
Abstract
MADS-box transcription factors are essential mediators of the vegetative and reproductive development of seed plants. Although MADS-box genes have been extensively characterized in angiosperms, their functions are not well-understood in gymnosperms, especially the Gnetales, due to an ambiguous phylogeny of seed plants. Here, we performed a genome-wide search for MADS-box genes in Gnetum luofuense and found 11 Type I and 38 Type II MADS-box members (i.e. three MIKC* and 35 MIKCc genes). The relative abundance of the Type I Mα and Type II MIKCC subgroups (including the DEF/GLO and TM8-like genes) were mainly contributed by tandem duplications. Comparisons of the gene expression levels among members of the MIKCc subgroup reveal that the DEF/GLO-like genes and several TM8-like genes were exclusively expressed in reproductive organs, whereas TM3-like, StMADS11-like and other TM8-like genes exhibited a broad expression pattern in both vegetative and reproductive organs in G. luofuense. In addition, 14 Type II MIKCc genes were found in the stem transcriptome of Ephedra equisetina and we made an attempt to assess the homology to the MIKCc genes within Gnetales. The results of this study provide valuable information for understanding the phylogenies and functions of MADS-box genes in seed plants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Members of the MADS-box family encode transcription factors (TFs) in seed plants (Garcia-Maroto et al. 2003; Gramzow and Theissen 2010; Gramzow et al. 2014; Masiero et al. 2011; Melzer et al. 2010) that likely originated from the common ancestor of extant eukaryotes (Alvarez-Buylla et al. 2000; Gramzow and Theissen 2010). There are two types of MADS-box genes—Type I (SERUM RESPONSE FACTOR, SRF-like) and Type II (MYOCYTE ENHANCER FACTOR, MEF-2 like). Type I MADS-box genes can be further divided into the Mα, Mβ, and Mγ groups based on phylogeny. Type II MADS-box genes are classified into two groups, namely MIKC* and MIKCC, depending on phylogeny and the length of their Keratin-like (K) domains (Kwantes et al. 2012; Parenicova et al. 2003). Type I MADS-box TFs are known to participate in female gametophyte as well as embryo and seed development (Colombo et al. 2008; Masiero et al. 2011; Wuest et al. 2010), whereas the MIKC-type TFs regulate almost all aspects of sporophytic and gametophytic development in seed plants (Gramzow and Theissen 2010; Gramzow et al. 2014; Smaczniak et al. 2012).
Order Gnetales comprises three extant genera—Ephedra, Welwitchia and Gnetum (Kubitzki 1990b; Price 1996). Gnetum contains approximately 40 species, including trees, shrubs and lianas distributed in pantropical forests (Kubitzki 1990a; Markgraf 1930; Price 1996). Previous phylogenetic studies based on molecular data revealed that South American, African, and Asian Gnetum constitute three major lineages (Hou et al. 2015; Won and Renner 2003, 2006). Except for African Gnetum, almost all extant species have bisexual but functionally unisexual structures—reproductive organs are typical male and female strobili that bear multiple layers of involucre collars (Fig. 1). Each collar of the female strobilus has one roll of fertile reproductive units whereas the male strobilus bears one roll of sterile ovules subtended by four to five rolls of microsporangia (Endress 1996; Jörgensen and Rydin 2015; Markgraf 1930). Phylogenies based merely on morphological data suggest that Gnetum is closely related to angiosperms (Crane 1985; Doyle and Donoghue 1986), but a growing body of molecular data suggest that this genus is a sister group to conifers (Ran et al. 2018; Wan et al. 2018; Wickett et al. 2014). This ambiguity in seed plant phylogeny warrants further investigation of the MADS-box TFs to better understand their functions in the reproductive evolution of Gnetales.

a Mature and developing female strobili of Gnetum luofuense. b Mature and developing male strobili of Gnetum luofuense. c Female strobili with secreted pollination droplets d Male strobili with secreted pollination droplets. White bars represent 10 mm
Previous studies of Gnetum MADS-box genes mainly focused on the diversity and functions of MIKCC members. For example, three genes, GpMADS1, GpMADS3 and GpMADS4 were found to participate in the development of female reproductive units in G. parvifolium (Shindo et al. 1999). Functions of 19 G. gnemon MIKCC genes (i.e. GGM1-GGM19) were inferred by comparing their expression profiles in leaves, female strobili and male strobili (Becker et al. 2003; Becker et al. 2000; Winter et al. 1999). GGM2, GGM3, GGM9, and GGM11 were found to form a quartet-like complex that determines the sexual identity of G. gnemon (Wang et al. 2010). In addition, a recent study using transcriptome data identified 35 MIKCC genes in G. gnemon, half of which were TM8-like genes (Gramzow et al. 2014). Furthermore, GpMADS3 regulates the transition from shoot meristem to floral primordium in a FLORICAULA/LEAFY-dependent manner (Shindo et al. 2001).
Despite these previous reports, our knowledge of Gnetum MADS-box genes is still limited. Firstly, the total number of Type I MADS-box genes is not known, presumably because they express at low levels, lack obvious mutant phenotypes, and may be functionally redundant in many plants (Gramzow and Theissen 2010; Gramzow et al. 2014; Masiero et al. 2011). Secondly, tandem duplications of TM3, SQUA, AGL6 and TM8 (Zhao et al. 2017) have been detected among subgroups of angiosperms, whereas those of the MIKCc members in Gnetum remain to be characterized. Thirdly, the functions of MIKCc genes—e.g., StMADS11, TM3 and TM8—in sex determination and the development of female and male strobili in G. gnemon have not been well understood, let alone their functions in other vegetative and reproductive organs, e.g. roots, stems, and seeds. Finally, studies of the MADS-box genes in other gnetalean genera, such as Ephedra and Welwitschia, are scare, and only five MADS-box TFs have been characterized in W. mirabilis thus far (Moyroud et al. 2017).
To address these questions, we surveyed Type I and Type II MADS-box genes in nuclear genome of G. luofuense (which was mis-identified as “G. montanum”, see details below)(Wan et al. 2018). The availability of genome makes it possible to accurately identify the MIKCc members in Gnetum, which potentially avoids the scenario that many pseudogenes were detected in conifers (Gramzow et al. 2014). We also conducted phylogenetic analyses on both types of MADS-box genes as well as the TM8 genes (MIKCC genes), which have not been extensively investigated in previous studies. These analyses were carried out using sequences derived from genome-wide screening of other seed plants and previous studies. Moreover, we analyzed the transcript profiles of the G. luofuense MIKCc genes in three vegetative and three reproductive tissues to infer their functions and evolutionary histories. In addition, a genome-wide search was performed to detect tandem duplications. Finally, we identified the MIKCc genes in Ephedra equisetina and determined their homology to those found in G. luofuense.
Results
Diversity of the MADS-Box Genes in G. luofuense and E. equisetina
This is the first time of conducting genome-wide search for MADS-box genes in the Gnetales. We identified a total of 49 candidate MADS-box genes in G. luofuense, including 11 Type I and 38 Type II genes, accounting for 22.4% and 77.6% of all MADS-box genes found, respectively (Figs. 2 and 3). The Type II G. luofuense MADS-box genes consisted of three MIKC* and 35 MIKCc members, which were further divided into 12 subgroups including StMADS11(“1” indicates gene numbers thereafter), GpMADS4 (2), GGM19 (1), AGL15 (2), DEF/GLO (4), B-sister (1), AGL6 (2), SQUA (1), AG (1), AG12 (1), TM3 (1), and TM8 (18). In addition, 14 MIKCc genes were identified in E. equisetina based on the transcriptome data and these were divided into ten subgroups, including StMADS11 (2), GpMADS4 (1), GGM19(1), GGM5 (1), DEF/GLO (3), B-sister (1), AGL6 (2), SQUA (1), TM3 (1), and TM8 (1)(Fig. 3). The elucidation of the numbers of MADS-box genes in G. luofuense and E. equisetna paves pathways for the subsequent analyses.

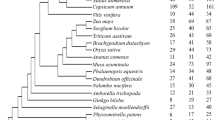
Phylogeny of Type I MADS-box genes and Type II MIKC*genes in G. luofuense (solid red dots), Arabidopsis thaliana and Oryza sativa plotted by the maximum likelihood method. Bootstrap values ≥50 are present on each node of the phylogeny. The accession information used for phylogenetic reconstructions was provided in the supplementary material

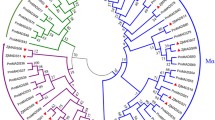
Phylogenetic relationships among the MIKCC genes plotted by the maximum likelihood method. The resulting phylogeny representing 16 MIKCC subgroups was built using nine seed plant species including Amborella trichopoda (Atr), Arabidopsis thaliana (Ath), Ephedra equisetina (Eeq), Gnetum gnemon (Ggn), Gnetum luofuense (Glu), G. parvifolium (Gpa), Pinus taeda (Pta), Oryza sativa (Osa) and Welwitchia mirabilis (Wmi). Bootstrap values ≥50 are given at each node. The MIKCC genes identified in G. luofuense and E. equisetina are indicated by red and yellow dots, respectively. The accession information used for phylogenetic reconstructions was provided in the supplementary material
Phylogenies of the G. luofuense and E. equisetina MADS-Box Genes
Phylogeny of Type I and Type II MIKC* Genes
To elucidate the evolution of MADS-box genes in G. luofuense and E. equisetna, we conducted three phylogenetic analyses for different types of MADS-box genes. In general, the deep divergence of the phylogenies based on Type I and Type II MADS-box genes in different seed plant groups all had low statistical support (see Figs. 2, 3 and 4). We identified eleven Type I MADS-box genes in G. luofuense (Fig. 2). The phylogeny of Type I genes reveals that seven Mα genes in G. luofuense clustered into one clade with a bootstrap value (BS) of 65, designated clade A, which was nested within a clade that consists of 24 Arabidopsis thaliana Mα genes and 11 Oryza sativa genes. Three G. luofuense Mβ genes formed a monophyletic clade (designated clade B), which is nested within a group of 21 and four Mβ genes in A. thaliana and O. sativa, respectively. In addition, one G. luofuense Mγ gene fell into a clade that comprised 15 A. thaliana and ten O. sativa Mγ genes. In addition, three G. luofuense MIKC* genes were identified, of which one formed an S-clade with three A. thaliana and two O. sativa MIKC* genes; whereas the other two G. luofuense MIKC* genes formed a P-clade with three A. thaliana MIKC* genes (BS = 97).

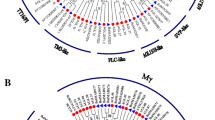
Phylogenetic relationships among TM8-like genes plotted using the maximum likelihood method. The phylogeny was built based on 18 TM8-like genes from G. luofuense, 21 from Gnetum gnemon, one from Ephedra equisetina (Eeq) and 23 from the following representative land plants: Amborella trichopoda (Atr), Cephalotaxus harringtonia (Cha), Cycas elongate (Cel), Ginkgo biloba (Gbi), Picea abies (Pab), Picea sitchensis (Psi), Pinus banksiana (Pba), Pinus contorta (Pco), Pinus pinaster (Ppi), Pinus taeda (Pta), Sciadopitys verticillata (Sve), Solanum lycopersicum (Sly) and Wollemia nobilis (Wno). Bootstrap values≥50 are given on each node. The accession information used for phylogenetic reconstructions was provided in the supplementary material. A scale bar was provided at the right corner
Phylogenies of the Type II MIKCc Genes
The MIKCC phylogeny corroborates the delimitation between gymnosperm and angiosperm MIKCc genes (Fig. 3). For example, the TM3-like genes were subdivided into two clades, clade A (BS = 59) contained one G. luofuense, one G. gnemon, one E. equisetina and three P. taeda genes; where a clade B consisted of six A. thaliana, one Amborella trichopoda and two O. sativa MIKCc genes (BS = 78). Another example shows that three SQUA-like genes, one from G. luofuense, one from G. gnemon and one from E. equisetina, were excluded from clade C (BS = 75), which contained four A. thaliana, one A. trichopoda and four O. sativa SQUA-like genes. Moreover, clade D (BS = 95)—comprised three AGL12-like genes from G. luofuense, G. gnemon and P. taeda—was sister to clade E (BS = 100), which comprised one A. thaliana, one A. trichopoda and two O. sativa AGL12-like genes. Five StMADS11-like genes in gymnosperms formed two paraphyletic groups, each was sister to clade F (BS = 76), which consisted of two A. thaliana, one A. trichopoda and three O. sativa StMADS11-like genes.
In general, these results evidenced the homology among the MIKCc genes in Gnetales (Fig. 3). For example, three AG-like genes—one from G. gnemon, one from G. luofuense and one from W. mirabilis—formed a clade (designated clade G, BS = 82), which segregated from that in P. taeda. The AGL6-like genes clustered into one group (designated clade H), which consisted of two G. luofuense, two G. gnemon, one G. parvifolium, one W. mirabilis and two E. equisetina AGL6-like genes. Nevertheless, one E. equisetina DEF/GLO-like gene fell into clade I (BS = 88), which comprised one A. thaliana, one A. trichopoda and two O. sativa genes. Moreover, 18 G. luofuense, three G. gnemon and one E. equisetina TM8-like genes clustered into clade J, which was separate from the two A. trichopoda genes. However, another phylogenetic analysis (Fig. 4) placed the TM8-like genes in one large clade—namely clade K (BS = 77), which contained 19 G. gnemon, 17 G. luofuense and one E. equisetina genes—and a small clade—designated clade L (BS = 100), which consisted of two G. gnemon and one G. luofuense TM8-like genes; these results support the splitting of TM8-like genes in the Gnetales.
Gene Expression and Tandem Duplication
The further comparisons of expression profiles among the detected MIKCc genes lead to better understand their potential roles in regulating reproductive and vegetative organs in G. luofuense. The expression profiles of the 35 G. luofuense MIKCC genes and their clustering based on RPKM (reads per kilobase per million mapped reads) values are summarized in Fig. 5a. Except for one AGL12-like gene, the G. luofuense MIKCC genes clustered into three groups. Group I contained one AGL6-like, one AG-like, two AGL15-like, one GpMADS4-like and two TM8-like genes that were exclusively and strongly expressed in reproductive tissues. We also found four DEF/GLO-like genes that were exclusively and highly expressed in male strobili. Group II had one AGL6-like, one SQUA-like, one GpMADS4-like, one B-sister-like, one GGM19-like and seven TM8-like genes that were expressed at high levels (RPKM values>12) in reproductive tissues but at low levels (RPKM values<12) in vegetative tissues. Finally, Group III contained one TM3-like, one StMADS11-like, and nine TM8-like genes that were all ubiquitously and strongly expressed in vegetative and reproductive organs (i.e. RPKM values>12). In addition, the AGL12-like genes were strongly expressed in both roots and male strobili. In addition, the assessment of tandem duplications is helpful of understanding the diversity of MADS-box genes in G. luofuense. Tandem duplications were identified among Type I and Type II MIKC members (Fig. 5b). Taken together, these data suggest at least one tandem duplication event that generated Mα-like and DEF/GLO-like genes but five events gave rise to TM8-like genes in G. luofuense.

a Expression patterns of MIKCc genes from six different tissues of G. luofuense. b Tandem duplications of MADS-box genes identified in the assembled scaffolds of the G.luofuense genome
Discussion
Type I MADS-Box Genes in Gnetum
A total of 11 G. luofuense Type I MADS-box genes were identified, more than those found in other conifers (with P. taeda as an exception) but less than those found in angiosperms (Table 1). Among the Type I MADS-box genes identified in G. luofuense, seven were Mα genes, three were Mβ genes and one was the Mγ gene (Fig. 2). The higher number of Mα than Mβ/Mγ genes was likely the result of tandem duplications (Fig. 5b). Type I MADS-box genes participate in endosperm and embryo development (Colombo et al. 2008; Day et al. 2008; Wuest et al. 2010) and control post-zygotic compatibility in angiosperms (Walia et al. 2009). Moreover, the Mα TFs preferably form heterodimers with the Mβ and Mδ proteins, suggesting their essential roles in stabilizing higher-order heterodimer complexes (Immink et al. 2009; Masiero et al. 2011). Besides, TFs of Type I MADS-box genes posses simpler structure than those of Type II MADS-box genes, they may be generated and degraded relatively fast in seed plants (Nam et al. 2004). Compared to those in angiosperms, less is known about the functions of gymnosperms Type I MADS-box genes. In general, Mα is expressed in various shoot tissues, whereas Mβ/Mδ expressions were detected in the buds, male cones, and embryos of conifer species (Gramzow et al. 2014). Further studies are required to investigate the functions of Type I MADS-box genes in Gnetum and the remaining gnetalean genera.
The MIKC* Genes in G. luofuense
Our results well illustrated that the three G. luofuense MIKC* genes could be subdivided into two clades (Fig. 2). This result is consistent with the phylogeny of MIKC* genes, which identified two major clades—the S- and P-clades (Gramzow et al. 2014). The MIKC* transcription factors also play an important role in the gametophytic and sporophytic development of bryophytes (Kwantes et al. 2012; Zobell et al. 2010). In conifers, P-clade MIKC* TFs have been shown to be broadly expressed in both female and male reproductive organs, whereas those in the S-clade are typically expressed exclusively in male reproductive organs (Gramzow et al. 2014). Thus, Type II MIKC* TFs may have a broader regulatory role in gymnosperm species but mainly regulate male gametophyte development, such as pollen maturation and pollen tube growth in angiosperms (Adamczyk and Fernandez 2009; Gramzow et al. 2014; Kwantes et al. 2012). More efforts are needed to survey the diversity and functions of MIKC* genes in the Gnetales.
Diversity of the MIKCC Genes in G. luofuense and E. equisetina
The total number of Type II MADS-box genes in G. luofuense was 38, including three MIKC* and 35 MIKCc genes (Fig. 3). The number of G. luofuense MIKC* genes was different from that of G. gnemon, whereas the latter is consistent with the published data (Gramzow et al. 2014) (Table 1). Moreover, 14 E. equisetina MIKCc genes were identified based on the transcriptome data of stem; this result is consistent with a previous study in which 14–16 Type II MADS-box genes were proposed in the most recent common ancestor of gymnosperms (Gramzow et al. 2014). In addition, E. equisetina and G. luofuense had ten and 12 MIKCc members, accounting for 62.5% and 75% of all 14 identified members, respectively. The diversity of the MIKCC genes in the Gnetales corroborates the ancestry of MADS-box genes prior to the diversification of land plants (Alvarez-Buylla et al. 2000; Gramzow and Theissen 2010). Further studies, especially those on the Type I and Type II MADS-box genes in Ephedra and Welwitschia, are required to further assess the diversity of the MIKCC genes in the Gnetales.
In Gnetum, we identified more Type II genes than Type I MADS-box genes. The scenario is consistent with the comparisons between the two type genes observed in several conifer species (Table 1). By contrast, more Type I than Type II MADS-box genes were observed in several angiosperms such as A. thaliana, Capsella rubella and Solanum tuberosum (Table 1). The relatively higher number of Type II MADS-box genes present in G. luofuense was presumably owing to the large genome size of gymnosperms like conifers (Gramzow et al. 2014). It is also possible that tandem duplications of MIKCC genes occurred more frequently in gymnosperms, which was reflected by the expansions commonly seen in StMADS11-like, TM3-like, and TM8-like groups (Gramzow et al. 2014). In the present study, DEF/GLO-like and TM8-like genes constituted the majority (62.6%) of MIKCC genes found in G. luofuense. At least one and five tandem duplication events in the DEF/GLO-like and TM8-like subgroups, respectively, were detected (Fig. 5b), suggesting the major contribution of tandem duplication to the expansion of these two subgroups.
Phylogenies of Gnetales MIKCC Genes
The two phenogenetic analyses of MIKCC genes yielded different results and the TM8-like genes had poor statistical support (Figs. 3 and 4), leading to confusions in the phylogenetic analysis of MIKCC genes based on extensive sampling of seed plants. Nevertheless, some valuable information can be inferred from these phylogenetic analyses. First, the MIKCC genes from gymnosperms and angiosperms could be clearly separated, which is consistent with previous studies (Becker et al. 2000; Carlsbecker et al. 2013; Chen et al. 2017; Gramzow et al. 2014; Melzer et al. 2010; Winter et al. 1999). In addition, the Gnetales MIKCC genes are likely closely related to those found in gymnosperms (Fig. 3). Thus, the MIKCC genes either form sister groups in subgroups (e.g. TM3, SQUA and AGL12), or constitute paraphyletic groups in subgroups (e.g. StMADS11 and AGL6) between Gnetales and other gymnosperms (Fig. 3). Here, the delimitation of MIKCC genes between gymnosperms and angiosperms is consistent with that reported in previous studies (Moyroud et al. 2017; Shindo et al. 1999; Winter et al. 1999).
We also assessed the homology of MIKCC genes from Gnetales to understand the evolution of MADS-box genes in seed plants. According to a previous study, the AGL6-like genes e.g. GGM11 from G. gnemon and WelAGL6 from W. mirabilis are homologous (Moyroud et al. 2017), which is consistent with that observed in this study. However, our results did not support the homology of MIKCC genes within Gnetales—we identified one AG-like gene, one AGL12-like gene, and two AGL15-like genes that were Gnetum-specific (Fig. 3). Moreover, some MIKCc genes in E. equisetina, G. luofuense, G. gnemon and W. mirabilis did not fully cluster into one monophyletic group, as we observed among the DEF/GLO-like, StMADS11-like and B-sister-like genes. Furthermore, the phylogenetic analysis based on extensive sampling of gymnosperms placed G. luofuense and G. gnemon TM8-like genes into separate clades (namely clades K and M), although this result had low statistic support (Fig. 4). Taken together, these results suggest that MIKCC group genes in the Gnetales seemingly have multiple origins, and more studies are required to validate such a statement.
Transcript Profiles of the MIKCC Genes in G. luofuense
In previous studies, the transcript profiles of G. gnemon and G. parvifolium MIKCC genes were only tested in leaves, female strobili and male strobili (Becker et al. 2000; Shindo et al. 1999; Winter et al. 1999). In the present study, we examined the expression of 35 MIKCC genes in various vegetative and reproductive tissues in G. luofuense. The cluster analysis based on gene expression data classified the MIKCC genes (except for AGL12) into three major groups (Fig. 5a), which exhibits slight differences from the results of previous tissue-specific gene expression analyses in other Gnetum species. For an example, two DEF/GLO-like (B-class) genes TnS000843651t08 and TnS000077603t01 were both specifically and strongly expressed in the male strobili of G. luofuense. An ortholog of the former i.e. GGM2 was ubiquitously expressed in the male strobili of G. gnemon, whereas an ortholog of the later, i.e. GGM15 was weakly expressed (Becker et al. 2000; Winter et al. 1999), probably because GGM15 was restrictedly expressed to the antherophores (Becker et al. 2003; Winter et al. 1999). In W. mirabilis, the expression levels of two class B genes, WelAPs/PI-1 and WelAPs/PI-2, were high in male strobili in early developmental stages but low in late developmental stages (Moyroud et al. 2017). This specific expression pattern of G. luofuense and W. mirabilis B genes in male reproductive tissues makes them reliable markers to distinguish between the male (where they are up-regulated) and female strobili (where they are down-regulated) in extant gymnosperms (Winter et al. 1999).
Besides the class B genes, one GGM3 from classes C and D, which is orthologous to the AG-like gene TnS000064931t01, was found to express at a high level in both female and male strobili in G. gnemon (Becker et al. 2003; Winter et al. 1999). GGM3 functions as a general promoter in theearlystages of nucellus, antherophores and female reproductive unit development in G. gnemon; while in later developmental stages, the expression of GGM3 is restricted to the outer envelopes of fertile and sterile ovules (Winter et al. 1999). In Cryptomeria japonica, the AGL6-like gene CjMADS14 is expressed in female and male strobili, suggesting its role in reproductive organ development (Katahata et al. 2014). Our results show that the G. luofuense genes TnS000229425t02 and TnS000222675t04—orthologs of the G. gnemon AGL6-like GGM9 and GGM11, respectively—were strongly expressed in both female and male strobili but not the vegetative reproductive organs (Fig. 5a). GGM9 was known to express throughout the sterile ovule primordium during early development but at a low level in sporogenic tissues and the antherophores in latter developmental stages (Becker et al. 2003; Winter et al. 1999). By contrast, GGM11 was expressed in the upper envelopes surrounding sterile ovules, and its expression increased with the development of sterile reproductive units (Becker et al. 2000, 2003; Winter et al. 1999). Furthermore, transcription factors GGM2, GGM3, GGM9 and GGM11 were found to form a quartet complex that participates in the sex determination of G. gnemon (Wang et al. 2010). A recent study has revealed that genes in the B and C/D classes originated 339 and 332 Mya, respectively, before the emergence of gymnosperms that occurred 305 Mya (Shen et al. 2019). Besides, the AGL6 genes originated 296 Mya and shared the most common ancestor with genes from classes A and E (Shen et al. 2019).
The transcript profiles of several MIKCc genes were different between G. luofuense and other Gnetum species. For example, the expression levels of B-sister-like gene TnS000495759t01, SQUA-like gene TnS013912449t01 and GGM19-like gene TnS000640815t04 were high in both the female and male strobili of G. luofuense (Fig. 5a), but their G. gnemon orthologs in correspondence i.e. GGM13, GGM18, and GGM19 were all weakly expressed in the female and male strobili of G. gnemon (Becker et al. 2000). Another study reported that the expression levels of GpMADS1 (an ortholog of the TM8-like gene TnS013912549t01), GpMADS3 (an ortholog of the AGL6-like gene TnS000229425t02) and GpMADS4 (an ortholog of the GpMADS4-like gene TnS000967423t06) were low or moderate in the female strobili of G. parvifolium (Shindo et al. 1999), which is different from what we observed in G. luofuense. Furthermore, we found that one GpMADS4-like gene, TnS000967423t06 (an ortholog of GGM7), was strongly expressed in both the female and male strobili of G. luofuense. This finding disagrees with that reported in a previous study of G. gnemon, which showed that this gene was strongly expressed in female strobilibut weakly expressed in male strobili (Becker et al. 2000). GGM7 expression was similar throughout the entire involucre but was relatively high in sterile ovules and antherophores in G. gnemon; in later developmental stages, GGM7 expression weakened and was restricted to the base of antherophores (Becker et al. 2003).
The TM8-like genes constituted nearly half of the MIKCc genes we identified in G. luofuense; they were also found to have undergone more frequent tandem duplications than other G. luofuense MIKCc genes (Fig. 5b). In addition, some TM8-like genes were found to be broadly expressed in both vegetative and reproductive organs, while others were exclusively expressed in the female and male strobili of G. luofuense (Fig. 5a). Our results show that TnS013912549t01, an ortholog of G. parvifolium GGM8 and GpMADS1, was strongly expressed in the female and male strobili of G. luofuense, consistent with the expression pattern of GGM8 in G. gnemon (Becker et al. 2000), but remarkably different from that of G. parvifolium GpMADS1, which was found to involve in the differentiation of three envelopes of female reproductive units and the initiation of their nucellus (Shindo et al. 1999). In addition, the TM8-like genes were found to involve in the development of arils (seed coats) in Ginkgo biloba and Taxus baccata (Lovisetto et al. 2012). In angiosperms, the TM8-like genes have been found to be widely expressed in the leaves, roots and seedlings of tomato (Hileman et al. 2006). Since the results revealed that the TM8-like genes are widely expressed in various tissues of G. luofuense, further studies are required to investigate their roles in regulating the development of vegetative and reproductive organs.
Besides regulating the development of reproductive organs, the MIKCc genes might also participate in the development of vegetative organs in G. luofuense, but this, so far, has not been well-illustrated in previous studies. For example, we found that the G. luofuense AGL12-like gene (an ortholog of G. gnemon GGM10, TnS000393325t08) was weakly expressed in male strobili but strongly expressed in roots, consistent with previous studies showing that MIKCc genes regulate the development of primary root meristems in A. thaliana (Burgeff et al. 2002; Tapia-López et al. 2008). Another study reported that the AGL12-like gene OsMADS26 was strongly expressed in the shoots and roots of O. sativa in response to stresses (Lee et al. 2008). In addition, we identified a TM3-like gene TnS000222675t05 (an ortholog of G. gnemon GGM1) that was widely and strongly expressed in the stems and leaves of G. luofuense. Our results are in line with previous studies showing that G. gnemon GGM1 was expressed in leaves, male strobili and female strobili (Becker et al. 2000; Winter et al. 1999). Previous studies have reported strong TM3-like gene expressions in root meristems, shoot meristems and organ primordia in Eucalyptus globules (Decroocq et al. 1999) and Ipomoea batatas (Kim et al. 2005). Moreover, we found StMADS11-like gene TnS000069483t10 was strongly expressed in both vegetative and reproductive organs of G. luofuense, which is different from the results of previous studies where GGM12 (an ortholog of G. gnemon TnS000069483t10) was found to express exclusively in leaves and male strobili (Becker et al. 2000) of G. gnemon. The StMADS11-like genes were found to be responsible for regulating the development of vascular bundles in the leaf and stem of Solanum tuberosum (Carmona et al. 1998). More studies are required to resolve the complex regulatory mechanism of MIKCc genes in the development of vegetative organs in Gnetum.
Methods
Plant Materials
Plant materials used for RNA sequencing (RNA-seq) were collected from two mature plants and a young seedling (2–3 years old) of G. luofuense grown in the Fairy Lake Botanical Garden (SZBG), Shenzhen, Guangdong, China (N22°34′49″, E114°10′26″). Before tissue collection, unfertilized female strobili were bagged to avoid contamination from pollen grains. The stems, leaves, and female cones at anthesis were collected from a young female G. luofuense plant, and the root tissues were harvested from a 2–3 years old offspring of this plant (voucher: XHMMT01). The male strobili were collected from a male individual growing next to the female individual (voucher: XHMMT10). The female individual, whose nuclear genome was sequenced, was initially identified as “G. montanum” (Wan et al. 2018). This identification was, however, controversial given the taxonomic treatment (Hou et al. 2016)—G. luofuense are characterized by broad, oval seeds that are 20–25 mm in length and 13–17 mm in diameter, with fleshy seed coats covered by silver scales and seed bases that are contracted into a 2–5 mm seed stipe. By contrast, G. montanum usually have smaller cylindric ovoid and/orcylindric seeds that are 16–20 mm in length and 7–11 mm in diameter and features a pronounced seed stipe of 3–5 mm. Gnetum luofuense is endemic to China in Guangdong, Hainan, Jiangxi and Hong Kong, whereas G. montanum is widely distributed throughout China (in Guangxi, Guizhouand Yunnan), India (in Assam and Sikkim), Burma, Thailand and Vietnam. Thereafter, we named all harvested plant materials G. luofuense. In addition, we collected the stem tissues from a wild Ephedra equisetina plant growing in Qinghai, China (N38°35′24″, E105°32′24″). RNA sequencing was carried out by SZBG, Shenzhen, Guangdong, China.
RNA Sequencing
The roots, stems, leaves, male strobili, female strobili, and seeds of G. luofuense as well as the stem tissues of E. equisetina were harvested and used for RNA-seq. Five biological replicates were harvested for each tissue and pooled before RNA-seq. Total RNA was extracted using the TRIzolre agent (Invitrogen, USA) and DNase I (Promega, USA) was used to remove DNA contamination. Seven libraries for RNA-seq were constructed using the NEB Next Ultra™ RNA Library Prep Kit (NEB, USA) and were sequenced on an Illumina Hiseq™ 2000 platform (with a 100-bp read length) by Novogene Co., Ltd. (Beijing, China).
Sequence Retrieval
Transcripts from different G. luofuense tissues and E. equisetina stems were mapped to the nuclear genome of G. luofuense (Wan et al. 2018) using TopHat v2.0.13 and Cufflinks version 2.1.1 with split reading permitted (Trapnell et al. 2009, 2010). The genome data of A. trichopoda were downloaded from the following website Ensembl_ftp://ftp.ensemblgenomes.org/pub/plants/release-25/plants/ to obtain sequences of MIKCc genes. To identify the conserved domains in MADS-box genes, we searched the transcripts from G. luofuense and E. equisetina using two Pfam models—SRF (PF00319) and K-box (PF01486)—using the hidden Markov model (HMM) software package HMMER (v3.1b2, http://hmmer.org)(E-value<1e−5)(Albert et al. 2013; Finn et al. 2011). To ensure the accuracy of search results, we manually checked all candidate MADS-box genes that contain the MADS (M) and/or Keratin-like (K) domains using the NCBI conserved domain database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi?). Sequences of Type I and Type II MADS-box genes in G. luofuense and E. equisetina were deposited in the supplementary dataset. Moreover, the sequences of MADS-box genes (i.e. the MIKCc genes) from G. gnemon and G. parvifolium were obtained from Shindo et al. 1999 and Winter et al. 1999, respectively. The sequences of MADS-box genes from Picea abies and W. mirabilis were downloaded from Carlsbecker et al. 2013 and Moyroud et al. 2017, respectively. The sequences of MADS-box genes from A. thaliana and O. sativa were retrieved from The Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org/browse/genefamily/MADSlike.jsp) and a previous study (Arora et al. 2007), respectively. In addition, the sequences of TM8-like genes in Cephalotaxus harringtonia, Cycas elongate, Ginkgo biloba, G. gnemon, Picea abies, Picea sitchensis, Pinus banksiana, Pinus contorta, Pinus pinaster, Pinus taeda, Sciadopitys verticillata, Solanum lycopersicum and Wollemia nobilis were obtained from Gramzow et al. 2014.
Expression Profiling and Detection of Tandem Duplication Events
The numbers of aligned reads were counted for each gene and were normalized to RPKM. The hierarchical clustering of expression patterns among the tested genes was performed using untransformed RPKM values using Cluster v3.0 (De Hoon et al. 2002). The hierarchical parameter was set to ‘correlation with spearman rank’ to compute similarity. Results of the cluster analysis were displayed as a heat map using Java TreeView v1.0.4 (Saldanha 2004). To identify tandem duplications among the MADS-box genes from G. luofuense, we searched all MADS-box genes against the nuclear genome of G. luofuense (=G. montanum Wan et al. 2018) using BLASTp under the following thresholds: identity>60, e-value<3e-25, the length of searched protein>50 amino acids, and distance between two adjacent genes<500 kb (Hanada et al. 2008).
Phylogenetic Analyses of MADS-Box Genes
All candidate MADS-box genes were searched against the Pfam database and highly conserved amino acid sequences were retained for phylogenetic analyses. We produced multiple alignments for the conserved sequences of MADS-box genes using MUSCLE v3.8.31 (Edgar 2004) and obtained an alignment matrix for Type I and a super matrix for Type II MADS-box genes. Moreover, a specific alignment of the TM8-like genes was also generated based on extensive sampling of G. gnemon, E. equisetina and other gymnosperms. RAxML-HPC2 v8.2.12 (Stamatakis 2014) implemented in web service CIPRES Gateway v.3.3 (www.phylo.org) was used to construct three maximum likelihood (ML) trees for Type I, Type II and TM8-like genes. Prior to the phylogenetic analyses, the best fit model for amino acid replacement was determined using ProtTest v3.2 (Abascal et al. 2005) and the LG + Γ model was used for all alignments according to the values of AICc (corrected Akaike Information Criterion). We performed rapid bootstrapping to search for trees with the highest score; statistical support for the three ML trees was derived from 1000 pseudo-replicates of simulated bootstraps.
Abbreviations
- K-domain:
-
Keratin-like domains
- MEF:
-
myocyte enhancer factor
- ML:
-
maximum likelihood
- TF:
-
transcription factors
- SRF:
-
serum response factor
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Adamczyk BJ, Fernandez DE (2009) MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol 149:1713–1723
Albert VA, Barbazuk WB, Der JP, Leebens-Mack J, Ma H, Palmer JD, Rounsley S, Sankoff D, Schuster SC, Soltis DE (2013) The Amborella genome and the evolution of flowering plants. Science 342:1241089
Alvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, Ditta GS, De Pouplana LR, Martínez-Castilla L, Yanofsky MF (2000) An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci U S A 97:5328–5333
Arora R, Agarwal P, Ray S, Singh AK, Singh VP, Tyagi AK, Kapoor S (2007) MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8:242
Becker A, Winter KU, Meyer B, Saedler H, Theissen G (2000) MADS-box gene diversity in seed plants 300 million years ago. Mol Biol Evol 17:1425–1434
Becker A, Saedler H, Theissen G (2003) Distinct MADS-box gene expression patterns in the reproductive cones of the gymnosperm Gnetum gnemon. Dev Genes Evol 213:567–572
Burgeff C, Liljegren SJ, Tapia-López R, Yanofsky MF, Alvarez-Buylla ER (2002) MADS-box gene expression in lateral primordia, meristems and differentiated tissues of Arabidopsis thaliana roots. Planta 214:365–372
Carlsbecker A, Sundstrom JF, Englund M, Uddenberg D, Izquierdo L, Kvarnheden A, Vergara-Silva F, Engstrom P (2013) Molecular control of normal and acrocona mutant seed cone development in Norway spruce (Picea abies) and the evolution of conifer ovule-bearing organs. New Phytol 200:261–275
Carmona MJ, Ortega N, Garcia-Maroto F (1998) Isolation and molecular characterization of a new vegetative MADS-box gene from Solanum tuberosum L. Planta 207:181–188
Chen F, Zhang XT, Liu X, Zhang LS (2017) Evolutionary analysis of MIKCc-type MADS-box genes in gymnosperms and angiosperms. Front Plant Sci 8:895
Colombo M, Masiero S, Vanzulli S, Lardelli P, Kater MM, Colombo L (2008) AGL23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J 54:1037–1048
Crane PR (1985) Phylogenetic analysis of seed plants and the origin of angiosperms. Ann Missouri Bot Gard 72:716–793
Day RC, Herridge RP, Ambrose BA, Macknight RC (2008) Transcriptome analysis of proliferating Arabidopsis endosperm reveals biological implications for the control of syncytial division, cytokinin signaling, and gene expression regulation. Plant Physiol 148:1964–1984
De Hoon, M., S. Imoto, and S. Miyano, 2002. Cluster 3.0. Human genome center, University of Tokyo, Tokyo, Japan
Decroocq V, Zhu X, Kauffman M, Kyozuka J, Peacock WJ, Dennis ES, Llewellyn DJ (1999) A TM3-like MADS-box gene from Eucalyptus expressed in both vegetative and reproductive tissues. Gene 228:155–160
Doyle JA, Donoghue MJ (1986) Seed plant phylogeny and the origin of angiosperms: an experimental cladistic approach. Bot Rev 52:321–431
Duan, Weike, et al. "Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage)." Molecular genetics and genomics 290.1 (2015): 239-255
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Endress PK (1996) Structure and function of female and bisexual organ complexes in Gnetales. Int J Pl Sci 157:113–125
Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:29–37
Garcia-Maroto F, Carmona MJ, Garrido JA, Vilches-Ferron M, Rodriguez-Ruiz J, Alonso DL (2003) New roles for MADS-box genes in higher plants. Biol Plant 46:321–330
Gramzow L, Theissen G (2010) A hitchhiker's guide to the MADS world of plants. Genome Biol 11:214
Gramzow L, Weilandt L, Theissen G (2014) MADS goes genomic in conifers: towards determining the ancestral set of MADS-box genes in seed plants. Ann Bot 114:1407–1429
Hanada K, Zou C, Lehti-Shiu MD, Shinozaki K, Shiu S-H (2008) Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol 148:993–1003
Hileman LC, Sundstrom JF, Litt A, Chen MQ, Shumba T, Irish VF (2006) Molecular and phylogenetic analyses of the MADS-box gene family in tomato. Mol Biol Evol 23:2245–2258
Hou C, Humphreys AM, Thureborn O, Rydin C (2015) New insights into the evolutionary history of Gnetum (Gnetales). Taxon 64:239–253
Hou C, Wikström N, Strijk J, Rydin C (2016) Resolving phylogenetic relationships and species delimitations in closely related gymnosperms using high-throughput NGS, sanger sequencing and morphology. Plant Syst Evol 302:1345–1365
Immink RGH, Tonaco IAN, de Folter S, Shchennikova A, van Dijk ADJ, Busscher-Lange J, Borst JW, Angenent GC (2009) SEPALLATA3: the 'glue' for MADS box transcription factor complex formation. Genome Biol 10:R24
Jörgensen A, Rydin C (2015) Reproductive morphology in the Gnetum cuspidatum group (Gnetales) and its implications for pollination biology in the Gnetales. Plant Ecol Evol 148:387–396
Katahata SI, Futamura N, Igasaki T, Shinohara K (2014) Functional analysis of SOC1-like and AGL6-like MADS-box genes of the gymnosperm Cryptomeria japonica. Tree Genet Genomes 10:317–327
Kim S-H, Hamada T, Otani M, Shimada T (2005) Isolation and characterization of MADS box genes possibly related to root development in sweetpotato (Ipomoea batatas L. lam.). J Plant Biol 48:387–393
Kubitzki, K. (1990a) Gnetaceae. In: K. U. Kramer and P. S. Green (eds), The families and genera of vascular plants. Springer, Berlin, Heidelberg, Germany. pp383–386
Kubitzki, K. (1990b) General traits of the Gnetales. In: K. U. Kramer and P. S. Green (eds), The families and genera of vascular plants. Springer, Berlin, Heidelberg. pp378–379
Kwantes M, Liebsch D, Verelst W (2012) How MIKC* MADS-box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Mol Biol Evol 29:293–302
Lee S, Woo Y-M, Ryu S-I, Shin Y-D, Kim WT, Park KY, Lee I-J, An G (2008) Further characterization of a rice AGL12 group MADS-box gene, OsMADS26. Plant Physiol 147:156–168
Lovisetto A, Guzzo F, Tadiello A, Toffali K, Favretto A, Casadoro G (2012) Molecular analyses of MADS-box genes trace back to gymnosperms the invention of fleshy fruits. Mol Biol Evol 29:409–419
Markgraf F (1930) Monographie der Gattung Gnetum Ser. 3. Bull Jard Bot Buitenzorg 10:407–511
Masiero S, Colombo L, Grini PE, Schnittger A, Kater MM (2011) The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 23:865–872
Melzer R, Wang YQ, Theissen G (2010) The naked and the dead: the ABCs of gymnosperm reproduction and the origin of the angiosperm flower. Semin Cell Dev Biol 21:118–128
Moyroud E, Monniaux M, Thevenon E, Dumas R, Scutt CP, Frohlich MW, Parcy F (2017) A link between LEAFY and B-gene homologues in Welwitschia mirabilis sheds light on ancestral mechanisms prefiguring floral development. New Phytol 216:469–481
Nam J, Kim J, Lee S, An G, Ma H, Nei M (2004) Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc Natl Acad Sci U S A 101:1910–1915
Parenicova L, de Folter S, Kieffer M, Horner DS, Favalli C, Busscher J, Cook HE, Ingram RM, Kater MM, Davies B, Angenent GC, Colombo L (2003) Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15:1538–1551
Price RA (1996) Systematics of the Gnetales: a review of morphological and molecular evidence. Int J Pl Sci 157:40–49
Ran JH, Shen TT, Wang MM, Wang XQ (2018) Phylogenomics resolves the deep phylogeny of seed plants and indicates partial convergent or homoplastic evolution between Gnetales and angiosperms. P Roy Soc B-Biol Sci 285:20181012
Saldanha AJ (2004) Java Treeview-extensible visualization of microarray data. Bioinformatics 20:3246–3248
Shen G, Yang C-H, Shen C-Y, Huang K-S (2019) Origination and selection of ABCDE and AGL6 subfamily MADS-box genes in gymnosperms and angiosperms. Biol Res 52:25
Shindo S, Ito M, Ueda K, Kato M, Hasebe M (1999) Characterization of MADS genes in the gymnosperm Gnetum parvifolium and its implication on the evolution of reproductive organs in seed plants. Evol Dev 1:180–190
Shindo S, Sakakibara K, Sano R, Ueda K, Hasebe M (2001) Characterization of a FLORICAULA/LEAFY homologue of Gnetum parvifolium and its implications for the evolution of reproductive organs in seed plants. Int J Pl Sci 162:1199–1209
Smaczniak C, Immink RG, Angenent GC, Kaufmann K (2012) Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139:3081–3098
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313
Tapia-López R, García-Ponce B, Dubrovsky JG, Garay-Arroyo A, Pérez-Ruíz RV, Kim S-H, Acevedo F, Pelaz S, Alvarez-Buylla ER (2008) An AGAMOUS-related MADS-box gene, XAL1 (AGL12), regulates root meristem cell proliferation and flowering transition in Arabidopsis. Plant Physiol 146:1182–1192
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Walia H, Josefsson C, Dilkes B, Kirkbride R, Harada J, Comai L (2009) Dosage-dependent deregulation of an AGAMOUS-LIKE gene cluster contributes to interspecific incompatibility. Curr Biol 19:1128–1132
Wan T et al (2018) A genome for gnetophytes and early evolution of seed plants. Nat Plants 4:82–89
Wang YQ, Melzer R, Theissen G (2010) Molecular interactions of orthologues of floral homeotic proteins from the gymnosperm Gnetum gnemon provide a clue to the evolutionary origin of 'floral quartets'. Plant J 64:177–190
Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA (2014) Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci U S A 111:4859–4868
Winter KU, Becker A, Munster T, Kim JT, Saedler H, Theissen G (1999) MADS-box genes reveal that gnetophytes are more closely related to conifers than to flowering plants. Proc Natl Acad Sci U S A 96:7342–7347
Won H, Renner SS (2003) Horizontal gene transfer from flowering plants to Gnetum. Proc Natl Acad Sci U S A 100:10824–18029
Won H, Renner SS (2006) Dating dispersal and radiation in the gymnosperm Gnetum (Gnetales) - clock calibration when outgroup relationships are uncertain. Syst Biol 55:610–622
Wuest SE, Vijverberg K, Schmidt A, Weiss M, Gheyselinck J, Lohr M, Wellmer F, Rahnenfuhrer J, von Mering C, Grossniklaus U (2010) Arabidopsis female gametophyte gene expression map reveals similarities between plant and animal gametes. Curr Biol 20:506–512
Zhao T, Holmer R, de Bruijn S, Angenent GC, van den Burg HA, Schranz ME (2017) Phylogenomic synteny network analysis of MADS-box transcription factor genes reveals lineage-specific transpositions, ancient tandem duplications, and deep positional conservation. Plant Cell 29:1278–1292
Zobell O, Faigl W, Saedler H, Munster T (2010) MIKC* MADS-box proteins: conserved regulators of the gametophytic generation of land plants. Mol Biol Evol 27:1201–1211
Acknowledgments
We thank Dr. Zheng Li from the University of Arizona (USA) for his constructive comments on an early version of this manuscript. We also thank the two reviewers of this manuscript for the valuable comments. We also thank Dr. Min Yang and Dr. Yiying Liao from Fairy Lake Botanical Garden, Shenzhen & Chinese Academy of Science for providing valuable technical and analytical assistance.
Funding
This work was funded by the Scientific Project of Shenzhen Urban Administration (201519) and a Major Technical Research Project from the Innovation of Science and Technology Commission of Shenzhen (JSGG20140515164852417). Additional funding was provided by the Scientific Research Program of the Sino-Africa Joint Research Centre (SAJL201607).
Author information
Authors and Affiliations
Contributions
Author Contributions: Conceptualization, C.H. and T.W.; Funding acquisition, T.W.; Investigation, L.L. and Z.L.; Methodology, Z.L.; Project administration, T.W.; Resources, Y.S. and T.W.; Software, L.L. and Z.L.; Supervision, Y.S. and T.W.; Writing – original draft, C.H.; Writing – review & editing, C.H. and T.W. All authors contributed to the drafts and gave final approval for publication.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by: Marcelo C. Dornelas
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hou, C., Li, L., Liu, Z. et al. Diversity and Expression Patterns of MADS-Box Genes in Gnetum luofuense—Implications for Functional Diversity and Evolution. Tropical Plant Biol. 13, 36–49 (2020). https://doi.org/10.1007/s12042-019-09247-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-019-09247-x