q Wang and P Jena, Nano Lett. 5, 1273 (2005)
H Wang, N Ding, X Zhao and C-M L Wu, J. Phys. D Appl. Phys. 51, 125303 (2018)
J Song, H Jiang, J Wu, Y Huang and K-C Hwang, Scr. Mater. 57, 571 (2007)
G Kim, J Park and S Hong, Chem. Phys. Lett. 522, 79 (2012)
X Shu-Wen, C Jian and Z Jun, Chin. Phys. Lett. 30, 103102 (2013)
M G Mashapa, N Chetty and S S Ray, J. Nanosci. Nanotechnol. 12, 7021 (2012)
M G Mashapa, N Chetty and S S Ray, J. Nanosci. Nanotechnol. 12, 7796 (2012)
H S Kang, J. Phys. Chem. B 110, 4621 (2006)
A Zobelli, C P Ewels, A Gloter, G Seifert, O Stephan, S Csillag and C Colliex, Nano Lett. 6, 1955 (2006)
J-X Zhao and Y-H Ding, J. Chem. Phys. 131, 014706 (2009)
R Sundaram, S Scheiner, A K Roy and T Kar, Phys. Chem. Chem. Phys. 17, 3850 (2015)
Y Lin, T V Williams and J W Connell, J. Phys. Chem. Lett. 1, 277 (2010)
A Nag, K Raindogia, K P S S Hembram, R Datta, U V Waghmare and C N R Rao, ACS Nano 4, 1539 (2010)
S-Y Xie, W Wang, K A S Fernando, X Wang, Y Lin and Y-P Sun, Chem. Commun. 29, 3670 (2005)
D Golberg, Y Bando, C C Tang and C Y Zhi, Adv. Mater. 19, 2413 (2007)
S Pal, S R C Vivekchand, A Govindaraj and C N R Rao, J. Mater. Chem. 17, 450 (2007)
X Wu, W An and X C Zeng, J. Am. Chem. Soc. 128, 12001 (2006)
T Ikuno, T Sainsbury, D Okawa, J M J Frechet and A Zettl, Solid State Commun. 142, 643 (2007)
A Maguer, E Leroy, L Bresson, E Doris, A Loiseau and C Mioskowski, J. Mater. Chem. 19, 1271 (2009)
J Cano Ordaz, C Anota, M S Villanueva and M Castroc, New. J. Chem. 41, 8045 (2017)
E C Anota, M S Villanueva, E Shakerzadeh and M Castro, Appl. Nanosci. 8, 455 (2018)
E Chigo Anota, M Salazar Villanueva, A Bautista Hernández, W Ibarra Hernández and M Castro, Appl. Phys. A 124, 590 (2018)
S Mukherjee and P Thilagar, Chem. Commun. 52, 1070 (2016)
N S Hosmane, Boron science: New technologies and applications (CRC Press, Francis & Taylor Group, Boca Raton, Florida, 2012)
Z J Leśnikowski, J. Med. Chem. 59, 7738 (2016)
R N Grimes, J. Chem. Educ. 81, 657 (2004)
J Aihara, J. Am. Chem. Soc. 100, 3339 (1978)
E C Anota, M S Villanueva, S Valdez and M Castro, Struct. Chem. 28, 1757 (2017), https://doi.org/10.1007/s11224-017-0953-8
E Chigo-Anota, G Cárdenas-Jirón, M Salazar Villanueva, A Bautista Hernández and M Castro, Physica E 94, 196 (2017)
Y Zhao and D G Truhlar, Theor. Chem. Acc. 120, 215 (2008)
P C Hariharan and J A Pople, Mol. Phys. 27, 209 (1974)
Y Zhang, A Wu, X Xu and Y Yan, J. Phys. Chem. A 111, 9431 (2007)
R Ghafouri and F Ektefa, Struct. Chem. 26, 507 (2015)
M Anafcheh and R Ghafouri, Physica E 56, 351 (2014)
M W Schmidt, K K Baldridge, J A Boatz, S T Elbert, M S Gordon, J H Jensen, S Koseki, N Matsunaga, K A Nguyen, S J Su, T L Windus, M Dupuis and J A Montgomery, J. Comput. Chem. 14, 1347 (1993)
M S Gordon and M W Schmidt, Theory and applications of computational chemistry: The first forty years edited by C E Dykstra, G Frenking, K S Kim and G E Scuseria (Elsevier, Amsterdam, 2005)
S-P Ju, Y-C Wang and T-W Lien, Nanoscale Res. Lett. 6, 160 (2011)
Y Li, Z Zhou, D Golberg, Y Bando, P V R Schleyer and Z Chen, J. Phys. Chem. C 112, 1365 (2008)
S Sinthika, E M Kumar, V J Surya, Y Kawazoe, N Park, K Iyakutti and R Thapa, Sci. Rep. 5, 17460 (2015)
R Saito, G Dressehaus and M S Dresselhaus, Physics properties of carbon nanotubes (World Scientific, New York, 1998)
R G Parr, L V Szentpály and S Liu, J. Am. Chem. Soc. 121, 1922 (1999)
M Anafcheh, R Ghafouri and N L Hadipour, Sol. Energy Mater. Sol. Cells 105, 125 (2012)
M Anafcheh and R Ghafouri, J. Cluster Sci. 25, 505 (2014)
R G Parr, R A Donnelly, M Levy and W E Palke, J. Chem. Phys. 68, 3801 (1978)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Anafcheh, M., Shahbaz, N. & Zahedi, M. N–N bond cleavage and ring expansion at the surface of exchange and substitutional antisite defective boron nitride nanotubes by boron cluster: A density functional theory study. Pramana - J Phys 93, 64 (2019). https://doi.org/10.1007/s12043-019-1825-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12043-019-1825-0